

Abstract
One of the fundamental tasks in biology is to identify the functions of all proteins to reveal the primary machinery of a cell. Knowledge of the subcellular locations of proteins will provide key hints to reveal their functions and to understand the intricate pathways that regulate biological processes at the cellular level. Protein subcellular location prediction has been extensively studied in the past two decades. A lot of methods have been developed based on protein primary sequences as well as protein-protein interaction network. In this paper, we propose to use the protein-protein interaction network as an infrastructure to integrate existing sequence based predictors. When predicting the subcellular locations of a given protein, not only the protein itself, but also all its interacting partners were considered. Unlike existing methods, our method requires neither the comprehensive knowledge of the protein-protein interaction network nor the experimentally annotated subcellular locations of most proteins in the protein-protein interaction network. Besides, our method can be used as a framework to integrate multiple predictors. Our method achieved 56% on human proteome in absolute-true rate, which is higher than the state-of-the-art methods.
Introduction
A cell is deemed to be the most basic construction unit of every living creature on this planet. Every living cell is composed of even more basic components, which are known as the subcellular compartments or subcellular organelles [1]. In general, there are two types of subcellular organelles, the membrane bounded subcellular compartments and the non-membrane bounded subcellular structures. The membrane bounded subcellular compartments can be roughly considered as isolated spaces surrounded by bio-membranes. For example, the mitochondria, the cell nucleus and the chloroplasts in green plants are all membrane bounded subcellular organelles. Some other subcellular structures, such as the ribosomes, the cytoskeleton and the centriole, which are non-membrane bounded, may also be recognized as subcellular organelles [2]. All these subcellular structures form a large dynamic system within a cell. The proteins and other macromolecules are synthesized, transferred and activated for their function within this system [3].
Changes in protein location are associated with a host of genetic disorders [4]. For example, the STAT3 (Signal Transducer and Activator of Transcription 3) should be directed to the nucleus in normal cells, while inappropriate nuclear relocation of STAT3 promotes oncogenesis through abnormal cell cycle progression, angiogenesis, and invasion of tissue [5]. Another example was in Zellweger syndrome. The mis-location of some peroxisomal proteins leads to dysfunctional fatty acid oxidation [6]. A third example was in glioma. A recent study showed that the GFRA4 (GDNF Family Receptor Alpha 4) are mis-located in the glioma. The artificial redirection of GFRA4 to the correct target results in a dramatic decrease in proliferation of glioma cells [7]. Therefore, the knowledge of accurate protein subcellular locations is of fundamental importance to both the life science and the drug industry.
There are several experimental methods that can determine the protein subcellular locations. For example, in yeast, the subcellular location of proteins can be visualized systematically by fusion of each ORF (Open Reading Frame) to the gene encoding GFP (Green Fluorescent Protein), either through transposon mutagenesis or PCR (Polymerase Chain Reaction) tagging [8], [9]. This technology requires the analysis of images, where a fully automated procedure is still not readily available [10]. Moreover, this technology is hardly feasible in humans and other mammals. In these organisms, immunolabeling and cell fractionation followed by tandem mass spectrometry were commonly applied [11], [12].
Unfortunately, all these experiments are costly and time consuming [13]. With the progress of proteome projects of many organisms, the number of known protein sequences has increased exponentially in the last two decades [14]. Experimental annotation of protein subcellular locations is too slow to catch up with the increment of protein sequences. A huge information gap between the protein sequences and their annotations has been created. Moreover, this gap is becoming wider with each passing day. To bridge this gap, many computational methods have been developed in the past few years to predict the protein subcellular locations from the primary sequences. These sequence-based methods generally fall into two categories: the signaling peptides based methods and the pseudo-amino acid composition based methods.
According to the cell biology, the proteins are usually synthesized in the cytosol and are transported to other subcellular compartments either during or after the translation [15]. The targets of the transportation are determined by the signaling peptides, which are short peptides that mostly reside in the N-terminus of protein sequences [15]. In some cases, these signals can also reside in the other parts of protein sequences. For example, the PTS1 (Peroxisomal Targeting Signal 1) peptides, which direct proteins to peroxisome, reside in the C-terminus of protein sequences [16]. If the signaling peptides can be found in the protein sequences, they can be used to predict their subcellular locations. Many impressive achievements have been made by finding the signaling peptide on the protein sequences [17]–[21].
However, due to the limitation of protein sequencing technology, the accuracy of the N-terminus of a protein sequence is not ideal, which restricted the application of signaling peptides based methods [22]. On the other hand, the subcellular location of a protein actually provides a micro physicochemical environment that should be compatible with the average physicochemical properties of a proteins surface [23], which was found to be related to the amino acid composition of a protein sequence [24]. Therefore, a large number of efforts have been made to predict the protein subcellular locations by using the pseudo-amino acid composition, which can be recognized as a universal numerical representation of the entire protein sequence [25], [26]. Several recent reviews have summarized the representative studies of this kind [27]–[29].
Rather than the above two sequence-based categories of methods, a number of sequence-based meta-predictors have been developed in the last few years. These works focused on developing voting schemes to combine the results of existing sequence-based predictors. Liu et al. proposed a weighted and adaptive voting scheme to integrate the prediction results of twelve independent predictors [30]. Laurila and Vihinen proposed the PROLocalizer method to combine over a dozen predictors based on signaling peptides analysis [31]. Park et al. developed an LDA (Linear Discriminative Analysis) based voting scheme to combine thirteen predictors [32]. Lin et al. proposed a minimalist ensemble algorithm that combined four predictors [33]. Magnus et al. proposed a voting scheme to combine four predictors for predicting protein subcellular locations in gram-negative bacteria [34]. By making use of the prediction results of existing predictors, these methods are different to the traditional ensemble classifiers, which create every module classifier in the ensemble [35]–[39].
From the system biology point of view, the proteins within a cell do not work independently. They interact with different proteins under different conditions. Because the physical interactions between a couple of proteins actually implied that the physical distance between interacting proteins is very close, the interacting proteins tend to localize within the same subcellular compartments [40], [41]. Furthermore, some proteins that lack of proper signaling peptides may be directed to its destination by a piggy-back mechanism, in which the signaling peptide is contained by the interacting partners of the protein instead of the protein itself [42], [43]. These facts implied that the protein-protein interaction information should be useful in predicting protein subcellular locations.
Several methods have been developed based on the protein-protein interactions in predicting protein subcellular locations. Scott et al. integrated protein-protein interaction as a module in their PSLT2 method to analyze the subcellular location in proteome-wide in yeast [44]. Lee et al. hybridized a group of network based features with pseudo-amino acid compositions in predicting protein subcellular locations [45]. Shin et al. developed a method to predict the protein subcellular locations from its interacting partners [46]. Mintz-Oron et al. used metabolic networks for enzyme localization prediction using constraint-based models [47]. Kumar and Ranganathan used statistical tests to analyze whether the interacting proteins would co-localize in both protein-protein interaction network and the metabolite-linked protein interaction network [48]. Jiang and Wu compared the performances of several different methods using protein-protein interaction networks and developed an ensemble classifier that can better identify subcellular locations on yeast protein-protein interaction network [49]. Mondal and Hu proposed the NetLoc method that can predict protein subcellular locations using four different types of protein networks [50]. In [45], [49], statistical inference based methods were employed to define a parameter for every interaction.
For almost all the existing methods, the protein-protein interaction network was used either as an independent predictor [44]–[46] or as a module classifier, whose results were further utilized in an ensemble [33]. Most of the existing studies require a complete and accurate protein-protein interaction network as the foundation of their method, and the experimental subcellular location annotations of most proteins in the protein-protein interaction network were usually required to improve the prediction performance.
In this paper, we propose to use the protein-protein interaction network as an infrastructure to integrate other sequence based predictors. The results of sequence-based predictors were combined on the protein-protein interaction network. When predicting the subcellular locations of a given protein, not only the protein itself, but also all its interacting partners were considered. These interacting partners were not treated equally, as a weight parameter was given to every interaction between the given protein and each partner. Unlike existing methods, our method does not require the comprehensive knowledge of the protein-protein interaction network. Moreover, given a protein, our method does not require the experimentally annotated subcellular locations of all its neighbors to predict its subcellular locations.
These characteristics of our method makes it possible to work on an incomplete and inaccurate protein-protein interaction network with only limited number of proteins annotated with subcellular locations. Besides, our method can be used as a framework to integrate multiple predictors. We demonstrate that, in human proteome, even with only one sequence based predictor, our method can improve its prediction performance with the help of protein-protein interaction data. Therefore, we can expect that our method can improve the prediction performance of most existing sequence-based predictors.
Materials and Methods
1 Protein-protein interactions dataset
The protein-protein interaction data were retrieved from BioGRID database version 3.2.96. The following filtering steps were carried out. (1) Only interactions between two human proteins were kept. (2) The interactions between two identical proteins were removed. (3) If some interactions appeared more than once in the dataset, only one interaction was kept. (4) The non-physical interactions were removed. After these filtering steps, there were 96967 interactions covering 13942 proteins remaining in the dataset. The dataset of this study can be obtained from the authors by email.
2 Experimental subcellular location annotations
The 13942 proteins were mapped to the UniProt database version 2013_07. These proteins in the protein-protein interaction network can be mapped to 18036 proteins in the UniProt database. The subcellular location annotations of these 18036 proteins were collected. If an annotation was marked as “Probable”, “By Similarity” or “Potential”, this annotation was discarded. The remaining subcellular location annotations were mapped to 11 different terms, including “Cell membrane”, “Cytoplasm”, “ER”, “Extracell”, “Golgi”, “Mitochondrion”, “Nucleus”, “Peroxisome”, “Lysosome”, “Endosome” and “Microsome”. The mapping was carried out following the keyword searching strategy as the state-of-the-art studies [51], [52]. Any subcellular location annotations that cannot be mapped to these terms were discarded.
To avoid ambiguous descriptions, we termed the 13942 proteins as the “BioGRID proteins” and the 18036 proteins as the “UniProt proteins”. If a BioGRID protein can be mapped to one or more than one UniProt proteins, the subcellular location of this BioGRID protein is the collection of all subcellular locations of all mapped UniProt proteins. Otherwise, the subcellular location of this BioGRID protein is “Unknown”. There were 6951 BioGRID proteins that can be annotated from the above procedure. Among 6951 proteins, there were 4879 proteins with only one subcellular location, 1709 proteins with two locations, 286 proteins with three locations, 50 proteins with four locations, 24 proteins with five locations and 3 proteins with six locations. According to the locative protein concept [52], this created 9493 locative proteins. The average multiplicity degree of the dataset was 1.37 [53]. The breakdown of the dataset in both the subcellular location multiplicity and the subcellular locations types can be found in Figure 1. According to these data, only about half proteins in the protein-protein interaction network have experimentally annotated subcellular locations. We used the results of sequence based predictors as the complementary and enhancements to the experimental annotations.
Figure 1. The summary of dataset.

(A) The number of locative proteins in different subcellular locations. There are 6951 proteins with experimentally annotated subcellular locations in the dataset. Because one protein may have more than one subcellular location, the number of locative proteins is 9493. (B) The number of proteins with different number of subcellular locations.
3 Sequence based subcellular location predictions
There are a number of existing methods that can predict protein subcellular locations based on primary sequences. We find that the Hum-mPLoc 2.0 [36] and Y-Loc [54] predictors provided the most convenient and reliable services that are suitable for our work. Therefore, two sequence based predictors were integrated in this work, Hum-mPLoc 2.0 [36] and Y-Loc [54]. According to Lin et al. [33], the integrated predictors should cover as many types of features as possible to improve the prediction performance in a meta-predictor [33]. These two predictors covered the pseudo-amino acid compositions, gene ontology annotations, evolutionary features and the signaling peptides features. Both predictors provided prediction results on the UniProt proteins, regardless to whether they have been experimentally annotated in the database. The subcellular location predictions of the BioGRID proteins were generated based on the results of these two predictors, respectively, as if they provided experimental annotations. If a predictor did not provide any prediction result for a protein, the result of this protein was recorded as “Unknown”. As the subcellular location terms in the two predictors are not identical to the 11 subcellular locations in this study, their location terms were mapped to the 11 subcellular location terms according to the biological definitions and the UniProt-GOA mapping [55].
4 Edge clustering coefficients
As indicated by existing studies, the reason why physical protein-protein interactions can be used to predict protein subcellular locations is that the physical locations of two interacting proteins are very close, which make them tend to localize within the same subcellular organelle [45], [46]. However, given two interacting proteins, it is difficult to infer that whether two proteins would have common subcellular locations directly from the protein-protein interaction network without knowing the subcellular locations of either protein. Fortunately, we find that ECC (Edge Clustering Coefficient) can be used as an indicator of whether two interacting proteins tend to have common subcellular locations.
ECC, which was originally proposed in the analysis of social networks [56], was employed in this study as an indicator of whether two interacting proteins tend to have common subcellular locations. According to Wang et al. [57], ECC can be used to describe the importance of an protein-protein interaction, as well as how close two interacting proteins are [57]. It had achieved many success in identifying essential proteins and protein complexes [57]–[59]. The definition of ECC can be described as follows.
For an interaction between two proteins pi and pj, the ECC of this interaction can be defined as follows:
| (1) |
where zi,j is the number of triangles that actually involve the edge connecting pi and pj in the network, di and dj the degrees of protein pi and pj in the network, respectively. The denominator means the number of triangles in which the edge connecting pi and pj may possibly participate at most.
5 Network based meta-predictor
Before we describe our network based method, we define some notations as follows. Let G = (V, E) be a PPI network, where V is the set of n vertices and E is the set of edges. Each vertex represents a protein (i.e. V = {p1, p2, …, pn}) and an edge Ei,j indicates that protein pi and protein pj has a physical interaction.
For every protein pi∈V (i = 1, 2, …, n), there are m possible subcellular locations (i.e. L = {l1, l2, …, lm}). In the current study, m = 11. Every protein can have one or more subcellular locations. The set of experimentally determined subcellular location of pi was denoted as SCL(pi). The set of Hum-mPLoc 2.0 predicted subcellular locations of pi was denoted as MP(pi). The set of Y-Loc predicted subcellular locations of pi was denoted as YC(pi). They are all subsets of L (i.e. SCL(pi) ⊆ L, MP(pi) ⊆ L and YC(pi) ⊆ L). If the experimental subcellular location does not exists, SCL(pi) = ∅. If the Hum-mPLoc 2.0 or Y-Loc cannot provide subcellular locations for protein pi, MP(pi) = ∅ or YC(pi) = ∅.
We defined the set of proteins with subcellular location lk (k = 1, 2, …, m) in experimental annotations (Sk(SCL)), Hum-mPLoc 2.0 predictions (Sk(MP)) or Y-Loc predictions Sk(YC) as:
| (2) |
where src ∈ {SCL, MP, YC}.
With all the above definitions, we now describe the network based method for predicting protein subcellular locations. Given a protein, its subcellular locations will be predicted based on the experimental and predicted subcellular locations of its interacting partners as well as the interactions between them. The sequence-based predictors can also provide predictions directly to the given protein. Our method considered all the above information to make the final predictions.
For every protein pu∈V, which has no experimental subcellular locations, we use the following steps to predict its subcellular locations.
Step 1: we find its neighbors in the PPI network. The set of the neighbors was denoted as NE(pu). We calculate the probability that observing any member of Sk(src) in NE(pu) as follows:
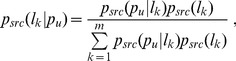 |
(3) |
where
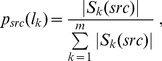 |
(4) |
| (5) |
src ∈ {SCL, MP, YC}, and |.| the cardinal of a set.
Step 2: the membership degree of a protein pu to a subcellular locations lk can be computed as
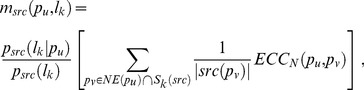 |
(6) |
where
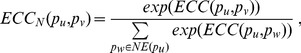 |
(7) |
exp(.) the exponential function, and src ∈ {SCL, MP, YC}. Here ECCN(pu, pv) ≠ ECCN(pv, pu), as we found that ECCN(pu, pv) can achieve better performance than ECC(pu, pv).
Step 3: for every src ∈ {SCL, MP, YC}, a set of subcellular locations can be predicted for protein pu, as follows:
| (8) |
where
| (9) |
NETsrc(pu) the set of predicted subcellular locations and C a parameter between 0 and 1.
In addition, if the following condition cannot be satisfied, we forced that NETsrc(pu) = ∅.
| (10) |
where θsrc is an integral parameter for every src∈{SCL, MP, YC}.
Step 4: the previous step provided three predictions: NETSCL(pu), NETMP(pu) and NETYC(pu). The sequence based predictors can also provide direct predictions on pu, as MP(pu) and YC(pu). We defined the following three sets SEQ(pu), NETSEQ(pu) and NET(pu):
| (11) |
| (12) |
| (13) |
The meaning of these definitions can be explained as follows. The SEQ(pu) is the prediction results from sequence information directly. The NETSEQ(pu) is the prediction results from network information with only the predicted subcellular location of the neighborhood proteins. The NET(pu) is also the prediction results from network but with both the predicted subcellular locations and the experimental subcellular locations of the neighborhood proteins.
For every lk∈L, lk belongs to the final predictions if and only if it satisfy either of the following two conditions: (1) lk∈NET(pu)∩SEQ(pu); (2) msrc(pu, lk) = uppersrc(pu, lk) for all src∈ {SCL, MP, YC}. This can be represented as follows:
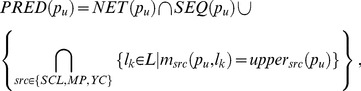 |
(14) |
where PRED(pu) is the final prediction results. When PRED(pu) = ∅, we used MP(pu) as PRED(pu), as Hum-mPLoc 2.0 performs better than Y-Loc.
6 Parameter calibrations
In eqn (8) and eqn (10), there are several parameters. We used a grid search method to find the best combination of these parameters. The parameter C in eqn (8) was searched from 0.5 to 0.95 with step 0.05. The parameter θSCL was searched from 1 to 20 with step 1. The parameter θMP and θYC was searched from 10 to 100 with step 10. Altogether 2000 trials were carried out. We finally achieved an optimized combination when C = 0.75, θSCL = 1 and θMP = θYC = 60.
7 Performance evaluations
Jackknife test has been widely used by many investigators to examine the quality of various predictors, as summarized in a recent review [27]. In this study, we also used jackknife test to evaluate the performance of our method. Because every protein may have one or more subcellular locations, using the traditional performance measures is difficult [53]. To measure the performance of a multi-label predictor, a set of statistical measures was established [53], [60], [61]. These statistical measures include aiming (AIM), coverage (CVR), accuracy (ACC), absolute-true rate (ATR) and absolute-false rate (AFR). They can be formulated as follows:
| (15) |
| (16) |
| (17) |
| (18) |
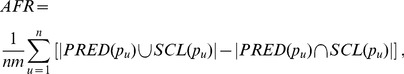 |
(19) |
where n and m are the total number of proteins and subcellular locations respectively, and
| (20) |
.
These measures can be interpreted briefly here. The AIM [53], which is also called “Precision”[60] or “Positive Predictive Value”[61], reflects the average ratio of correctly predicted subcellular locations over all predicted locations. The CVR [53], which is also termed as “Recall” [60] or “Sensitivity”[61], reflects the average ratio of the correctly predicted subcellular locations over the real locations. The ACC reflects the average ratio of correctly predicted subcellular locations over the total locations including the predicted and the real ones [53], [60], [61]. The ATR [53], [61], which is also called “Subset-accuracy”[60], reflects the ratio of proteins without either over-predicted locations or under-predicted locations. The AFR [53], which is also termed as “Hamming-Loss”[60], [61], is the average ratio of over-predicted locations and under-predicted locations over the total number of possible locations. Unlike the previous four measures, which are all the higher the better, a lower AFR value indicates a better prediction performance. A more comprehensive discussion of these measures can be found in several reviews [53], [60], [61].
The performance measures, which were utilized by Hum-mPLoc 2.0, were based on the locative protein concept [36]. However, according to a recent review [53], the prediction performance for each subcellular location based on locative protein concept is inconsistent with the ATR measure [53]. Therefore, we only use the multi-label performance measures, such as AIM, CVR and ATR, as the performance measures in our study.
Results and Discussion
1 Correlation between ECC and common subcellular locations
Given two protein pi and pj, which are connected by an edge Eij in the protein-protein interaction network, we defined the co-localization score (Qij) as follows:
| (21) |
where SCL, pi and pj have the same meanings as in the Method section. The numerator of eqn (21) is the number of subcellular locations that pi and pj both localized to. The denominator of eqn (21) is the number of subcellular locations that at least one of pi and pj localized to. When pi and pj have identical subcellular locations, Qij = 1. When pi and pj have no common subcellular location, Qij = 0. When pi and pj have some common subcellular locations, but not identical subcellular locations, the value of Qij indicates the fraction of the number of common subcellular locations over the total number of subcellular locations of pi and pj (i.e. 0 < Qij < 1). Therefore, Qij can indicate whether pi and pj tend to have the same subcellular locations. When pi or pj have no subcellular location annotations, Qij cannot be computed.
For every edge Eij that connects two proteins with subcellular location annotations, a Qij, as well as an ECC(pi,pj), can be computed. We plotted the average Qij as a function of the average ECC(pi,pj) in different ranges, such as [0,0.1), [0.1,0.2), …, [0.9,1.0). As shown in Figure 2, when 0≤ECC(pi,pj)<0.1, the average Qij is about 0.41. When 0.1≤ECC(pi,pj)<0.2, the average Qij is about 0.48. The average Qij continues to increase along with the ECC. When 0.9≤ECC(pi,pj)<1, the average Qij reaches 0.77. When ECC(pi,pj) varies in different ranges, the linear correlation coefficient between the average Qij and the average ECC(pi,pj) is 0.96.
Figure 2. The relationship between ECC and co-localization scores.

For every pair of interacting proteins with experimentally annotated subcellular locations, the ECC of their interactions and the co-localization score were computed. These interactions were divided into ten groups according to their ECC values. The first group contained the interactions with ECC value between 0 and 0.1.The second group contained the interactions with ECC value between 0.1 and 0.2. The third group contained the interactions with ECC value between 0.2 and 0.3, and so forth. The average values of ECC and co-localization score were computed for every group. The horizontal axis of this figure is the average value of ECC. The vertical axis of this figure is the average value of co-localization score. Ten dots were plotted on this figure to represent the ten groups of interactions. A straight line was generated using simple linear regression method to represent the linear relationship between the average ECC and the average co-localization score.
With the above observations, it is reasonable to use ECC as an indicator to whether two interacting proteins tend to have common subcellular locations.
2 Performance analysis
The prediction performance of our method was estimated using jackknife test on our dataset. In order to compare prediction performances, the performance of Hum-mPLoc 2.0 and Y-Loc was also estimated on the same dataset.
As shown in Table 1, the ATR of our method achieves 56.0%, while the ATR of Hum-mPLoc 2.0 is 51.4% and Y-Loc 47.4%. The ACC of our method achieves 70.0%, while the ACC of Hum-mPLoc 2.0 is 67.1% and Y-Loc 59.8%. The only measure that our method is slightly lower than Hum-mPLoc 2.0 is the CVR. Our method is 74.9% in CVR and Hum-mPLoc 2.0 is 75.4%. Since the ATR is the most strict and harsh measure of a predictor that deals with multi-label data [53], [62], the prediction performance of our method is better than both integrated methods.
Table 1. Comparison of prediction performances.
3 Examples of better predictions
Here, we provide some examples that our method gives better predictions than Hum-mPLoc 2.0. Because when our method cannot make predictions based on network information, we used the Hum-mPLoc 2.0 predictions as the final results, it is important to look into the details that how our method use network information to improve the prediction results of Hum-mPLoc 2.0.
The first example is the BioGRID protein 107454. It can be mapped to UniProt protein P11802, which has two experimentally annotated subcellular locations, “Cytoplasm” and “Nucleus”, in the UniProt database. Hum-mPLoc 2.0 provided only one result “Nucleus” based on sequence information. Our method, based on the network information, supplied the “Cytoplasm” location.
The second example is the BioGRID protein 107479. It can be mapped to UniProt protein P49715, which has only one experimentally annotated subcellular location, “Nucleus”, in the UniProt database. Hum-mPLoc 2.0 provided two subcellular location predictions, including “Cytoplasm” and “Nucleus”. Our method, based on the network information, removed the “Cytoplasm” location.
The third example is the BioGRID protein 107693. It can be mapped to UniProt protein P25067, which has only one experimentally annotated subcellular location, “Extracell”, in the UniProt database. However, Hum-mPLoc 2.0 provided “Cytoplasm” as its prediction result. Our method, based on the network information, corrected this result to “Extracell”.
There are a number of examples like the above three that our method actually provided better predictions than Hum-mPLoc 2.0. These better predictions can be achieved by supplying the extra predictions, removing the redundant predictions or correcting the wrong predictions.
4 Improving the prediction performance of single sequence based predictor
In the above results, we integrated two sequence based predictors. Actually, our method can work with only one sequence based predictor. Without optimizing the parameters, we directly applied our method with only Y-Loc predictor. The ATR is 48.6%, which is higher than the 47.4% of Y-Loc predictor independently. Again, without optimizing parameters, we directly applied our method with only Hum-mPLoc 2.0. Our method can achieve 54.9% in ATR, which is also higher than the 51.4% of Hum-mPLoc 2.0 predictor. A comprehensive performance can be found in Table 2. These results imply that our method can be used as a common approach to improve most existing sequence based predictors.
Table 2. Performance improvements for every single predictor.
| Predictor | AIMa | CVRb | ACCc | ATRd | AFRe |
| Hum-mPLoc 2.0 | 75.7% | 75.4% | 67.1% | 51.4% | 7.4% |
| Hum-mPLoc 2.0 + PPIf | 79.1% | 72.0% | 68.4% | 54.9% | 6.8% |
| Y-Loc | 72.4% | 61.0% | 59.8% | 47.4% | 8.4% |
| Y-Loc + PPIg | 73.2% | 61.1% | 60.5% | 48.6% | 8.2% |
AIM is Aiming, as defined in eqn (15);
CVR is Coverage, as defined in eqn (16);
ACC is Accuracy, as defined in eqn (17);
ATR is Absolute-True-Rate, as defined in eqn (18);
AFR is Absolute-False-Rate, as defined in eqn (19);
These performance values were obtained without optimizing parameters. “+PPI” means using the current method with only Hum-mPLoc 2.0;
These performance values were obtained without optimizing parameters. “+PPI” means using the current method with only Y-Loc.
5 Iterative prediction
Given that our method can be applied on single sequence based predictor and can improve its performance. It is interesting to investigate a tricky case, whether our method can be iteratively applied to further improve the prediction performance, as once a sequence based predictor was integrated into our method, the whole predictor can be integrated again as if it is another sequence based predictor.
The iteration process was carried out as follows. In the first round of iteration, the method was identical to all the above, integrating two sequence based predictors. From the second round of iteration, the output from the last round would be used as the only integrated predictor in the method. Table 3 shows the prediction performance of the first four rounds. From the second round of iteration, the prediction performance would not change anymore. The performance in the second round of iteration is slightly better than the first round. This result is expected as the protein-protein interaction network should not be able to improve the prediction performance without a limitation. Because the iterative predictions actually use the information of indirect neighbors in the protein-protein interaction network, this result also implies that it is unnecessary to consider the indirect neighbors in our method.
Table 3. Performances of iterative prediction.
6 Some remarks on the current method
Now, let us have a global view of the framework. We have illustrated an information flow chart in Figure 3. The input of the whole framework is only the protein sequences. There are three phases of the whole process. In the first phase, several sequence-based predictors, like Y-Loc and Hum-mPLoc 2.0, make subcellular location predictions. In the second phase, these prediction results are collected and then annotated on a protein-protein interaction network. In the final phase, the annotated protein-protein interaction network is analyzed to produce the final prediction results. Our work focuses only on the second and the third phase, but there is no restriction of what kind of predictors are used in the first phase. Therefore, our method can be used as a wrapper to combine and enhance every existing sequence-based predictor without modifying the predictors themselves. This is the main advantage of current framework.
Figure 3. The information flow chart of the whole framework.

The input of the framework is only the protein sequences. There are three phases in the whole process. (A) In the first phase, several existing sequence-based predictors give prediction results using only protein sequences. In the current study, these sequence-based predictors include the Y-Loc predictor and the Hum-mPLoc 2.0 predictor. The number n is 2. (B) In the second phase, the prediction results of the first phase were collected and then annotated on a protein-protein interaction network. (C) In the third phase, the annotated protein-protein interaction network was analyzed and the network-based prediction results were generated.
Although we only treat these sequence-based predictors as black-boxes, which require only protein sequences as input and give subcellular locations as output, we need to remind the readers that some characters of these sequence-based predictors should not be ignored. Some existing sequence-based predictors may use the input protein sequences to generate or to derive other types of features by querying the public databases. For example, the GO annotations, which were generated covertly in some predictors, may cause some potential bias in the results.
In the second phase, there is a problem regarding the protein-protein interaction network. It is well known that the protein subcellular localizations were used as an approach to detect the protein-protein interactions. Therefore, from a view of machine learning, directly using these interactions may cause over-fitting problem. However, this problem does not exist in the current study. Among 96967 protein-protein interactions, there are only 255 interactions that are supported solely by co-localization experiments. These interactions make less than 0.3% of the whole dataset. After manually removing these interactions, there is no observable difference in most of the performance measures. The only observable difference is that the coverage (CVR) increased from 74.9% to 75.0%. Therefore, the risk of over-fitting in the current study can be eliminated.
The final thought regarding this framework is how to characterize the protein-protein interactions. Theoretically, there should be infinite number of measures that could characterize a protein-protein interaction with a number. However, in the current framework, a feasible measure must satisfy the following conditions: (1) it can be calculated solely from the network, as the other types of knowledge may be inconsistent or unavailable to the protein-protein interaction network; (2) it must be highly correlated with the probability that the interacting proteins share subcellular locations; (3) the first two conditions must be satisfied even only incomprehensive and inaccurate protein-protein interaction networks are available, as the knowledge of protein-protein interaction network is still not comprehensive or accurate. As far as we know, the ECC is the only choice we have for this task. Therefore, we believe that, to some extent, ECC is an optimal choice in characterizing the protein-protein interactions in this study.
Conclusions
In this paper, we proposed a method that can predict protein subcellular locations using protein-protein interaction network as well as the results of existing sequence based predictors. Unlike existing method using a voting scheme to integrate other existing predictors or the other PPI network based methods, the protein-protein interaction network does not provide predictions results all by its own, it works as an infrastructure to coordinate the prediction results of interacting proteins from sequence based predictors.
We applied our method in the human proteome and protein-protein interaction networks. The results shows that our method can improve the sequence based predictions. Since our method can integrate any number of sequence based predictors, this method could serve as a common approach to combine the results of existing methods and improve the prediction performance of almost every existing sequence based methods.
Acknowledgments
We would like to thank Prof. Jianxin Wang for helpful discussions on applying edge clustering coefficients. We would also like to thank Hong Kong Scholar Program for providing collaboration opportunities for the authors.
Funding Statement
This work was supported by National Science Foundation of China (NSFC 61005041), a grant from the Research Grants Council of the Hong Kong Special Administrative Region, China (CityU 122511), Specialized Research Fund for the Doctoral Program of Higher Education (SRFDP 20100032120039), Tianjin Science Foundation (No. 12JCQNJC02300), China Postdoctoral Science Foundation (2012T50240 and 2013M530114) and the Seed Foundation of Tianjin University (No. 60302006 and 60302024). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Reece JB, Campbell NA (2011) Campbell biology/Jane B. Reece... [et al.]. Boston: Benjamin Cummings: imprint of Pearson.
- 2.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, et al.. (2007) Molecular Biology of the Cell. 5th ed. Garland Science. 1392 p. [Google Scholar]
- 3. Wickner W, Schekman R (2005) Protein Translocation Across Biological Membranes. Science 310: 1452–1456. [DOI] [PubMed] [Google Scholar]
- 4. Munkres KD, Benveniste K, Gorski J, Zuiches CA (1970) Genetically induced subcellular mislocation of Neurospora mitochondrial malate dehydrogenase. Proc Natl Acad Sci USA 67: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reich NC, Liu L (2006) Tracking STAT nuclear traffic. Nat Rev Immunol 6: 602–612. [DOI] [PubMed] [Google Scholar]
- 6. Dodt G, Braverman N, Wong C, Moser A, Moser HW, et al. (1995) Mutations in the PTS1 receptor gene, PXR1, define complementation group 2 of the peroxisome biogenesis disorders. Nat Genet 9: 115–125. [DOI] [PubMed] [Google Scholar]
- 7. Lee K, Byun K, Hong W, Chuang H-Y, Pack C-G, et al. (2013) Proteome-wide discovery of mislocated proteins in cancer. Genome Res 23: 1283–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, et al. (1999) Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature 402: 413–418. [DOI] [PubMed] [Google Scholar]
- 9. Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, et al. (2003) Global analysis of protein localization in budding yeast. Nature 425: 686–691. [DOI] [PubMed] [Google Scholar]
- 10.Coelho LP, Kangas JD, Naik AW, Osuna-Highley E, Glory-Afshar E, et al.. (2013) Determining the subcellular location of new proteins from microscope images using local features. Bioinformatics: btt392. [DOI] [PMC free article] [PubMed]
- 11. Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, et al. (2010) Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 28: 1248–1250. [DOI] [PubMed] [Google Scholar]
- 12. Gilchrist A, Au CE, Hiding J, Bell AW, Fernandez-Rodriguez J, et al. (2006) Quantitative proteomics analysis of the secretory pathway. Cell 127: 1265–1281. [DOI] [PubMed] [Google Scholar]
- 13. Murphy RF, Boland MV, Velliste M (2000) Towards a systematics for protein subcelluar location: quantitative description of protein localization patterns and automated analysis of fluorescence microscope images. Proc Int Conf Intell Syst Mol Biol 8: 251–259. [PubMed] [Google Scholar]
- 14. The UniProt Consortium (2012) Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Research 41: D43–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Transfer of proteins across membranes. I. Presence of proteolytically processed, unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma (1975) J Cell Biol. 67: 835–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gould SG, Keller GA, Subramani S (1987) Identification of a peroxisomal targeting signal at the carboxy terminus of firefly luciferase. J Cell Biol 105: 2923–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2: 953–971. [DOI] [PubMed] [Google Scholar]
- 18. Emanuelsson O (2002) Predicting protein subcellular localisation from amino acid sequence information. Brief Bioinformatics 3: 361–376. [DOI] [PubMed] [Google Scholar]
- 19. Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786. [DOI] [PubMed] [Google Scholar]
- 20. Nakai K, Horton P (1999) PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci 24: 34–36. [DOI] [PubMed] [Google Scholar]
- 21. Imai K, Nakai K (2010) Prediction of subcellular locations of proteins: where to proceed? Proteomics 10: 3970–3983. [DOI] [PubMed] [Google Scholar]
- 22. Reinhardt A, Hubbard T (1998) Using neural networks for prediction of the subcellular location of proteins. Nucleic Acids Res 26: 2230–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cedano J, Aloy P, Pérez-Pons JA, Querol E (1997) Relation between amino acid composition and cellular location of proteins. J Mol Biol 266: 594–600. [DOI] [PubMed] [Google Scholar]
- 24. Andrade MA, O’Donoghue SI, Rost B (1998) Adaptation of protein surfaces to subcellular location. J Mol Biol 276: 517–525. [DOI] [PubMed] [Google Scholar]
- 25. Chou K-C, Shen H-B (2008) Cell-PLoc: a package of Web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc 3: 153–162. [DOI] [PubMed] [Google Scholar]
- 26. Casadio R, Martelli PL, Pierleoni A (2008) The prediction of protein subcellular localization from sequence: a shortcut to functional genome annotation. Brief Funct Genomic Proteomic 7: 63–73. [DOI] [PubMed] [Google Scholar]
- 27. Chou K-C (2011) Some remarks on protein attribute prediction and pseudo amino acid composition. J Theor Biol 273: 236–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chou K-C, Shen H-B (2007) Recent progress in protein subcellular location prediction. Anal Biochem 370: 1–16. [DOI] [PubMed] [Google Scholar]
- 29. Du P, Li T, Wang X (2011) Recent progress in predicting protein sub-subcellular locations. Expert Rev Proteomics 8: 391–404. [DOI] [PubMed] [Google Scholar]
- 30. Liu J, Kang S, Tang C, Ellis LBM, Li T (2007) Meta-prediction of protein subcellular localization with reduced voting. Nucleic Acids Res 35: e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Laurila K, Vihinen M (2011) PROlocalizer: integrated web service for protein subcellular localization prediction. Amino Acids 40: 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Park S, Yang J-S, Jang SK, Kim S (2009) Construction of functional interaction networks through consensus localization predictions of the human proteome. J Proteome Res 8: 3367–3376. [DOI] [PubMed] [Google Scholar]
- 33. Lin J-R, Mondal AM, Liu R, Hu J (2012) Minimalist ensemble algorithms for genome-wide protein localization prediction. BMC Bioinformatics 13: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Magnus M, Pawlowski M, Bujnicki JM (2012) MetaLocGramN: A meta-predictor of protein subcellular localization for Gram-negative bacteria. Biochim Biophys Acta 1824: 1425–1433. [DOI] [PubMed] [Google Scholar]
- 35. Li L, Zhang Y, Zou L, Li C, Yu B, et al. (2012) An ensemble classifier for eukaryotic protein subcellular location prediction using gene ontology categories and amino acid hydrophobicity. PLoS ONE 7: e31057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shen H-B, Chou K-C (2009) A top-down approach to enhance the power of predicting human protein subcellular localization: Hum-mPLoc 2.0. Anal Biochem 394: 269–274. [DOI] [PubMed] [Google Scholar]
- 37. Chou K-C, Wu Z-C, Xiao X (2012) iLoc-Hum: using the accumulation-label scale to predict subcellular locations of human proteins with both single and multiple sites. Mol Biosyst 8: 629–641. [DOI] [PubMed] [Google Scholar]
- 38. Wu Z-C, Xiao X, Chou K-C (2011) iLoc-Plant: a multi-label classifier for predicting the subcellular localization of plant proteins with both single and multiple sites. Mol Biosyst 7: 3287–3297. [DOI] [PubMed] [Google Scholar]
- 39. Wu Z-C, Xiao X, Chou K-C (2012) iLoc-Gpos: a multi-layer classifier for predicting the subcellular localization of singleplex and multiplex Gram-positive bacterial proteins. Protein Pept Lett 19: 4–14. [DOI] [PubMed] [Google Scholar]
- 40. Schwikowski B, Uetz P, Fields S (2000) A network of protein-protein interactions in yeast. Nat Biotechnol 18: 1257–1261. [DOI] [PubMed] [Google Scholar]
- 41. Gandhi TKB, Zhong J, Mathivanan S, Karthick L, Chandrika KN, et al. (2006) Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38: 285–293. [DOI] [PubMed] [Google Scholar]
- 42. Glover JR, Andrews DW, Rachubinski RA (1994) Saccharomyces cerevisiae peroxisomal thiolase is imported as a dimer. Proc Natl Acad Sci USA 91: 10541–10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cai CX, Linsenmayer TF (2001) Nuclear translocation of ferritin in corneal epithelial cells. J Cell Sci 114: 2327–2334. [DOI] [PubMed] [Google Scholar]
- 44. Scott MS, Calafell SJ, Thomas DY, Hallett MT (2005) Refining protein subcellular localization. PLoS Comput Biol 1: e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee K, Chuang H-Y, Beyer A, Sung M-K, Huh W-K, et al. (2008) Protein networks markedly improve prediction of subcellular localization in multiple eukaryotic species. Nucleic Acids Res 36: e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shin CJ, Wong S, Davis MJ, Ragan MA (2009) Protein-protein interaction as a predictor of subcellular location. BMC Syst Biol 3: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mintz-Oron S, Aharoni A, Ruppin E, Shlomi T (2009) Network-based prediction of metabolic enzymes’ subcellular localization. Bioinformatics 25: i247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kumar G, Ranganathan S (2010) Network analysis of human protein location. BMC Bioinformatics 11 Suppl 7S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jiang JQ, Wu M (2012) Predicting multiplex subcellular localization of proteins using protein-protein interaction network: a comparative study. BMC Bioinformatics 13 Suppl 10S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mondal AM, Hu J (2010) NetLoc: Network based protein localization prediction using protein-protein interaction and co-expression networks. 2010 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). pp. 142–148.
- 51. Park K-J, Kanehisa M (2003) Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics 19: 1656–1663. [DOI] [PubMed] [Google Scholar]
- 52. Chou K-C, Shen H-B (2006) Predicting Eukaryotic Protein Subcellular Location by Fusing Optimized Evidence-Theoretic K-Nearest Neighbor Classifiers. J Proteome Res 5: 1888–1897. [DOI] [PubMed] [Google Scholar]
- 53. Chou K-C (2013) Some remarks on predicting multi-label attributes in molecular biosystems. Mol Biosyst 9: 1092–1100. [DOI] [PubMed] [Google Scholar]
- 54. Briesemeister S, Rahnenführer J, Kohlbacher O (2010) YLoc--an interpretable web server for predicting subcellular localization. Nucleic Acids Res 38: W497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dimmer EC, Huntley RP, Alam-Faruque Y, Sawford T, O’Donovan C, et al. (2012) The UniProt-GO Annotation database in 2011. Nucleic Acids Res 40: D565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Radicchi F, Castellano C, Cecconi F, Loreto V, Parisi D (2004) Defining and identifying communities in networks. PNAS 101: 2658–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang J, Li M, Wang H, Pan Y (2012) Identification of essential proteins based on edge clustering coefficient. IEEE/ACM Trans Comput Biol Bioinform 9: 1070–1080. [DOI] [PubMed] [Google Scholar]
- 58. Peng W, Wang J, Wang W, Liu Q, Wu F-X, et al. (2012) Iteration method for predicting essential proteins based on orthology and protein-protein interaction networks. BMC Syst Biol 6: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang J, Peng X, Li M, Pan Y (2013) Construction and application of dynamic protein interaction network based on time course gene expression data. Proteomics 13: 301–312. [DOI] [PubMed] [Google Scholar]
- 60. Tsoumakas G, Katakis I (2007) Multi-Label Classification: An Overview. International Journal of Data Warehousing and Mining 3: 1–13. [Google Scholar]
- 61. Du P, Xu C (2013) Predicting multisite protein subcellular locations: progress and challenges. Expert Rev Proteomics 10: 227–237. [DOI] [PubMed] [Google Scholar]
- 62. Lin W-Z, Fang J-A, Xiao X, Chou K-C (2013) iLoc-Animal: a multi-label learning classifier for predicting subcellular localization of animal proteins. Mol Biosyst 9: 634–644. [DOI] [PubMed] [Google Scholar]