

Background: Pladienolide B is a complex natural product that potently inhibits pre-mRNA splicing.
Results: The same molecular features of pladienolide B are required for the drug's effects on cell growth, morphology, and splicing.
Conclusion: Simplified synthesis and modification of active pladienolide B is possible.
Significance: Pladienolide B analogs can be used to study the relationship between splicing and cancer cell function.
Keywords: Alternative Splicing, Drug Action, Enzyme Inhibitors, Nucleus, RNA Splicing
Abstract
Pladienolide B (PB) is a potent cancer cell growth inhibitor that targets the SF3B1 subunit of the spliceosome. There is considerable interest in the compound as a potential chemotherapeutic, as well as a tool to study SF3B1 function in splicing and cancer development. The molecular structure of PB, a bacterial natural product, contains a 12-member macrolide ring with an extended epoxide-containing side chain. Using a novel concise enantioselective synthesis, we created a series of PB structural analogs and the structurally related compound herboxidiene. We show that two methyl groups in the PB side chain, as well as a feature of the macrolide ring shared with herboxidiene, are required for splicing inhibition in vitro. Unexpectedly, we find that the epoxy group contributes only modestly to PB potency and is not absolutely necessary for activity. The orientations of at least two chiral centers off the macrolide ring have no effect on PB activity. Importantly, the ability of analogs to inhibit splicing in vitro directly correlated with their effects in a series of cellular assays. Those effects likely arise from inhibition of some, but not all, endogenous splicing events in cells, as previously reported for the structurally distinct SF3B1 inhibitor spliceostatin A. Together, our data support the idea that the impact of PB on cells is derived from its ability to impair the function of SF3B1 in splicing and also demonstrate that simplification of the PB scaffold is feasible.
Introduction
An essential step in expression of human genes is pre-mRNA splicing, the process by which intron sequences are removed from gene transcripts to create functional mRNA for protein translation. Splicing is facilitated and regulated by the spliceosome, a highly dynamic macromolecular complex that assembles de novo at each intron from five small nuclear ribonucleoproteins (snRNPs)3 and dozens of additional proteins. Increasing evidence connects mutations in components of the spliceosome to various types of cancer and points to the splicing machinery as a target for new anticancer drugs. A recent example is the core spliceosome component SF3B1 and myelodysplastic syndrome, a heterogeneous group of diseases caused by abnormal proliferation of hematopoietic stem cells. Whole exome sequencing showed that 75% of an myelodysplastic syndrome subtype had mutations in SF3B1, which cluster in a particular region of the protein (1–3). SF3B1 mutations are also present in cancers of several other tissues (4–6).
Interestingly, SF3B1 is also the target of pladienolide B (PB), a natural product with potent cytotoxicity and antitumor activity both in cancer cell lines and mouse xenograft models (7–9). In cell culture, mutation of a single amino acid in SF3B1 confers resistance to PB, making it likely that its cytotoxicity is directly related to SF3B1 function (9), although how these activities are connected to splicing inhibition is not known. Two other natural products, herboxidiene (GEX1A) and FR901464 (and the related molecules spliceostatin A (SSA) and meayamycin), also interact with SF3B1 and have similar cytotoxic effects (10–12). In vitro studies indicate that both PB and FR901464 analogs SSA and meayamycin interfere with the role of SF3B1 in stabilizing the addition of U2 snRNP to the spliceosome and identifying the intron branch point sequence (13–16). However, it has been difficult to use structure activity relationship studies to define the precise mechanism by which these compounds interact with SF3B1 because of their complex structures.
Novel concise enantioselective syntheses (17, 18) have now opened the door to systematic structure activity relationship studies for PB and herboxidiene. In this study, we identify structural features of PB that are responsible for inhibition of human in vitro splicing. We find that the same features are responsible for a wide ranging set of phenotypic effects in cells, eliminating the hypothesis that splicing inhibition and cellular phenotypes arise from different parts of the same molecule and further underscoring the link between inhibition of the spliceosome and the cellular response to PB. There are also several positions in the molecule that can be modified with no change in activity. Our data point toward more straightforward synthetic pathways and modifications in PB, which will be key to dissecting the function of its target SFB1 in the spliceosome and studying the relationship between splicing pathways and cancer cell growth. They may also lead to structurally less complex new PB analogs that are better tuned to selectively target cancer cell growth and increase the therapeutic potential of the drug.
EXPERIMENTAL PROCEDURES
Synthesis of PB Structural Analogs
The synthesis of PB structural analogs will be described elsewhere (see supplemental materials for NMR, FTIR, and mass spectrometry analysis of compounds).4
In Vitro Splicing Reactions
Pre-mRNA substrate was derived from the adenovirus major late transcript. A [32P]UTP body-labeled G(5′)ppp(5′)G-capped substrate was generated by T7 run-off transcription followed by gel purification. Nuclear extract was prepared as previously described (19) from HeLa cells grown in DMEM/F-12 1:1 and 5% (v/v) newborn calf serum. For splicing reactions, 10 nm pre-mRNA substrate was incubated with 60 mm potassium glutamate, 2 mm magnesium acetate, 2 mm ATP, 5 mm creatine phosphate, 0.05 mg ml−1 tRNA, and 50% (v/v) HeLa nuclear extract at 30 °C.
Denaturing Gel Analysis
RNA was extracted from in vitro splicing reaction and separated on a 15% (v/v) denaturing polyacrylamide gel. 32P-Labeled RNA species were visualized by phosphorimaging and quantified with ImageQuant software (Molecular Dynamics). Splicing efficiency is the amount of mRNA relative to total RNA and normalized to a dimethyl sulfoxide (DMSO) control reaction. IC50 values for inhibitors are the concentrations of inhibitor that cause 50% decrease of splicing efficiency, which were derived from averaged plots of splicing efficiency versus compound concentration from 3–6 independent assays.
Native Gel Analysis
Splicing reactions were set up as described above and incubated 30 °C for 4–30 min. Time point samples were kept on ice until all samples were ready for analysis. 10 μl of splicing reactions were mixed with 10 μl of native gel loading buffer (20 mm Trizma base, 20 mm glycine, 25% (v/v) glycerol, 0.1% (w/v) cyan blue, 0.1% (w/v) bromphenol blue, 1 mg ml−1 heparin sulfate) and incubated at room temperature for 5 min before loading onto a 2.1% (w/v) low melting temperature agarose gel. Gels were run at 72 V for 3.5 h, dried onto Whatman paper, and exposed to phosphorimaging screens, which were digitized with a Typhoon Scanner (Molecular Dynamics).
Cytological Profiling
For the cytological profiling data, HeLa cells were treated with 2 nm to 67 μm of PB, PB analogs, or herboxidiene for ∼20 h, followed by staining, automated imaging, and processing as described by Schulze et al. (22).
Immunostaining
1 × 105 HeLa cells were cultured with 0.01–1 μm of drug for 4 h in chamber slides, acetone-fixed, and blocked with 1% BSA. The cells were then DAPI-stained, incubated with anti-SC35 (1:500; BD Biosciences), and labeled with Alexa Fluor®568-conjugated anti-mouse IgG (1:500; Invitrogen). Images were acquired with a Leica EPI fluorescence microscope.
Relative speckle sizes were obtained in an unbiased manner by importing cell images into ImageJ and running a script that used the auto default threshold algorithm and “analyze particles” routine to select DAPI-stained nuclei in the blue channel. For each nucleus, the script then used the auto MaxEntropy threshold and “analyze particles” routine to select speckles in the red channel and calculate the speckle area. The distribution of speckle sizes from 20 to 40 nuclei from each condition was plotted with the Acula.com box plot generator and compares to DMSO control by a nonparametric Mann-Whitney U value test.
Monitoring Endogenous Splicing Changes by Semiquantitative RT-PCR
HeLa cells were treated for 4 h with DMSO or 0.01–1 μm drug. Total RNA was isolated from 1–2 × 106 HeLa cells with TRI reagent LS (Molecular Research Center, Inc.) according to the manufacturer's instructions. After DNase treatment, 1 μg of total RNA was reverse-transcribed by MMLV reverse transcriptase in a 20-μl reaction. 10-μl PCRs were performed with a reaction mixture containing 1 μl of the RT reaction, 200 μm dNTPs, 0.2 μm of each forward and reverse primer, ∼1 unit Taq polymerase, and 1× Taq buffer. PCR conditions were 2 min at 94 °C followed by 30 cycles of 30 s at 94 °C, 30 s at 55–60 °C, 1 min at 60 °C, and a final elongation of 10 min at 68 °C. PCR products were separated on a 6% native polyacrylamide gel, stained with ethidium bromide, imaged, and quantified using the Quantity One software (Bio-Rad).
Primer sequences are as follows: RBM5ex17: forward, 5′-CGGCTGTAGTGCCCAGAGT-3′, and reverse, 5′-TTGCGAGTTGGGTCATAAT-3′, 58 °C annealing temperature (13); CCNA2: forward, 5′-AACTTCAGCTTGTGGGCACT-3′, and reverse, 5′-AAAGGCAGCTCCAGCAATAA-3′, 60 °C annealing temperature; SF3A1: forward, 5′-CCAAATTCCAGGAACGTG-3′, and reverse, 5′-AGCTCCTCTGGCGTGGTG-3′, 55 °C annealing temperature (20); U6: forward, 5′-CGCTTCGGCAGCACATATAC-3′, and reverse, 5′-GAATTTGCGTGTCATCCTT-3′, 60 °C annealing temperature (21).
RESULTS
Synthesis of PB Structural Analogs
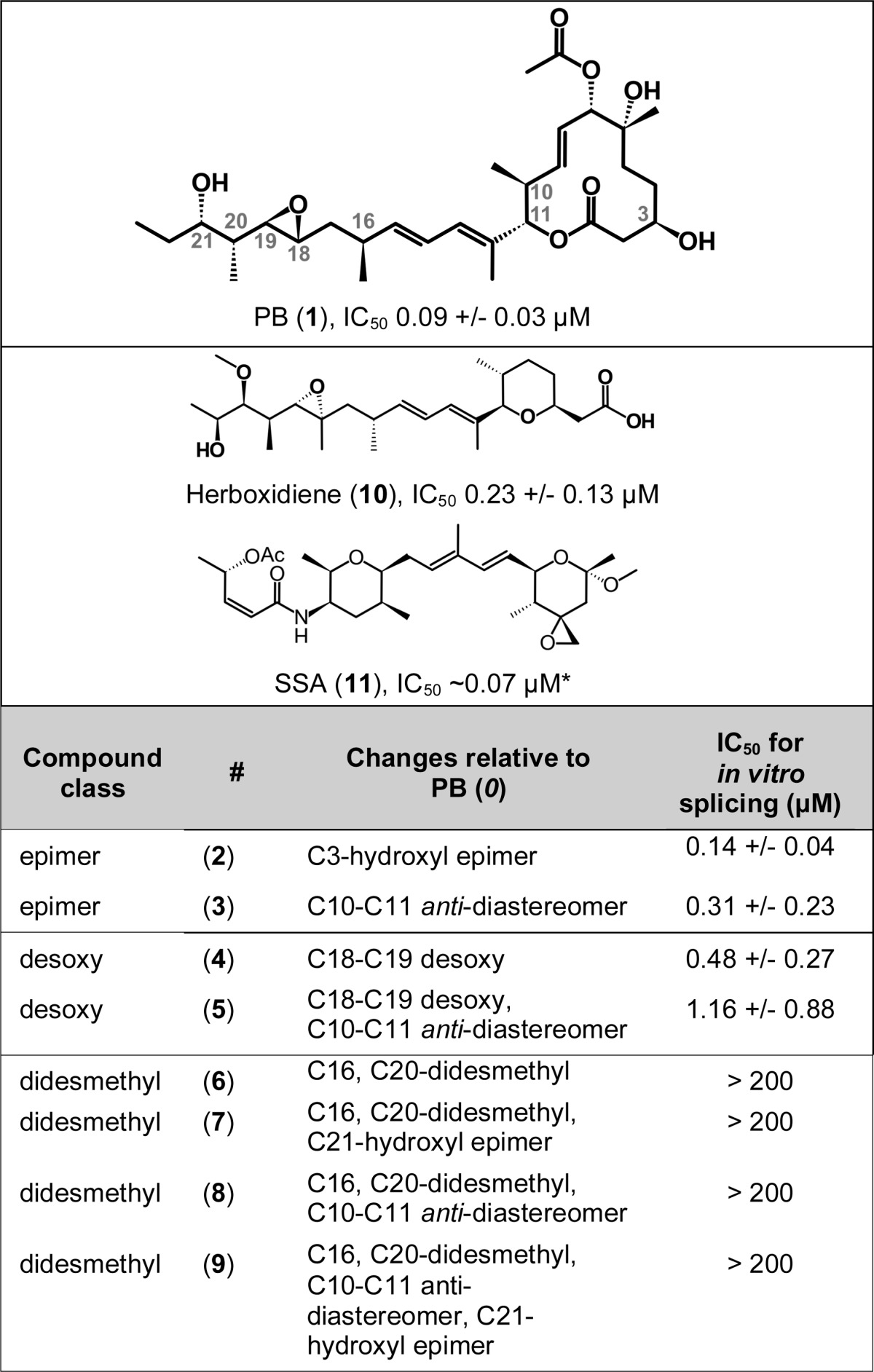
Although it is established that PB targets the spliceosomal core protein SF3B1 and inhibits pre-mRNA splicing, the structural elements of PB responsible for this biological activity are unknown. PB is structurally complex with multiple stereocenters, and the design of less complex PB structural variants is of interest for several reasons, including enabling the creation of probes for studying spliceosome function. To probe the structural elements of PB that are dispensable for splicing inhibition, we utilized our recent total synthesis of PB (1) (17) and herboxidiene (10) (17, 18) to synthesize a number of key analogs as depicted in Table 1 and in the supplemental materials. The details of the synthesis will be described elsewhere.4 We first synthesized C3-hydroxyl epimer (2) and C10-C11 anti-diastereomer (3). Furthermore, we synthesized PB analogs where specific functional groups were deleted from the side chain of PB. These analogs include preparation of C18-C19 desoxy PB (4), C18-C19 desoxy PB containing C10-C11 anti-diastereomer (5), C16, C20-didesmethyl PB (6), C16, C20-didesmethyl with C10-C11 anti-diastereomer PB (8), and their combination with C21-hydroxyl epimer (7, 9). All test compounds were purified by HPLC to >95% purity. Notably, the synthetic pathway for the desoxy compounds, which have a distinct retention time, would not allow for formation of epoxide.
TABLE 1.
Chemical structure of compounds tested in this study
IC50 refers to the concentration required to reduce in vitro splicing by half compared to DMSO control and is based on the averaged values of 3–6 individual experiments. *, The IC50 for SSA was obtained from Ref. 14.
Structural Requirements for PB Splicing Inhibition in Vitro
To test the effects of the PB analogs on splicing inhibition, we added the compounds to in vitro splicing reactions containing a synthetic pre-mRNA substrate, ATP, and nuclear extract from HeLa cells. The reaction is monitored by denaturing PAGE to separate the substrate and product mRNA, and splicing efficiency is quantified as the percentage of pre-mRNA converted to mRNA. In this system, DMSO alone has no effect on splicing (Fig. 1A, lane 1), whereas PB (1) inhibits splicing with an IC50 of 0.1 μm (Fig. 1, A, lanes 2–7, and B). Herboxidiene (10) also inhibits splicing in this system with the same IC50 (Fig. 1, B and C, lanes 29–34). The PB analogs showed different potencies, allowing us to classify several PB molecular features with respect to their contribution to in vitro splicing inhibition.
FIGURE 1.

Impact of PB analogs on in vitro splicing. A, denaturing gel analysis of RNA isolated from splicing reactions incubated with the indicated concentrations of dimethyl sulfoxide (DMSO), PB, PB analogs (epimer, desoxy, and didesmethyl), and herboxidiene (herb). Identities of bands are schematized to the left as (from top to bottom) lariat intermediate, pre-mRNA, mRNA, and 5′ exon intermediate. The free lariat is indicated by an asterisk. B, quantification of normalized splicing efficiency versus inhibitor concentration for the splicing reactions shown in A and C.
With all the C16-C20 didesmethyl compounds (6–9), absence of a pair of methyl groups flanking an epoxy moiety resulted in a loss of splicing inhibition, indicating that one or both methyl groups are key to splicing inhibition by PB (Fig. 1, C, lanes 1–28, and D). Removal of the epoxy group (desoxy 4, 5) reduces splicing inhibition by more than 5-fold, indicating that the epoxy group contributes to splicing inhibition by PB but is not absolutely required (Fig. 1, A, lanes 20–31, and B). Epimerization of the hydroxyl group at C3 in the ring (2), as well as of the C10-C11 linkage between the “arm” and macrolide ring (3), has no significant effect on splicing inhibition (Fig. 1, A, lanes 8–19, and B), which means that the conformation of these stereocenters does not contribute to splicing inhibition by PB.
In addition to assaying splicing chemistry, we also tested the effect of the compounds on spliceosome assembly. Spliceosome assemble on intron substrates via an ordered series of intermediate complexes. A subset of these complexes (H/E, A, B, and C) can be visualized by native gel analysis of the same in vitro splicing reactions described above. H/E and A complexes form as early intermediates that convert to B and then to C complex, at which point the splicing reaction is catalyzed. As before, DMSO alone has no effect, and spliceosomes assemble over time in the normal progression from H/E → A → B → C complex (Fig. 2A, lanes 2–5). With increasing concentrations of PB (1) (Fig. 2A, lanes 6–11) or herboxidiene (10) (Fig. 2B, lanes 29–34), spliceosome assembly appears to halt before B and C complex formation at a pre-mRNA-containing complex that migrates near the position of the early intermediate known as A complex. This A-like complex is indistinguishable from the one that forms in the presence of SSA under the same experimental conditions (14). Because the amount of the complex does not increase commensurate with the loss of higher order complexes, the results are consistent with a model in which drug treatment results with a less stable A complex that is not able to proceed to the next assembly step (13–15).
FIGURE 2.

Native gel analysis of spliceosome assembly. A and B, the results shown are from time course analysis of splicing reactions in HeLa nuclear extract (NE) in 1% DMSO or 30-min time points of splicing reactions with the indicated concentrations of PB, PB analogs (epimer, desoxy, and didesmethyl), and herboxidiene (herb). The identities of complexes are denoted with assembly occurring in the following order: H/E → A → B → C. DMSO, dimethyl sulfoxide.
With the PB analogs, the effects on spliceosome assembly directly correspond to the effects on mRNA production. The didesmethyl compounds (6–9), which do not inhibit splicing, also do not interfere with spliceosome assembly (Fig. 2B, lanes 1–28). C3 and C10-C11 epimers (2, 3) give a block at an A-like complex at the same concentrations as PB (Fig. 2A, lanes 12–23). With the compounds lacking the epoxide (4, 5), the block at the A-like complex is not complete, and a small amount of higher order assembly still occurs (Fig. 2A, lanes 24–37), which correlates with the decrease, but not complete loss, of splicing chemistry in their presence. The similar effects of active analogs and PB indicate that they are likely inhibiting splicing assembly by the same mechanism: interference with U2 snRNA branch site recognition/progression of the A complex intermediate (15).
Cellular Changes Produced by PB and Analogs Correlate with Splicing Inhibition
We hypothesized that the effect of PB in cells is a function of its ability to inhibit pre-mRNA splicing. To address this subject, we used a cytological profiling assay to monitor the effect of PB and analogs on HeLa cells (22). In the assay, HeLa cells cultured in 384-well plates were treated with test compounds in a range from nanomolar to micromolar concentration or with DMSO alone for 20 h and then stained with fluorescent probes targeting actin (phalloidin), tubulin (α-tub), total DNA (Hoechst), newly synthesized DNA (EdU), and phosphohistone H3 (α-pHH3). The cells were imaged through automated microscopy, and a phenotypic profile for each compound was created using an algorithm that measured 244 cellular features based on the staining patterns, which provides information pertaining to cell number, cytoskeletal structure, nuclear size and morphology, DNA replication, and mitosis (22). The profiles were compared by cluster analysis and plotted as differences in feature value relative to DMSO treatment versus compound concentration.
Treatment of HeLa cells with PB (1) results in a distinct profile relative to DMSO even at low nanomolar concentrations (Fig. 3A). The profile is primarily a result of decreases in signal for features associated with DNA synthesis and mitosis and an increase signal for features corresponding to nuclear size and shape. The C3 and C10–11 epimer compounds (2, 3) give the same strong profile as PB, which is consistent with its nearly identical inhibition of splicing chemistry and spliceosome assembly in vitro (Fig. 3A). At higher concentrations (>∼100 nm), the desoxy compounds (4, 5) also give a very similar profile, but they show little change relative to DMSO at lower concentrations (Fig. 3A). This result is consistent with the desoxy compounds behaving in a manner similar to PB but with lower potency. The cytological profile of herboxidiene (10) strongly resembles that of PB (Fig. 3A). The subtle differences in the herboxidiene profile are likely due to assay variability rather than to a significantly different response. Notably, treatment with the didesmethyl compounds (6–9) even at the highest concentration of 67 μm yields few minimal phenotypic differences in HeLa cells in the assay (Fig. 3A), consistent with the inability of these analogs to inhibit in vitro splicing and spliceosome assembly. This result suggests that the PB scaffold does not have strong off target effects, and is consistent with PB selectivity for SF3B1.
FIGURE 3.

Cellular effects of PB and analogs. A, cytological profiles of HeLa cells treated with increasing concentrations of PB, PB analogs (epimer, desoxy, and didesmethyl), and herboxidiene (herb) (2 nm to 67 μm) and SSA (0.02 nm to 0.67 μm). Each row represents a different drug condition, whereas columns indicate the change in different cell parameters relative to DMSO treatment. Blue represents an increase in the parameter, yellow represents a decrease, and black indicates no difference. Color intensity indicates the magnitude of the difference. Features associated with DNA synthesis (EdU), mitosis (pHH3), and nuclear size/shape are highlighted. B, cluster analysis of cytological profiles of SF3B1 inhibitors with those produced by compounds with known molecular targets/cellular effects.
We also compared the PB (1) profile with that of SSA (11), and they are nearly indistinguishable (Fig. 3A). Because SSA is a splicing inhibitor with the same molecular target in the spliceosome, this result further strengthens the hypothesis that the changes in HeLa cells are due to the ability of PB to inhibit splicing through SF3B1. Remarkably, PB/SSA profiles do not cluster with those produced by compounds that cause cell death or apoptosis, which shows distinctly different changes in the measured parameters (Fig. 3B) (22). Instead, they most closely cluster with profiles of compounds that cause a G1 cell cycle arrest (Fig. 3B), consistent with previous studies showing that PB and SSA arrest cell cycle progression during the G1 phase and the G2/M transition (7, 10, 23). The result may reflect a higher sensitivity of intron(s) in gene(s) required for cell cycle progression with inhibition of SF3B1 by PB, which has been reported for SSA (13).
Nuclear Speckle Morphology Changes with PB Treatment
In addition to the automated cytological profiling, we also examined the morphology of nuclear speckles in HeLa cells treated 4 h with PB or analogs (Fig. 4A). Nuclear speckles are cellular bodies that contain transcription and pre-mRNA processing components (24). Knockdown of PB target SF3B1 causes a “mega-speckles” phenotype in which the speckles coalesce into large bodies (25). PB treatment of cells also induces formation of mega-speckles (8). Using immunofluorescence of SFRS2 (also called SC35), a common speckle marker, we observed mega-speckles in cells treated with PB (1) from 0.01 to 1 μm, which were absent in DMSO-treated control cells (Fig. 4, A and B). In line with the cytological profiling data, treatment with a didesmethyl compound (6) does not cause mega-speckles to form, and treatment with a desoxy (4) compound results in an intermediate phenotype, with fewer mega-speckles present relative to the same concentration of PB (Fig. 4, A and B). This result underscores the correlation between in vitro splicing inhibition and the cellular response to the drugs, i.e., the molecular features that are responsible for loss of splicing and block in spliceosome assembly are required in the same degree for changes in HeLa cell appearance. Mega-speckles also appear in cells treated with herboxidiene (10), but higher concentration of the drug is required (Fig. 4A).
FIGURE 4.

Changes in nuclear speckle morphology. A, SFRS2 (SC35) immunofluorescence (magenta) over DAPI stain (blue) of HeLa cells treated with DMSO or 0.1–1 μm of the indicated compounds for 4 h. B, box plots of relative speckle size from 20–40 nuclei images for cells treated with DMSO or 1 μm of the indicated compounds. Nonparametric Mann-Whitney U value tests show that differences between treatment and DMSO treatment are highly significant for PB (**, p < 0.001) and moderately significant (*, p < 0.05) for the desoxy compound. C, immunofluorescence images as described in A of cells treated with DMSO or 0.67–67 μm of the indicated cell cycle inhibitor. herb, herboxidiene; DMSO, dimethyl sulfoxide.
Finally, because PB (1) gave cytological profiles similar to cell cycle inhibitors (Fig. 3B), we also looked at speckle morphology in cells treated with the cell cycle inhibitors etoposide (topoisomerase II inhibitor) and genistein (tyrosine kinase inhibitor). Although the overall appearance of treated cells changed with respect to DMSO, the compounds did not cause formation of mega-speckles (Fig. 4C). This result indicates that mega-speckles do not form simply in response to cell cycle inhibition, but to another effect of PB treatment.
Impact of PB Modifications on Splicing in Cells
To further corroborate the relationship between the cellular phenotype produced by PB and its impact on splicing, we used RT-PCR to examine splicing of three genes in HeLa cells treated with increasing concentrations of the drug. As a control for RNA isolation and RT-PCR activity, we also examined the intron-less U6 mRNA, which was not affected by treatment with any of the compounds, even at the highest concentration of 1 μm (Fig. 5D). For the genes RBM5 and CCNA2, we designed primers to assay for skipping of selected exons that exhibited alternative splicing changes in cells treated with SSA (13) (Fig. 5, A and B). We see similar changes in exon skipping with PB (1) treatment: increased skipping of exon 16 in RMB5 and increased skipping of exon 5 in CCNA2. However, the effect does not directly correlate with the amount of the drug, in that the highest concentration of PB resulted in a smaller change in exon skipping relative to lower concentrations. A desoxy compound (4) also increased exon skipping in both genes, but higher amounts of the drug were required to obtain the same magnitude of change. A didesmethyl compound (6), which was inactive in all previous assays, did not affect CCNA2 splicing. Surprisingly, it did alter RBM5 splicing, although the increase in exon 16 skipping was not as large as with PB or the desoxy compound. This result suggests that the didesmethyl compound still affects SF3B1 at some level and that the effect can impact certain splicing events. We did not detect splicing changes for either gene in cells treated with herboxidiene (10) (data not shown).
FIGURE 5.

Splicing changes in cells treated with PB analogs. Semiquantitative RT-PCR analysis of RNA isolated from cells treated with increasing drug concentrations for RBM5 exon 16 inclusion (A), CCNA2 exon 5 inclusion (B), SF3A1 intron 6 removal (C), and U6 snRNA (D). The positions of different cDNA products and molecular weight markers are diagrammed at the right and left of each gel image. The percentage of exon skipping for RBM5 and CCNA2 is indicated below each lane.
Notably, not all splicing events appear affected by PB treatment. For example, we did not detect a decrease in splicing of the intron between exons 6 and 7 of SF3A1 in cells treated with any of the compounds (Fig. 5C), which had previously been observed upon knockdown of the spliceosome proteins in the SF3A complex of the U2 snRNP (20). Together these results show that in cells PB and analogs do not affect every intron at the same level and that changes in splicing of endogenous transcripts with drug treatment are likely modulated by more than SF3B1 activity alone. Splicing consensus sequences, particularly the branch point signal, probably play a role, as has been shown for SSA (13). Additionally, indirect effects through splicing of other factors and differential compound stability or cellular uptake may also come into play. Still, there is a general trend that cellular splicing events appear to be most sensitive to the strongest in vitro splicing inhibitors.
DISCUSSION
PB exhibits a remarkable combination of both antitumor activity and splicing inhibition, which gives it potential as lead compound for a new class of anticancer drug. PB is a member of a family of several pladienolide natural products from Streptomyces platensis, all of which share a complex macrolide ring structure (7, 26). The other family members and a handful of synthetic analogs have varying effects on cell growth and splicing, which provides some clues as to which molecular features of PB may be important for activity (7, 8, 27–29). In this study, we used an expanded series of PB analogs to directly test the requirement of PB features for in vitro splicing inhibition and effects in cells and identified both dispensable and indispensable functional groups.
Our results clearly show that one or both of the methyl groups at C16 and C20 are key to inhibition of splicing and most effects of PB on cells. This finding is in line with previous reports of compounds created on a simplified PB scaffold that lacked these methyl groups and that had >10,000-fold less activity in splicing and cell growth (28). Although we have not yet evaluated each position independently, previous studies suggest that both of them contribute to PB activity. The presence of an additional hydroxyl group at C20 in pladienolide F results in an ∼10-fold drop in cytotoxicity (7). Similarly, addition of a hydroxyl group at the position equivalent to C16 in the herboxidiene analog GEX1Q2 lowers activity in cell growth assays considerably (30). In the context of loss of the acetyl group at C7, the presence of an additional hydroxyl group at C16 in pladienolide E or at C20 in pladienolide G has a drastic effect on activity. Absence of the acetyl group at C7 alone in pladienolide A reduces cell growth inhibition by ∼250-fold relative to PB, but this reduction is further exacerbated ∼1,000-fold with pladienolide E and >50,000-fold with pladienolide G (7). However, it is notable that a hydroxyl group is also present at C16 in two other PB analogs (pladienolide D and E7107), both of which mimic the effects of PB on splicing and cells with similar potencies (8, 15). Also, isomerization of the C16-C17 bond in the context of FD-895, another related natural product with an additional hydroxyl group at C17 (31), has limited effect on the cytotoxicity of the compound (29, 32). More studies focusing on each position independently will be required to clear up the contribution of functional groups to PB activity at these sites.
Some feature(s) of the macrolide ring distal to the arm linkage appears to contribute to the effects of PB on spliceosome assembly and thus splicing inhibition. As noted above, absence of the acetyl group at C7 in pladienolide A reduces activity (7). Combined loss of side groups at C3, C7 and C10 abrogates bioactivity completely (29). Interestingly, herboxidiene, which has a less complex ring structure but maintains an acetyl group, inhibits splicing in vitro to the same extent as PB. Its cytological profile is very similar to PB, and the slight differences can be attributed to assay variability and clustering parameters. Notably, herboxidiene is less potent in cellular splicing assays and in impacting nuclear speckles. More work will be needed to determine whether differences between PB and herboxidiene change activity toward SF3B1 or instead alter its uptake or stability in cells. Notably, our results with the C3 epimers show that at least the stereochemistry at this position in the macrolide ring is not important for splicing inhibition.
Absence of the epoxide at C18-C19 resulted in only a 10-fold loss of splicing inhibition or cell activity. Because PB, herboxidiene, and FR901464 share an epoxy group, it was proposed to be a required feature of a pharmacophore common to the compounds (33). Based on that pharmacophore model, a set of compounds termed sudomycins was synthesized on an FR901464-like scaffold with an epoxide, and these compounds exhibit only limited cytotoxicity and effects on splicing (33, 34). Our data indicate that the epoxide contributes to PB activity but is not absolutely required. Corroborating this observation, a recent report provides evidence that changing the epoxide of FD-985 to a cyclopropane does not drastically alter its cytotoxicity but instead appears to stabilize the compound (29). Furthermore, recent papers identified an analog of FR901464, termed spliceostatin B, and two members of the thailanstatin family of natural products that resemble FR901464 and that do not contain the epoxide (35, 36). All three compounds are considerably less cytotoxic than their epoxide-containing counterparts. Thailanstatins B and C were also shown to inhibit splicing of a reporter gene but required ∼10-fold higher concentrations. These findings parallel our results with the PB desoxy compounds.
PB exhibits a remarkable combination of both antitumor activity and splicing inhibition, which gives it potential as lead compound for a new class of anticancer drug (37, 38). Our current structure-activity relationship studies demonstrate that creating less complex PB analogs that retain bioactivity is quite feasible. As it turns out, the C3-hydroxyl stereochemistry is not critical to spliceosome inhibition, which indicates that the hydroxyl group can be replaced with isosteric functionalities, and even gem-dimethyl groups may be accommodated. Similarly, C10-C11 stereochemistry is not critical to activity, and it can be eliminated with the incorporation of gem-dimethyl groups. Furthermore, C18-C19 desoxy PB (4, 5) retains good potency, which indicates that stable PB analogs without labile epoxide functionality can be developed (29). Finally, because the activity of herboxidiene resembles PB in many aspects, the macrolide ring of PB may also be simplified. Thus, the design of structurally less complex and more stable PB analogs promises to greatly facilitate medicinal chemistry efforts toward PB analog development for further in-depth studies.
The cytostatic effect of PB along with the discovery of SF3B1 as its molecular target suggests a relationship between splicing and cancer cell growth (8). The relationship is strengthened by the observation that PB inhibits splicing of cell cycle genes (8) and by the identification of an SF3B1 mutation that abrogate the ability of PB to halt cell growth (9). Our data further underscore the link by demonstrating that the ability of PB to inhibit splicing in vitro is tied to its ability to impact a diverse set of cellular phenotypes. However, it is important to note that changes in cell growth do not correlate with a general inhibition of splicing of all introns but are more likely to result from more subtle changes in alternative splicing of a key set of transcripts. This is evident from the differential effects that we observe in cellular splicing for different genes and from the difference in drug potency for in vitro versus in vivo effects, which also been noted for SSA, another SF3B1 targeting drug (13, 39). Presumably, our in vitro splicing substrate is not as dependent on robust SF3B1 activity, and more drug is required to impact its splicing, whereas certain splicing events in cells are very sensitive. These sensitive events are likely to correlate with alternative splicing choices, and the cellular effect of PB analogs will depend both on how many splicing events they modulate and on how changes in isoform ratios of the affected genes impact phenotype. So, even though the didesmethyl PB analogs affect some splicing events in cells, the absence of an effect on cytological profiles and nuclear structure suggests off target effects that would hamper development of PB as a potential cancer drug will be limited. Ultimately, how the important features of PB that we identified contribute to its activity is still an open question. It may be that they affect its affinity for SF3B1, its ability to interfere with SF3B1 function, or its stability in extracts and cells. Future studies addressing these possibilities will be key for understanding the mechanisms of PB splicing inhibition and its chemotherapeutic promise.
Acknowledgments
We thank the UCSC Chemical Screening Center and B. Abrams from the UCSC Life Sciences Microscopy Center for technical support. We also thank M. Yoshida at the RIKEN Advanced Science Institute for providing SSA.
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA136762 (to M. S. J.) and 1S10RR022455. This work was also supported by the University of California Cancer Research Coordinating Committee, the California Institute for Quantitative Biosciences (QB3), and the United States Department of State.

This article contains supplemental materials.
A. K. Ghosh, D. D. Anderson, K. A. Effenberger, and M. S. Jurica, manuscript in preparation.
- snRNP
- small nuclear ribonucleoprotein
- PB
- pladienolide B
- SSA
- spliceostatin A
- DMSO
- dimethyl sulfoxide.
REFERENCES
- 1. Papaemmanuil E., Cazzola M., Boultwood J., Malcovati L., Vyas P., Bowen D., Pellagatti A., Wainscoat J. S., Hellstrom-Lindberg E., Gambacorti-Passerini C., Godfrey A. L., Rapado I., Cvejic A., Rance R., McGee C., Ellis P., Mudie L. J., Stephens P. J., McLaren S., Massie C. E., Tarpey P. S., Varela I., Nik-Zainal S., Davies H. R., Shlien A., Jones D., Raine K., Hinton J., Butler A. P., Teague J. W., Baxter E. J., Score J., Galli A., Della Porta M. G., Travaglino E., Groves M., Tauro S., Munshi N. C., Anderson K. C., El-Naggar A., Fischer A., Mustonen V., Warren A. J., Cross N. C., Green A. R., Futreal P. A., Stratton M. R., Campbell P. J. (2011) Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N. Engl. J. Med. 365, 1384–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Quesada V., Conde L., Villamor N., Ordóñez G. R., Jares P., Bassaganyas L., Ramsay A. J., Beà S., Pinyol M., Martínez-Trillos A., López-Guerra M., Colomer D., Navarro A., Baumann T., Aymerich M., Rozman M., Delgado J., Giné E., Hernández J. M., González-Díaz M., Puente D. A., Velasco G., Freije J. M., Tubío J. M., Royo R., Gelpí J. L., Orozco M., Pisano D. G., Zamora J., Vázquez M., Valencia A., Himmelbauer H., Bayés M., Heath S., Gut M., Gut I., Estivill X., López-Guillermo A., Puente X. S., Campo E., López-Otín C. (2012) Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 44, 47–52 [DOI] [PubMed] [Google Scholar]
- 3. Yoshida K., Sanada M., Shiraishi Y., Nowak D., Nagata Y., Yamamoto R., Sato Y., Sato-Otsubo A., Kon A., Nagasaki M., Chalkidis G., Suzuki Y., Shiosaka M., Kawahata R., Yamaguchi T., Otsu M., Obara N., Sakata-Yanagimoto M., Ishiyama K., Mori H., Nolte F., Hofmann W. K., Miyawaki S., Sugano S., Haferlach C., Koeffler H. P., Shih L. Y., Haferlach T., Chiba S., Nakauchi H., Miyano S., Ogawa S. (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69 [DOI] [PubMed] [Google Scholar]
- 4. Harbour J. W., Roberson E. D., Anbunathan H., Onken M. D., Worley L. A., Bowcock A. M. (2013) Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 45, 133–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ellis M. J., Ding L., Shen D., Luo J., Suman V. J., Wallis J. W., Van Tine B. A., Hoog J., Goiffon R. J., Goldstein T. C., Ng S., Lin L., Crowder R., Snider J., Ballman K., Weber J., Chen K., Koboldt D. C., Kandoth C., Schierding W. S., McMichael J. F., Miller C. A., Lu C., Harris C. C., McLellan M. D., Wendl M. C., DeSchryver K., Allred D. C., Esserman L., Unzeitig G., Margenthaler J., Babiera G. V., Marcom P. K., Guenther J. M., Leitch M., Hunt K., Olson J., Tao Y., Maher C. A., Fulton L. L., Fulton R. S., Harrison M., Oberkfell B., Du F., Demeter R., Vickery T. L., Elhammali A., Piwnica-Worms H., McDonald S., Watson M., Dooling D. J., Ota D., Chang L. W., Bose R., Ley T. J., Piwnica-Worms D., Stuart J. M., Wilson R. K., Mardis E. R. (2012) Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Biankin A. V., Waddell N., Kassahn K. S., Gingras M. C., Muthuswamy L. B., Johns A. L., Miller D. K., Wilson P. J., Patch A. M., Wu J., Chang D. K., Cowley M. J., Gardiner B. B., Song S., Harliwong I., Idrisoglu S., Nourse C., Nourbakhsh E., Manning S., Wani S., Gongora M., Pajic M., Scarlett C. J., Gill A. J., Pinho A. V., Rooman I., Anderson M., Holmes O., Leonard C., Taylor D., Wood S., Xu Q., Nones K., Fink J. L., Christ A., Bruxner T., Cloonan N., Kolle G., Newell F., Pinese M., Mead R. S., Humphris J. L., Kaplan W., Jones M. D., Colvin E. K., Nagrial A. M., Humphrey E. S., Chou A., Chin V. T., Chantrill L. A., Mawson A., Samra J. S., Kench J. G., Lovell J. A., Daly R. J., Merrett N. D., Toon C., Epari K., Nguyen N. Q., Barbour A., Zeps N., Kakkar N., Zhao F., Wu Y. Q., Wang M., Muzny D. M., Fisher W. E., Brunicardi F. C., Hodges S. E., Reid J. G., Drummond J., Chang K., Han Y., Lewis L. R., Dinh H., Buhay C. J., Beck T., Timms L., Sam M., Begley K., Brown A., Pai D., Panchal A., Buchner N., De Borja R., Denroche R. E., Yung C. K., Serra S., Onetto N., Mukhopadhyay D., Tsao M. S., Shaw P. A., Petersen G. M., Gallinger S., Hruban R. H., Maitra A., Iacobuzio-Donahue C. A., Schulick R. D., Wolfgang C. L., Morgan R. A., Lawlor R. T., Capelli P., Corbo V., Scardoni M., Tortora G., Tempero M. A., Mann K. M., Jenkins N. A., Perez-Mancera P. A., Adams D. J., Largaespada D. A., Wessels L. F., Rust A. G., Stein L. D., Tuveson D. A., Copeland N. G., Musgrove E. A., Scarpa A., Eshleman J. R., Hudson T. J., Sutherland R. L., Wheeler D. A., Pearson J. V., McPherson J. D., Gibbs R. A., Grimmond S. M. (2012) Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491, 399–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mizui Y., Sakai T., Iwata M., Uenaka T., Okamoto K., Shimizu H., Yamori T., Yoshimatsu K., Asada M. (2004) Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J. Antibiot. 57, 188–196 [DOI] [PubMed] [Google Scholar]
- 8. Kotake Y., Sagane K., Owa T., Mimori-Kiyosue Y., Shimizu H., Uesugi M., Ishihama Y., Iwata M., Mizui Y. (2007) Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 3, 570–575 [DOI] [PubMed] [Google Scholar]
- 9. Yokoi A., Kotake Y., Takahashi K., Kadowaki T., Matsumoto Y., Minoshima Y., Sugi N. H., Sagane K., Hamaguchi M., Iwata M., Mizui Y. (2011) Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 278, 4870–4880 [DOI] [PubMed] [Google Scholar]
- 10. Kaida D., Motoyoshi H., Tashiro E., Nojima T., Hagiwara M., Ishigami K., Watanabe H., Kitahara T., Yoshida T., Nakajima H., Tani T., Horinouchi S., Yoshida M. (2007) Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 3, 576–583 [DOI] [PubMed] [Google Scholar]
- 11. Hasegawa M., Miura T., Kuzuya K., Inoue A., Won Ki S., Horinouchi S., Yoshida T., Kunoh T., Koseki K., Mino K., Sasaki R., Yoshida M., Mizukami T. (2011) Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem. Biol. 6, 229–233 [DOI] [PubMed] [Google Scholar]
- 12. Gao Y., Vogt A., Forsyth C. J., Koide K. (2013) Comparison of splicing factor 3b inhibitors in human cells. Chembiochem 14, 49–52 [DOI] [PubMed] [Google Scholar]
- 13. Corrionero A., Miñana B., Valcárcel J. (2011) Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 25, 445–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roybal G. A., Jurica M. S. (2010) Spliceostatin A inhibits spliceosome assembly subsequent to prespliceosome formation. Nucleic Acids Res. 38, 6664–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Folco E. G., Coil K. E., Reed R. (2011) The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 25, 440–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Albert B. J., McPherson P. A., O'Brien K., Czaicki N. L., Destefino V., Osman S., Li M., Day B. W., Grabowski P. J., Moore M. J., Vogt A., Koide K. (2009) Meayamycin inhibits pre-messenger RNA splicing and exhibits picomolar activity against multidrug-resistant cells. Mol. Cancer Ther. 8, 2308–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghosh A. K., Anderson D. D. (2012) Enantioselective total synthesis of pladienolide B. A potent spliceosome inhibitor. Org. Lett. 14, 4730–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghosh A. K., Li J. (2011) A stereoselective synthesis of (+)-herboxidiene/GEX1A. Org. Lett. 13, 66–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tanackovic G., Krämer A. (2005) Human splicing factor SF3a, but not SF1, is essential for pre-mRNA splicing in vivo. Mol. Biol. Cell 16, 1366–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Brien K., Matlin A. J., Lowell A. M., Moore M. J. (2008) The biflavonoid isoginkgetin is a general inhibitor of Pre-mRNA splicing. J. Biol. Chem. 283, 33147–33154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schulze C. J., Bray W. M., Woerhmann M. H., Stuart J., Lokey R. S., Linington R. G. (2013) “Function-first” lead discovery. Mode of action profiling of natural product libraries using image-based screening. Chem. Biol. 20, 285–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakajima H., Hori Y., Terano H., Okuhara M., Manda T., Matsumoto S., Shimomura K. (1996) New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J. Antibiot. 49, 1204–1211 [DOI] [PubMed] [Google Scholar]
- 24. Spector D. L., Lamond A. I. (2011) Nuclear speckles. Cold Spring Harb. Perspect. Biol. 3, a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Allende-Vega N., Dayal S., Agarwala U., Sparks A., Bourdon J. C., Saville M. K. (2013) p53 is activated in response to disruption of the pre-mRNA splicing machinery. Oncogene 32, 1–14 [DOI] [PubMed] [Google Scholar]
- 26. Sakai T., Sameshima T., Matsufuji M., Kawamura N., Dobashi K., Mizui Y. (2004) Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. I. Taxonomy, fermentation, isolation and screening. J. Antibiot. 57, 173–179 [DOI] [PubMed] [Google Scholar]
- 27. Müller S., Mayer T., Sasse F., Maier M. E. (2011) Synthesis of a pladienolide B analogue with the fully functionalized core structure. Org. Lett. 13, 3940–3943 [DOI] [PubMed] [Google Scholar]
- 28. Gundluru M. K., Pourpak A., Cui X., Morris S. W., Webb T. R. (2011) Design, synthesis and initial biological evaluation of a novel pladienolide analog scaffold. Medchemcomm 2, 904–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Villa R., Kashyap M. K., Kumar D., Kipps T. J., Castro J. E., La Clair J. J., Burkart M. D. (2013) Stabilized cyclopropane analogs of the splicing inhibitor FD-895. J. Med. Chem. 56, 6576–6582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sakai Y., Tsujita T., Akiyama T., Yoshida T., Mizukami T., Akinaga S., Horinouchi S., Yoshida M., Yoshida T. (2002) GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. II. The effects on cell cycle progression and gene expression. J. Antibiot. 55, 863–872 [DOI] [PubMed] [Google Scholar]
- 31. Seki-Asano M., Okazaki T., Yamagishi M., Sakai N., Takayama Y., Hanada K., Morimoto S., Takatsuki A., Mizoue K. (1994) Isolation and characterization of a new 12-membered macrolide FD-895. J. Antibiot. 47, 1395–1401 [DOI] [PubMed] [Google Scholar]
- 32. Villa R., Mandel A. L., Jones B. D., La Clair J. J., Burkart M. D. (2012) Structure of FD-895 revealed through total synthesis. Org. Lett. 14, 5396–5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lagisetti C., Pourpak A., Goronga T., Jiang Q., Cui X., Hyle J., Lahti J. M., Morris S. W., Webb T. R. (2009) Synthetic mRNA splicing modulator compounds with in vivo antitumor activity. J. Med. Chem. 52, 6979–6990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fan L., Lagisetti C., Edwards C. C., Webb T. R., Potter P. M. (2011) Sudemycins, novel small molecule analogues of FR901464, induce alternative gene splicing. ACS Chem. Biol. 6, 582–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu X., Biswas S., Tang G. L., Cheng Y. Q. (2013) Isolation and characterization of spliceostatin B, a new analogue of FR901464, from Pseudomonas sp. No. 2663. J. Antibiot. 66, 555–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X., Biswas S., Berg M. G., Antapli C. M., Xie F., Wang Q., Tang M. C., Tang G. L., Zhang L., Dreyfuss G., Cheng Y. Q. (2013) Genomics-guided discovery of thailanstatins A, B, and C as pre-mRNA splicing inhibitors and antiproliferative agents from Burkholderia thailandensis MSMB43. J. Nat. Prod. 76, 685–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hong D. S., Kurzrock R., Naing A., Wheler J. J., Falchook G. S., Schiffman J. S., Faulkner N., Pilat M. J., O'Brien J., Lorusso P. (2013) A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest. New Drugs 10.1007/s10637-013-0046-5 [DOI] [PubMed] [Google Scholar]
- 38. Eskens F. A., Ramos F. J., Burger H., O'Brien J. P., Piera A., de Jonge M. J., Mizui Y., Wiemer E. A., Carreras M. J., Baselga J., Tabernero J. (2013) Phase I, pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 19, 6296–6304 [DOI] [PubMed] [Google Scholar]
- 39. Furumai R., Uchida K., Komi Y., Yoneyama M., Ishigami K., Watanabe H., Kojima S., Yoshida M. (2010) Spliceostatin A blocks angiogenesis by inhibiting global gene expression including VEGF. Cancer Sci. 101, 2483–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]