

Background: The metabolism of non-growing microbes is poorly understood.
Results: In a nitrogen-starved and non-growing photoheterotrophic bacterium, metabolic flow was diverted to mobilize electrons for H2 production.
Conclusion: During starvation bacteria decouple their metabolism from biosynthesis.
Significance: An understanding of metabolic activities of non-growing cells can be used to engineer improved biocatalysts.
Keywords: Bacterial Metabolism, Biofuel, Metabolic Engineering, Metabolic Regulation, Metabolic Tracers, Nitrogenase, Transcriptomics, Bacterial Starvation, Hydrogen Gas, Metabolic Flux Analysis
Abstract
When starved for nitrogen, non-growing cells of the photosynthetic bacterium Rhodopseudomonas palustris continue to metabolize acetate and produce H2, an important industrial chemical and potential biofuel. The enzyme nitrogenase catalyzes H2 formation. The highest H2 yields are obtained when cells are deprived of N2 and thus use available electrons to synthesize H2 as the exclusive product of nitrogenase. To understand how R. palustris responds metabolically to increase H2 yields when it is starved for N2, and thus not growing, we tracked changes in biomass composition and global transcript levels. In addition to a 3.5-fold higher H2 yield by non-growing cells we also observed an accumulation of polyhydroxybutyrate to over 30% of the dry cell weight. The transcriptome of R. palustris showed down-regulation of biosynthetic processes and up-regulation of nitrogen scavenging mechanisms in response to N2 starvation but gene expression changes did not point to metabolic activities that could generate the reductant necessary to explain the high H2 yield. We therefore tracked 13C-labeled acetate through central metabolic pathways. We found that non-growing cells shifted their metabolism to use the tricarboxylic acid cycle to metabolize acetate in contrast to growing cells, which used the glyoxylate cycle exclusively. This shift enabled cells to more fully oxidize acetate, providing the necessary reducing power to explain the high H2 yield.
Introduction
The vast majority of studies on microbial physiology and metabolism have been performed under conditions of nutrient excess. However, in most natural environments microbes are frequently starved for critical growth nutrients. While some mechanisms by which bacteria cope with starvation have been well characterized, such as cell differentiation and the stringent response, the diverse metabolic strategies employed by starving microbes is still poorly understood (1).
Purple nonsulfur bacteria, like Rhodopseudomonas palustris, are well known for their ability to grow photoheterotrophically, wherein light is used for energy and organic compounds are used for carbon and electrons. R. palustris will consume a variety of fermentation products, including acetate and volatile fatty acids (e.g. butyrate). When growing, R. palustris metabolizes these compounds via acetyl-CoA and the glyoxylate shunt (2). The glyoxylate shunt bypasses the lower tricarboxylic acid (TCA)2 cycle, thereby retaining the two carbons in acetyl-CoA for biosynthesis that would otherwise be lost as CO2. When starved for nutrients like nitrogen but supplied with light and organic carbon, R. palustris can maintain a relatively high rate of metabolic activity in a non-growing state for months or longer (3). Under these conditions, it is assumed that R. palustris repeatedly energizes and cycles electrons through a H+-pumping electron transfer chain to generate ATP for cell maintenance in a process called cyclic photophosphorylation.
R. palustris has served as a model organism for studies of hydrogen gas (H2) production (4). H2 is an important chemical used in industries such as ammonia production and petroleum refining. It can also be used as a transportation fuel in H2-powered vehicles. Nearly all H2 currently used by our society is derived from fossil fuels. However, H2 can be produced in a sustainable manner through a variety of methods, including biologically. R. palustris produces H2 through the photosynthetic conversion of compounds that are abundant in agricultural waste, such as acetate. R. palustris makes H2 via the enzyme, nitrogenase. Nitrogenase is best known for converting N2 gas into NH4+ but H2 is an additional obligate product of the reaction. If access to N2 gas as a substrate is prevented, nitrogenase produces H2 as its sole product. Most work has been done with cells expressing Mo-nitrogenase. R. palustris also encodes alternative iron (Fe-) and vanadium (V-)nitrogenases. These consume more reducing power and produce more H2 per molecule of N2 fixed than Mo-nitrogenase (5).
R. palustris regulatory mutants have been constructed that synthesize active nitrogenase and make H2 while growing under an Ar atmosphere with NH4+ as the sole nitrogen source (2, 6–8). Under these growth conditions, the CO2-fixing Calvin cycle competes with nitrogenase for electrons (2, 6). Genetically disrupting Calvin cycle activity increased the H2 yield on all carbon sources tested (2). However, even the most electron-rich substrate tested, butyrate, gave a H2 yield that was only 25% of the theoretical maximum as most of the electrons were used for biosynthesis. Thus, the main process that competes for electrons against H2 production is biosynthesis. Indeed, when wild-type R. palustris growth was prevented by replacing N2 with Ar, H2 yields from acetate reached 43% of the theoretical maximum yield (9). This high H2 yield and the ability to remain metabolically active for long periods of time makes starving R. palustris intriguing from both applied and fundamental perspectives.
Here we set out to understand the physiology of non-growing R. palustris cells that were starved for nitrogen, since it is under these conditions that the highest H2 yields are observed. Excreted products, biomass composition, and global transcript abundances were monitored as R. palustris transitioned to and remained in a non-growing state. 13C-tracer studies were performed to identify the central metabolic pathways that provide reductant for H2 production. Together, these data inform on microbial responses to nitrogen starvation and provide a basis for engineering strategies to improve H2 production traits.
EXPERIMENTAL PROCEDURES
Chemicals, Strains, and Culture and Suspension Conditions
Sodium [1-13C]acetate and [U-13C]acetate were purchased from Cambridge Isotope Laboratories (Andover, MA). R. palustris wild-type strain CGA009 was used for all experiments. CGA009 is defective in uptake hydrogenase activity (10). All cultures and suspensions were incubated at 30 °C in front of 60 watt incandescent light bulbs. Strains were grown in a chemically defined medium, NFM (11), with N2 gas as the sole nitrogen source in sealed, anaerobic 160-ml serum vials that were stirred with stir bars or in 27-ml anaerobic tubes. We generated suspensions of non-growing cells by flushing mid-exponential phase cultures (0.4–0.5 OD660) with Ar for 10 min while stirring, thereby replacing the N2.
Determining Biomass Composition
Cells were grown in 60 ml of NFM with 20 mm unlabeled acetate. Duplicate vials of cell suspensions were chilled on ice at 0, 12, 30, 50, 90, 140, and 180 h of nitrogen starvation, and fractioned to quantify optical density, soluble compounds (e.g. acetate, α-ketoglutartate [αKG]), gasses, and biomass components. Acetate and αKG were quantified by HPLC as described (12). H2 and CO2 were quantified by GC as described (6, 9). Total cell protein was quantified by the bicinchoninic acid assay (Thermo Fisher Scientific, Rockford, IL) as described (6). Dry cell weights (DCW) were determined as described (13) by filtering 25 ml of cell suspension through 0.22-micron PVDF filters (Millipore, Billerica, MA). Polyhydroxybutyrate (PHB) was then hydrolyzed and extracted as crotonic acid by boiling the filters and cells in 1 ml of pure sulfuric acid in screw-cap glass test tubes. Extracts were diluted with 4 ml water, centrifuged, filtered, and diluted 10-fold with water. Crotonic acid was quantified by HPLC as described (14). Total RNA and glycogen were quantified from 5 ml of cell suspension as described (13) except a glucose oxidase assay kit (Sigma-Aldrich) was used to quantify glucose from glycogen. Trehalose was quantified by centrifuging 5 ml of cell suspension, resuspending in 0.4 ml of 50 mm sodium acetate buffer (pH 5.2), lysing cells by sonication, boiling for 5 min, incubating with 0.04 units of trehalase (Sigma-Aldrich) for 20 h at 37 °C, diluting with 0.4 ml of 250 mm Tris-HCl (pH 7.5), and then quantifying the resulting glucose using a glucose oxidase assay kit. Fatty acids were quantified as methyl esters from ∼7 ml of cell suspension as described (15) using a Shimadzu GC-2014 with a 15 m SHRXI-5MS column (Columbia, MD). The electron content of biomass excluding PHB was determined from OD660 values at the onset of starvation using an experimentally determined conversion factor for growing cells of 625 mg DCW L−1 OD660−1 and a biomass molecular formula of CH1.8N0.18O0.38 (16), which would yield 5 available electrons per carbon. An increase in biomass electron content of 1.2 was then applied based on the average increase in colony forming units after starvation (see “Results”).
RNA-seq Using R. palustris-specific Selective Primers
RNA was extracted and purified from 5 ml of the same cell suspensions and time points used for tracking the biomass composition, as described previously (17). The R. palustris-specific selective primers v2 were based on the genome sequences of six R. palustris strains (CGA009, TIE-1, HaA2, BisB5, BisB18, and BisA53). We used a pool of 1,203 selective primers with no perfect match to any rRNA genes (e.g. 5S, 16S, and 23S rRNA). An additional set of 200 primers that were responsible for the majority of rRNA-priming events in test libraries, was further removed, leaving a final set of 1,003 R. palustris-specific selective primers v2 (supplemental Table S1). For first strand cDNA synthesis, 500 ng of total RNA was mixed with 1 μl of 100 μm R. palustris-specific selective primers v2 in a 5-μl reaction volume, and incubated at 65 °C for 5 min. After snap chilling on ice, 2 μl of 5× First-Strand Buffer (Invitrogen, Carlsbad, CA), 2 μl of 10 mm dNTP mix (New England BioLabs, Ipswich, MA), 0.5 μl of 0.1 m DTT, 0.5 μl of SuperScript III Reverse Transcriptase (Invitrogen) were added, and the mixture was further incubated at 40 °C for 90 min. To generate the second strand, 45.5 μl of water, 15 μl of 5× Second Strand Buffer (Invitrogen), 1.5 μl of 10 mm dNTP mix, 0.5 μl of Escherichia coli DNA Ligase (Invitrogen, 10 units/μl), 2 μl of DNA Polymerase I (Invitrogen, 10 units/μl), and 0.5 μl of RNase H (Invitrogen, 2 units/μl) were added to the reaction, and incubated at 16 °C for 2 h. The reaction was stopped by adding 25 μl of 20 mm EDTA, pH 8.0. Double-stranded cDNA was then purified by using the MinElute PCR Purification Kit (Qiagen, Valencia, CA) using 12 μl of Buffer EB. Finally, the cDNA library was constructed using the Ovation Ultralow Library System (NuGEN, San Carlos, CA) following the manufacturer's instructions. All cDNA libraries were sequenced with an Illumina HiSeq at the University of Washington High Throughput Genomics Center, and raw sequencing data were processed by with the Xpression pipeline (18). Reads that mapped to the reference genome sequence of R. palustris CGA009 were categorized as (i) uniquely mapped (aligned with ≤ 2 mismatches to a single genomic locus), (ii) partially mapped (aligned with > 2 mismatches or one gaps to a single genomic locus), (iii) non-uniquely mapped (aligned to the 5S, 16S, and 23S rRNA genes, or multiple genomic locus), and (iv) unmapped (no alignment to a reference genome). Only the uniquely mapped reads were subjected to further analysis. The number of reads overlapping each gene was recorded and normalized based on reads per kilobase per million uniquely mapped reads (RPKM). We used the statistical software DESeq (19) to evaluate whether, for a given region, an observed difference in read counts between tx and t0 was significant. Features were considered to be differentially expressed from levels observed immediately prior to nitrogen starvation if they had a p value of ≤0.001 and with fold change ratios ≥ 4. The resulting data has been deposited in NCBI Gene Expression Omnibus (20) under the GEO series accession number GSE51825.
Definition of the Theoretical Maximum H2 Yield
The maximum amount of H2 that can be produced from electrons made available when an organic substrate is fully oxidized to CO2. For example, the theoretical maximum H2 yield from acetate is 4 mol H2/mol acetate according to Equations 1–3.
13C-Labeling Experiments and Mass Spectrometry
1 ml of 10 ml-starter cultures grown in NFM acetate were used to inoculate 50 ml of NFM with 10 mm unlabeled acetate in 160-ml anaerobic serum vials with an N2 headspace. When a cell density of 0.4–0.5 OD660 was reached the headspace was replaced with Ar to halt growth and cells were incubated for ∼100 h during which time PHB accumulated. Suspensions were then decanted into 50-ml conical tubes in an anaerobic chamber, centrifuged, washed once with anaerobic phosphate buffer (12.5 mm NaH2PO4, 12.5 mm K2HPO4), resuspended in 1 ml of phosphate buffer, and then transferred into anaerobic 160-ml serum vials containing 56 ml of NFM with 10 mm of either [1-13C]acetate (three biological replicates), [U-13C]acetate (1 cell suspension), or unlabeled acetate (4 biological replicates). Cell suspensions were then flushed with Ar for 10 min while stirring. Unlabeled cell suspensions were then sampled and later harvested (260 h after receiving [13C]acetate; 360 h after starvation was initiated) to determine the changes in acetate, αKG, CO2, H2 and PHB. Labeled cell suspensions were sampled periodically (∼every 2 days) to track optical densities, acetate consumption, and H2 production before being quenched. Individual cell suspensions were quenched 216, 260, and 283 h after receiving [13C]acetate (316, 360, and 384 h of starvation; the suspension with [U-13C]acetate was harvested after 316 h of starvation). Cell suspensions were quenched as described (19) by decanting cell suspensions into 200 ml of 60% methanol with 10 mm ammonium acetate (pH 7.2) at −40 °C. Quenched suspensions were centrifuged at 14,300 rcf in a rotor pre-chilled to −20 °C. Quenched samples were not pooled (21). The supernatant was discarded and the remaining cell pellet was frozen in liquid N2 and stored at −80 °C until the extraction procedure. Metabolites were extracted from cell pellets three times with 78 °C ethanol as described (21), dried under N2 at 55 °C, and then were stored at −80 °C. Samples were resuspended in water and the metabolites analyzed by LC-MS/MS as described (21). Despite the larger culture volumes, we only observed a portion of those metabolites and fragments that were observed using the same methods on nitrogen-starved B. subtilis (21). Even so, we observed sufficient metabolite labeling patterns to resolve key fluxes in the R. palustris metabolic network and with enough redundancy to perform statistical analyses.
13C-Metabolic Flux Analysis
Flux values were estimated from the measured acetate uptake rate and carbon product formation rates and from metabolite mass isotopomer distributions using the software suite, 13CFLUX2 (22). Data from the single [U-13C]acetate-labeled culture were included as additional constraints using a relatively large S.D. (0.025) for each mass isotopomer to put more weight on the data from [1-13C]acetate-labeled cultures. The average S.D. for all mass isotopomers from the [1-13C]acetate-labeled cultures was 0.015. All mass isotopomer data were fit against a single metabolic model as described (23). The metabolic model was based on that previously described for R. palustris (6), but excluded biosynthetic outputs, included αKG, PHB, and glycogen outputs, and included inputs of unlabeled metabolites from biomass degradation pathways. Various modifications of this model were explored as described in the results (e.g. omitting malic enzyme, constraining malate dehydrogenase directionality). For the final model the fitting algorithm was initiated using 1300 different arrangements of starting values chosen at random using the 13CFLUX2 program, multifit. The fitting algorithm was applied to other models using at least 300 different starting value arrangements. Standard deviations for flux values were determined using the 13CFLUX2 linearized statistical analysis program, fwdsim.
RESULTS
Electron Flow during Nitrogen Starvation
R. palustris was grown anaerobically in mineral medium with N2 gas as the sole nitrogen source and acetate as the carbon source. Light was provided as the energy source. When cells reached the mid-logarithmic phase of growth, we removed the N2 by flushing the headspace of the sealed growth vessels with Ar. Entire cultures were then harvested for analysis of excreted products, biomass composition, and global transcript levels at various intervals for a week (180 h) following the initiation of nitrogen starvation. Starved cells consumed acetate and produced hydrogen and CO2 (Fig. 1). These non-growing cells also produced αKG, as reported previously (9). H2 production was most rapid at the beginning of the experiment then declined considerably after 100 h (Fig. 1). Similar trends and timing were observed for CO2 production and acetate consumption (Fig. 1). As the production of H2 and CO2 declined, the rate of αKG production increased after 50 h (Fig. 1). Similar to previous observations (9), H2 and αKG together accounted for about half of the electrons consumed (H2: 45 ± 5% and αKG: 1.7 ± 1.6%; S.D.; n = 11; across time points from 12 h to 180 h). Including cell suspensions from other experiments, the H2 yield was sometimes observed to exceed 50% of the theoretical maximum yield (6 out of 16 suspensions), reaching 66% in one case.
FIGURE 1.

H2 and CO2 production and acetate consumption was rapid during the first 50–100 h of nitrogen starvation and then slowed as αKG was excreted. Each data point is representative of a single sacrificed culture. Units are total μmoles normalized for the OD660 at the onset of nitrogen starvation to account for differences in cell density between cell suspensions.
We hypothesized that the remaining electrons could have been incorporated into internal storage products. The genome sequence of R. palustris (24) suggests that it can make several different storage products including glycogen, trehalose, and PHB. We observed a large accumulation of PHB upon nitrogen starvation (Fig. 2). Before starvation, no PHB was detected. Upon starvation, PHB accumulated and eventually accounted for over 30% of the DCW and 35 ± 6% of the electrons consumed. PHB accumulation followed a similar trend to that of acetate consumption and H2 and CO2 production, with the most rapid production occurring in the first 50 h of starvation. Glycogen and trehalose showed a brief accumulation after starvation began, but levels never accounted for more than 2% of the DCW (Fig. 2). Protein, RNA, and lipids showed less dramatic fluctuations in response to starvation (Fig. 2), although the phospholipid fatty acid composition changed. There was an overall shift from unsaturated fatty acid to saturated fatty acids and cyclopropane fatty acids. C18:1 fatty acids decreased from 75 to 59% of the total fatty acid composition by 180 h of starvation while C16:0 fatty acids increased from 10% to 20% (Fig. 3A). This decrease in C18 fatty acids could have contributed acetyl-CoA to central metabolism and perhaps to the synthesis of C20:0 fatty acids which accumulated to nearly 2% of the cellular fatty acids by 180 h (Fig. 3B). Nitrogen-starved cells also produced C17 and C19 cyclopropane fatty acids, together accounting for 5–6% of the fatty acid composition by 180 h (Fig. 3B).
FIGURE 2.

Polyhydroxybutyrate is synthesized in response to nitrogen starvation. Each data point is representative of a single sacrificed culture. Units are total mg normalized for the OD660 at the onset of nitrogen starvation to account for differences in cell density between cell suspensions.
FIGURE 3.
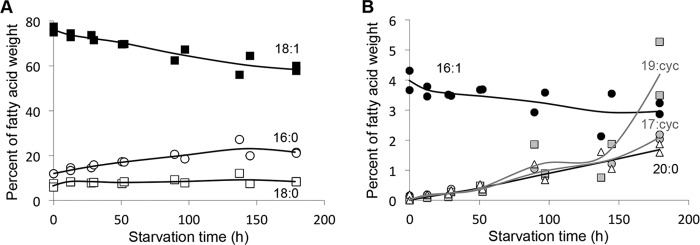
Fatty acid composition changes in favor of unsaturated fatty acids and cyclopropane fatty acids in response to nitrogen starvation. Graphs show fatty acids accounting for >6% (A) or <6% (B) of the total fatty acid dry cell weight. Each data point is representative of a single sacrificed culture.
We also took into account electrons used to make new cells after nitrogen starvation. Surprisingly, the OD of cultures increased 2.0 ± 0.1-fold (S.D.; n = 6) after removing N2. However, this OD increase was not simply due to cell division. Colony forming unit counts indicated that cell numbers only increased 1.2 ± 0.1-fold (S.D.; n = 4). We determined that the large OD increase was due to PHB accumulation. A mutant of R. palustris that is incapable of PHB synthesis3 only showed a 1.2 ± 0.2-fold (S.D.; n = 4) increase in OD upon nitrogen starvation. This brief increase in biomass was estimated to account for 25 ± 5% of the electrons consumed.
Taken together we determined that electrons from consumed acetate are distributed between H2, αKG, PHB, and a 1.2-fold increase in cells. These products account for 106 ± 12% (S.D.; n = 11; across time points from 12 h to 180 h) of the electrons consumed. The carbon balance, including measurements of CO2, shows that 99 ± 15% (S.D.; n = 11) of the acetate carbon consumed is accounted for in these products. Thus we identified all major endpoints of electron flow from acetate during nitrogen starvation.
Transcriptome Analysis Did Not Reveal Significant Metabolic Changes
We also isolated RNA from cells at the same time points as above to analyze the transcriptome of nitrogen-starved cells (supplemental Table S2). General trends that emerged were (i) up-regulation of genes involved in scavenging nitrogen, (e.g. the V- and Fe-nitrogenases) and (ii) down-regulation of core house-keeping and biosynthetic genes (e.g. ribosomal proteins, photosynthetic proteins) (Fig. 4).
FIGURE 4.

Transcript levels for genes encoding Mo-nitrogenase (A) and alternative nitrogenases (B and C) increased in response to nitrogen starvation while most genes for core physiological processes, such as those encoding ribosomal proteins (D), photosynthetic proteins (E), and central metabolic enzymes (F) showed lower transcript levels or no change. Genes encoding lower TCA cycle enzymes isocitrate dehydrogenase (RPA3834), αKG dehydrogenase (RPA0185–90), and succinyl-CoA synthetase (RPA0190–1) are shown as black-outlined symbols with dark blue lines. Genes encoding glyoxylate shunt enzymes, malate synthase (RPA4216) and isocitrate lyase (RPA4394) are shown as white symbols with black dotted lines (F). Red/Blue: ≥ 4-fold increase (Red) or decrease (Blue) in transcript level versus t0 with p value of ≤ 0.001. Gray: < 4-fold change in transcript level versus t0 or p value of > 0.001.
The transcriptomic dataset may also provide insight into the observed metabolic and cell composition changes noted above. RPA0895 and RPA2160, which encode 3-oxoacyl-ACP reductases, could be involved in the shift toward unsaturated fatty acids (supplemental Table S2). These genes were up-regulated in response to nitrogen starvation in contrast to most other genes involved in fatty acid synthesis (e.g. those encoding acetyl-CoA carboxylase; RPA0508, 2435–6; supplemental Table S2). Most notably RPA2160 transcript levels increased >60-fold by 180 h. RPA3082 was the only one of three potential cyclopropane-fatty-acyl-phospholipid synthase genes (RPA0924, 2569, 3082) that showed significantly higher transcript levels (supplemental Table S2), especially from 90 h onwards, coinciding with the increase in cyclopropane fatty acids.
Transcript levels for core metabolic processes such as ATPase (RPA0175–9 and RPA0843–7), NADH-ubiquinone dehydrogenase (RPA4252–64), and transhydrogenase (RPA4181–2) decreased upon nitrogen starvation (supplemental Table S2). Fig. 4F illustrates that most genes encoding central metabolic enzymes exhibited either lower transcript levels or no differential expression. The only two central metabolic genes that showed consistently higher transcript levels were RPA1329, encoding one of two fumarases, and RPA1331, encoding malate quinone oxidoreductase, although p values were above the threshold for all time points (supplemental Table S2). These lower transcript levels for metabolic genes are consistent with the slower metabolism of nitrogen-starved cells. The acetate uptake rate (which dictates an upper-limit for all downstream flux values) by non-growing cells in this study was 0.042 ± 0.007 μmol mg DCW−1 h−1 whereas growing cells consumed acetate at 2.0 ± 0.0 μmol mg DCW−1 h−1 (6).
Although there was some consistency between transcriptomic and physiological data it was clear that the transcriptomic data did not provide a complete picture of the physiological response to nitrogen starvation. For example, genes potentially involved in PHB synthesis (acetyl-CoA acetyltransferases, RPA0513, 175, and 3715; 3-hydroxybutyrl-CoA dehydrogenase, RPA4748; and PHB synthase, RPA2501) did not show significant transcript level changes (supplemental Table S2) despite prolific PHB accumulation. If not for the PHB measurements, one might misinterpret the transcriptomic dataset to indicate PHB degradation. A potential PHB depolymerase gene, RPA0565 showed up to 5-fold higher transcript levels in response to nitrogen starvation (supplemental Table S2).
More importantly, the transcriptomic data did not indicate how reductant was generated to explain the high H2 yields. Previously we determined that a R. palustris Calvin cycle mutant growing on acetate uses half of its reducing power to produce H2 at about 14.5% of theoretical maximum yield and the other half for biosynthesis (6). Thus, if biosynthesis were prevented, we would only expect there to be enough reducing power diverted from biosynthesis to produce H2 at 29% of theoretical maximum yield. However, we observed that nitrogen-starved cells produce H2 at levels up to 66% of the theoretical maximum yield. Thus, additional reducing power must be generated in nitrogen-starved cells. To determine how this additional reducing power was generated we turned to 13C-labeling experiments.
Nitrogen Starvation Causes a Shift in Metabolic Flux from the Glyoxylate Shunt to the TCA Cycle
Distribution of metabolic fluxes at branch points can be inferred from the unique patterns of 12C and 13C that each branch will produce when an organism is fed a 13C-labeled substrate (25). We previously used such techniques to determine metabolic fluxes during photoheterotrophic growth using the labeling patterns in proteinaceous amino acids (2, 6). However, nitrogen-starved cells do not synthesize amino acids and protein at a sufficient level for this approach. Thus, we examined the labeling patterns in the organic acids and sugar phosphates of central metabolism (21). We initiated these experiments with cells that had already been subjected to 100 h of nitrogen starvation, and thus were synthesizing little PHB, prior to receiving either [1-13C]acetate or [U-13C]acetate. We chose to incubate cells with [13C]acetate after the PHB accumulation phase to avoid any possible metabolic changes during this transition that could impact labeling patterns.
The first major branchpoint during the metabolism of acetate in R. palustris is the split to the glyoxylate shunt versus the lower tricarboxylic acid (TCA) cycle at isocitrate. The flux distribution at this branchpoint could have a large influence on reductant availability since the lower TCA cycle generates two reducing equivalents (e.g. 2 NAD(P)H) whereas the glyoxylate shunt does not generate any. The glyoxylate shunt flux would result in double-labeled succinate, malate, and αKG from [1-13C]acetate whereas TCA cycle flux would result in single-labeled succinate and malate and a mixture of single and double-labeled αKG (Fig. 5). A high percentage of succinate (82.5%) was single-labeled while only a small percentage (4.9%) was double-labeled (Fig. 5) strongly indicating a large TCA cycle flux with very little participation from the glyoxylate shunt. High proportions of both single- and double-labeled αKG (inferred from measurements of free glutamate) also indicated a high TCA cycle flux (Fig. 5). On its own, the high proportion of double-labeled malate (∼37%) suggested a large involvement from the glyoxylate shunt. However, even double-labeled malate can only give rise to single-labeled succinate through the action of the TCA cycle (Fig. 5). Thus, a pathway other than the glyoxylate shunt must have generated the double-labeled malate.
FIGURE 5.

Labeling patterns that would be generated from [1-13C]acetate by TCA cycle flux alone (blue) and glyoxylate cycle flux alone (red). These patterns are expected to be the dominant patterns resulting from the activity of these two cycles but can be influenced by other metabolic activities such as those shown in Fig. 7. Succinate is a symmetrical molecule such that a 13C-carboxyl group could represent either the first or last carbon. Green boxes show mass isotopomer distributions from metabolites from non-growing cells. αKG labeling patterns were inferred from free glutamate. The gray box shows a mass isotopomer distribution obtained from growing cells in a previous study that had near-exclusive glyoxylate shunt flux (6).
We then examined the complete mass isotopomer data set using 13CFLUX2 for two reasons: (i) to look for other activities that could generate the double-labeled malate while taking into account the labeling patterns observed in other metabolites and (ii) to quantify metabolic fluxes and thereby the contribution of the TCA cycle toward H2 production. The equations used to determine metabolic fluxes from labeling patterns describe both an isotopic and metabolic steady state (which can be satisfied by a pseudo-metabolic steady state in batch culture). Cell suspensions were harvested at different time points so that we could confirm isotopic steady state by comparing the mass isotopomer distributions (MIDs). Most of the MIDs from the three [1-13C]acetate-fed cultures were similar across time points (individual mass isotopomers showed S.D. ≤ 10% of the mean), indicating an isotopic steady state. There were a few mass isotopomers that showed a larger variation across time points that may have influenced the determination of flux values (4 out of 42 mass isotopomers had a S.D. between 10 and 16% of the mean). However, there are enough redundant measurements to provide confidence in our main conclusions. Suspensions were harvested after the PHB accumulation phase to avoid a possible metabolic shift occurring at this time. After PHB accumulation, we also observed constant rates of acetate consumption and H2 production up to 384 h indicating a pseudo-metabolic steady state.
To look for pathways that could explain the double-labeled malate, we started with the metabolic model depicted in Fig. 6. This model also takes into the account the possibility of metabolites entering central metabolism from the degradation of unlabeled biomass macromolecules. Arbitrary flux values were repeatedly optimized to generate a simulated data set that fit the experimentally determined MIDs and product formation rates (i.e. CO2, αKG, and PHB). The best flux solutions (i.e. that resulted in a minimal difference between the simulated and measured data sets) indicated massive cycling between malate, pyruvate, PEP, and oxaloacetate in either direction (Fig. 7A). This cycling would result in the exchange of an unlabeled carboxyl with 13CO2 (Fig. 7A; 13CO2 is the dominant form of CO2 in the cell suspensions since it is derived from the labeled-carboxyl of [1-13C]acetate). To explore other possibilities, we then ran the fitting algorithm using a model lacking malic enzyme (malate → pyruvate + CO2) to prevent the cycling noted above. An equally good solution was reached in which the double-labeled malate was explained by a high decarboxylating/carboxylating exchange flux between malate and PEP, involving malate dehydrogenase (Fig. 7B). When both the malic enzyme reaction and malate dehydrogenase exchange flux were removed from the model, malate patterns could still be largely explained by gluconeogenic exchange fluxes producing malate from patterns generated in the non-oxidative pentose phosphate pathway. Thus, there are several solutions that can explain the double-labeled malate. Importantly, none of these solutions involved the glyoxylate shunt as this activity would also generate double-labeled succinate.
FIGURE 6.

Metabolic flux distributions determined by fitting simulated mass isotopomer and extracelluar flux values to measured values using 13CFLUX2. When growing (A), R. palustris uses the glyoxylate shunt to conserve carbon and electrons for biosynthesis. When not growing (B), R. palustris oxidizes acetate via the TCA cycle. The extra reducing power generated by the TCA cycle under non-growing conditions is coupled to H2 production. Values for net fluxes have units of mole % of the acetate uptake rate ± S.D. and are reflected in arrow thickness. Dotted lines indicate values that were < 3 mol% of the acetate uptake rate. Large gray arrows indicate fluxes that could not be resolved but that best fits suggested were large in magnitude. Enlarged arrowheads indicate the direction of net flux. Actual acetate uptake rates were 2.0 ± 0.0 μmol mg DCW−1 h−1 (growing) and 0.042 ± 0.007 μmol mg DCW−1 h−1 (non-growing) (6). Unlabeled inputs of CO2, OAA, Pyr, R5P, Suc, αKG, G6P, and AcCoA from macromolecule degradation pathways are not shown for clarity but ranged between 0 and 4 mol% of the acetate uptake rate. Data from growing cells were reported previously (6).
FIGURE 7.

Fluxes between PEP, OAA, Mal, and Pyr are poorly resolved due to the possibility of multiple routes for generating double-labeled malate. In both A and B, single-labeled malate is decarboxylated to single-labeled PEP in Step 1 but by different routes. In Step 2, PEP is carboxylated with 13CO2 that derived from the 13C-carboxyl of acetate, the main source of CO2 in the closed system, as it is processed through decarboxylation reactions such as those in the lower TCA cycle (Fig. 6). These carboxyl exchange reactions generate double-labeled malate. These two particular scenarios represent futile cycles that result in the net expenditure of ATP. Other scenarios are possible that do not represent futile cycles, such as the reverse of the cycle depicted in panel A.
Unlike the multiple flux solutions that could explain the double-labeled malate, all the tested models consistently showed a nearly exclusive involvement of the lower TCA cycle relative to the glyoxylate shunt. This trend is in stark contrast to the nearly exclusive use of the glyoxylate shunt observed in growing cells (Fig. 6). The reducing power from the high TCA cycle flux was enough to fully explain the large H2 yield (Fig. 8). Other potential sources of reducing power, such as the oxidative pentose phosphate pathway exhibited negligible flux (Fig. 6). While we are not confident in all aspects of the flux map (Fig. 6, gray arrows), we note that the final solution balanced electrons to within 97% (Fig. 8), even though electron balance was not a constraint used by the fitting algorithm.
FIGURE 8.
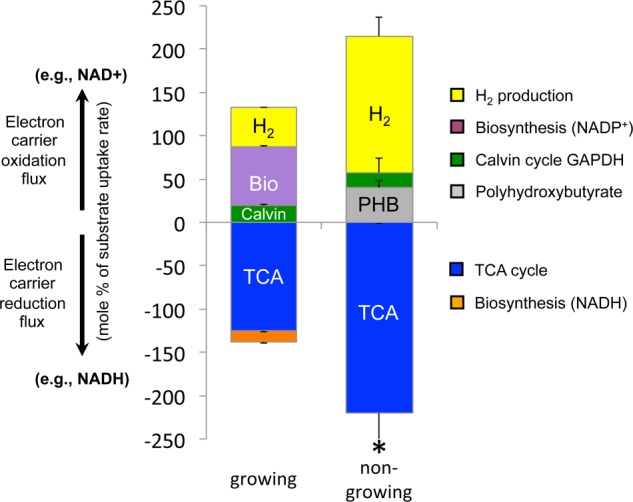
Fluxes through redox reactions are balanced under growing and non-growing conditions. Positive bars are fluxes through reactions that oxidize electron carriers whereas negative bars are fluxes through reactions that reduce electron carriers (generate reducing power). The collective bars on either side of the x axis should be equal if electrons are balanced. The sum of oxidation reaction fluxes was 98% of the sum of reducing fluxes for growing cells and 97% for non-growing cells. Error bars represent standard deviations. * indicates that the standard deviation was larger than the mean because of participation of malate dehydrogenase in a poorly resolved cycle (Fig. 6). The contribution of malate dehydrogenase to reducing power was determined by subtracting the malic enzyme flux value from the malate dehydrogenase flux value (i.e. to determine the amount of flux needed to maintain the other flux values in the TCA cycle). The flux values for malate dehydrogenase and malic enzyme in the poorly resolved cycle were otherwise too large to make a meaningful comparison of redox fluxes. Data from growing cells were reported previously (6).
DISCUSSION
Here we determined that when R. palustris growth is prevented by nitrogen starvation, acetate is metabolized through the TCA cycle rather than through the glyoxylate shunt. This TCA cycle flux results in a higher production of CO2 and reducing power than in growing cells (Fig. 6). The reducing power must be oxidized to maintain metabolic flow. Oxidation of reducing power is primarily coupled to H2 production, resulting in a 3.5-fold increase in H2 yield over growing cells (Fig. 8). It is currently not clear why the H2 production slowed to a third of the initial rate after 50–100 h of starvation (Fig. 1). It is unlikely that the slow rate was due to inhibition of nitrogenase by accumulated H2 because resuspending cells in fresh medium under Ar after 100 h (i.e. for 13C-labeling experiments) did not restore the early rapid H2 production typically observed upon nitrogen starvation (Fig. 1). The slow H2 production rate is tied to a slower rate of metabolism overall, as acetate uptake and CO2 production also slowed (Fig. 1). We speculate that R. palustris might first respond to limiting nitrogen by rapidly filling up its PHB carbon stores and then switch to a lower metabolic rate. The highest PHB level we observed (∼30% DCW) may represent a physiological maximum for R. palustris as it is an order of magnitude above levels typically reported for growing cells (26) and comparable to one of the highest values reported for R. palustris (27).
The large increase in electron-rich PHB in the first few days of starvation (Fig. 2) is also likely an important electron sink that was not reflected in our flux maps (Figs. 6 and 8) because we conducted 13C-labeling experiments after PHB synthesis had slowed. PHB synthesis is a known response to nitrogen limitation in R. palustris (28) and other bacteria (29). PHB was also synthesized when R. palustris and other purple nonsulfur bacteria were starved for phosphorous (26) or sulfur (30), but under those conditions PHB synthesis was greatly favored over H2 production. R. palustris also produced cyclopropane fatty acids when starved for nitrogen. Although the role of these fatty acids is unclear in bacteria, they are usually made in response to stressors (e.g. acid shock in E. coli (31)) and may be involved in coordinating stress responses. For example, a potential cyclopropane fatty acid synthase was shown to be needed for the proper transcriptional activation of a stress response to singlet oxygen in Rhodobacter sphaeroides (32).
The most notable discovery from our study was the metabolism of acetate via the TCA cycle instead of via the glyoxylate shunt. The glyoxylate shunt is canonically employed during growth on acetate to bypass the lower TCA cycle, which would otherwise result in the loss of the two acetate carbons as CO2. Lesser known, the glyoxylate shunt is also activated in bacteria such as E. coli during growth on glucose, for example to prevent unnecessary carbon loss via the TCA cycle when glucose is limiting or to prevent excess production of reducing power via the TCA cycle when other oxidative pathway fluxes are high (33). A few studies have followed the metabolism of 13C-labeled acetate in growing cells. During aerobic respiration on acetate by Corynebacterium glutamicum, 25% of isocitrate was metabolized via the glyoxylate shunt while 75% was diverted through the lower TCA cycle (34). It was speculated that the relatively high C. glutamicum TCA cycle fluxes were needed to generate sufficient reductant for respiration to support the high ATP demands of gluconeogenesis. Similar results were observed in E. coli respiring with acetate, with 30% of flux from isocitrate going to the glyoxylate shunt and 70% through the lower TCA cycle (35). A 13C-labeling study of Geobacter metallireducens showed high TCA cycle flux from acetate while respiring with Fe3+ with zero flux through the glyoxylate shunt (36). However, this depiction was misleading as there is nothing in the genome sequence to suggest that the glyoxylate shunt exists in G. metallireducens and rather pyruvate synthase is likely critical to assimilate acetate. The high reductant demand of respiration is not an issue for R. palustris under photosynthetic conditions. Indeed when we grew R. palustris photosynthetically with 13C-acetate we observed a nearly complete absence of TCA cycle flux with acetate metabolized almost exclusively via the glyoxylate shunt (6). When R. palustris is starved for nitrogen, we conclude that there are few remaining sinks for carbon in the absence of biosynthesis and with PHB already accumulated. Thus instead of conserving carbon via the glyoxylate shunt the TCA cycle is activated to oxidize acetate to CO2. Oxidation of carbon sources to CO2 may be a widespread bacterial response to nitrogen deprivation, even in non-photosynthetic, respiring bacteria. 13C-metabolic flux analysis of nitrogen-starved B. subtilis fed with glucose also showed elevated fluxes through CO2-producing oxidative pathways (37).
The transcriptomic data informed on several physiological aspects of the starving cells. The increase in transcript levels for nitrogen scavenging genes is consistent with previous microarray observations of R. palustris strains during growth with inefficient nitrogenase isozymes that likely induced a state of nitrogen starvation (5). The decrease in transcript levels for genes encoding biosynthetic and central metabolic enzymes is also consistent with expectations for non-growing cells. For example, transcript and protein levels were shown to decrease with decreasing growth rate in E. coli (38). However, we also observed instances where metabolic shifts could not have been predicted from the transcriptomic dataset, namely PHB synthesis and the branching of flux through the glyoxylate shunt and TCA cycle.
Transcripts for PHB synthesis genes were not differentially regulated despite PHB accumulation. This observation suggests that these genes are constitutively expressed and thus are regulated post-transcriptionally, similar to reports for other bacteria (29, 39). The surprising increase in expression of PHB depolymerase despite PHB accumulation may be indicative of simultaneous PHB synthesis and degradation. These concurrent opposing activities have been noted in other bacteria and can allow for PHB to be remodeled into other polyhydroxyalkanoates (29).
The glyoxylate shunt isocitrate lyase gene can be transcriptionally regulated in R. palustris. Transcript levels were 11-fold lower during growth on succinate (which does not require the glyoxylate shunt), compared with growth on acetate (based on microarray data comparison between GPL3954 [GSM798294 and GSM798299] and GSE5194 [GSM116900 and GSM116886]; data accessible in the NCBI GEO database (20)). However, during nitrogen starvation we did not observe such a decrease in isocitrate lyase transcript levels despite the low glyoxylate shunt flux (Fig. 4F). Thus, the shift of flux from the glyoxylate shunt to the TCA cycle must not be transcriptionally regulated under these conditions. The TCA cycle enzyme, isocitrate dehydrogenase has a higher affinity for isocitrate (Km: 0.009 mm (40)) than isocitrate lyase does (Km: 0.136 mm (41)) and thus could simply outcompete isocitrate lyase for isocitrate. In E. coli, isocitrate dehydrogenase activity is also inhibited by phosphorylation of the enzyme (42). If similar post-translational regulation is used in R. palustris then we can infer that isocitrate dehydrogenase is primarily in an unphosphorylated state during nitrogen starvation. However, biochemical or proteomic analyses would have to be performed to confirm that both enzymes are present and able to compete for isocitrate.
The discrepancies between our transcriptomic and fluxomic data demonstrate the need for cautious interpretations of metabolic trends from transcriptomic datasets. Our data and that obtained from nitrogen-starved B. subtilis (37), demonstrate that bacteria can have low transcript levels for metabolic genes and yet remain metabolically active. In the case of R. palustris, metabolic activity can persist in the absence of growth for months (3). This metabolic activity during starvation involved a constant input of energy (light) and electrons (acetate). Thus it is not clear whether our results can be extended to suggest that dormant microbes that exhibit little to no detectable rRNA in natural environments may still contribute metabolic activity to a community (43). However, conditions of constant energy, carbon, and electron inputs under otherwise growth-limiting conditions is certainly relevant to industrial bioprocesses where starvation is used to favor product formation over biosynthesis. Thus, newly configured tools for directly analyzing the metabolism of starving cells (37) will be crucial to understand and optimize their industrial attributes.
Acknowledgments
We thank Colin Lappala for assistance with RNA purification and RNA-seq analysis. We also thank Dr. Amy Schaefer and other members of the Harwood laboratory for fruitful discussions.
Experimental aspects of this research were supported equally by the Division of Chemical Sciences, Geosciences, and Biosciences, Office of Basic Energy Sciences, U.S. Department of Energy (DOE), through Grant DE-FG02-05ER15707 and by the Office of Science (BER), U.S. Department of Energy, through Grant DE-FG02-07ER64482. Experimental work was also funded through the European Commission 7th Framework project BaSysBio (LSHG-CT-2006-037469). United States National Science Foundation Grant MCB-11457304 (to C. S. H.) and Office of Science (BER), U.S. Department of Energy Grant DE-SC0008131 (to J. B. M.) provided support for data analysis and manuscript preparation.
The resulting data has been deposited in NCBI Gene Expression Omnibus under the GEO series accession number GSE51825.

This article contains supplemental Tables S1 and S2.
Y. Oda, J. B. McKinlay, and C. S. Harwood, unpublished data.
- TCA
- tricarboxylic acid
- Ace
- acetate
- AcCoA
- acetyl-CoA
- BPG
- 1,3-bisphosphoglycerate
- Cit/Ict
- citrate/isocitrate
- DCW
- dry cell weight
- αKG
- α-ketoglutartate
- E4P
- erythrose-4-phosphate
- FBP
- fructose-1,6-bisphosphate
- F6P
- fructose-6-phosphate
- G6P
- glucose-6-phosphate
- GAP
- glyceraldehyde-3-phosphate
- H2
- hydrogen gas
- Mal
- malate
- MalDH
- malate dehydrogenase
- MID
- mass isotopomer distribution
- OAA
- oxaloacetate
- R5P
- pentose-5-phosphate
- PEP
- phosphoenolpyruvate
- PEPCK
- phosphoenolpyruvate carboxykinase
- PEPC
- phosphoenolpyruvate carboxylase
- PEP syn
- phosphoenolpyruvate synthetase
- PG6
- 6-phosphogluconate
- 3PG
- 3-phosphoglycerate
- PHB
- polyhydroxybutyrate
- PYR
- pyruvate
- R1,5P
- ribulose-1,5-bisphosphate
- S7P
- sedoheptulose-7-phosphate
- Suc
- succinate.
REFERENCES
- 1. Lennon J. T., Jones S. E. (2011) Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 9, 119–130 [DOI] [PubMed] [Google Scholar]
- 2. McKinlay J. B., Harwood C. S. (2011) Calvin cycle flux, pathway constraints, and substrate oxidation state together determine the H2 biofuel yield in photoheterotrophic bacteria. mBio 2, 10.1128/mBio.00323-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gosse J. L., Engel B. J., Hui J. C., Harwood C. S., Flickinger M. C. (2010) Progress toward a biomimetic leaf: 4,000 h of hydrogen production by coating-stabilized nongrowing photosynthetic Rhodopseudomonas palustris. Biotechnol. Prog. 26, 907–918 [DOI] [PubMed] [Google Scholar]
- 4. McKinlay J. B., Harwood C. S. (2010) Photobiological production of hydrogen gas as a biofuel. Curr. Opin. Biotechnol. 21, 244–251 [DOI] [PubMed] [Google Scholar]
- 5. Oda Y., Samanta S. K., Rey F. E., Wu L., Liu X., Yan T., Zhou J., Harwood C. S. (2005) Functional genomic analysis of three nitrogenase isozymes in the photosynthetic bacterium Rhodopseudomonas palustris. J. Bacteriol. 187, 7784–7794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McKinlay J. B., Harwood C. S. (2010) Carbon dioxide fixation as a central redox cofactor recycling mechanism in bacteria. Proc. Natl. Acad. Sci. U.S.A. 107, 11669–11675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rey F. E., Heiniger E. K., Harwood C. S. (2007) Redirection of metabolism for biological hydrogen production. Appl. Environ. Microbiol. 73, 1665–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adessi A., McKinlay J. B., Harwood C. S., De Philippis R. (2012) A Rhodopseudomonas palustris nifA* mutant produces H2 from NH4+-containing vegetable wastes. Int. J. Hydrog. Energy 37, 15893–15900 [Google Scholar]
- 9. Huang J. J., Heiniger E. K., McKinlay J. B., Harwood C. S. (2010) Production of hydrogen gas from light and the inorganic electron donor thiosulfate by Rhodopseudomonas palustris. Appl. Environ. Microbiol. 76, 7717–7722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rey F. E., Oda Y., Harwood C. S. (2006) Regulation of uptake hydrogenase and effects of hydrogen utilization on gene expression in Rhodopseudomonas palustris. J. Bacteriol. 188, 6143–6152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim M., Harwood C. S. (1991) Regulation of benzoate-CoA ligase in Rhodopseudomonas palustris. FEMS Microbiol. Lett. 83, 199–203 [Google Scholar]
- 12. McKinlay J. B., Zeikus J. G., Vieille C. (2005) Insights into Actinobacillus succinogenes fermentative metabolism in a chemically defined growth medium. Appl. Environ. Microbiol. 71, 6651–6656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKinlay J. B., Shachar-Hill Y., Zeikus J. G., Vieille C. (2007) Determining Actinobacillus succinogenes metabolic pathways and fluxes by NMR and GC-MS analyses of 13C-labeled metabolic product isotopomers. Metab. Eng. 9, 177–192 [DOI] [PubMed] [Google Scholar]
- 14. Karr D. B., Waters J. K., Emerich D. W. (1983) Analysis of poly-3-hydroxybutyrate in Rhizobium japonicum bacteroids by ion-exclusion high-pressure liquid chromatography and UV detection. Appl. Environ. Microbiol. 46, 1339–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Y., Beisson F., Pollard M., Ohlrogge J. (2006) Oil content of Arabidopsis seeds: The influence of seed anatomy, light and plant-to-plant variation. Phytochemistry 67, 904–915 [DOI] [PubMed] [Google Scholar]
- 16. Carlozzi P., Sacchi A. (2001) Biomass production and studies on Rhodopseudomonas palustris grown in an outdoor, temperature controlled, underwater tubular photobioreactor. J. Biotechnol. 88, 239–249 [DOI] [PubMed] [Google Scholar]
- 17. Hirakawa H., Oda Y., Phattarasukol S., Armour C. D., Castle J. C., Raymond C. K., Lappala C. R., Schaefer A. L., Harwood C. S., Greenberg E. P. (2011) Activity of the Rhodopseudomonas palustris p-coumaroyl-homoserine lactone-responsive transcription factor RpaR. J. Bacteriol. 193, 2598–2607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Phattarasukol S., Radey M. C., Lappala C. R., Oda Y., Hirakawa H., Brittnacher M. J., Harwood C. S. (2012) Identification of a p-coumarate degradation regulon in Rhodopseudomonas palustris by Xpression, an integrated tool for prokaryotic RNA-seq data processing. Appl. Environ. Microbiol. 78, 6812–6818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anders S., Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Edgar R., Domrachev M., Lash A. E. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rühl M., Rupp B., Nöh K., Wiechert W., Sauer U., Zamboni N. (2012) Collisional fragmentation of central carbon metabolites in LC-MS/MS increases precision of 13C metabolic flux analysis. Biotechnol. Bioeng. 109, 763–771 [DOI] [PubMed] [Google Scholar]
- 22. Weitzel M., Nöh K., Dalman T., Niedenführ S., Stute B., Wiechert W. (2013) 13CFLUX2-high-performance software suite for 13C-metabolic flux analysis. Bioinformatics 29, 143–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwender J., Shachar-Hill Y., Ohlrogge J. B. (2006) Mitochondrial metabolism in developing embryos of Brassica napus. J. Biol. Chem. 281, 34040–34047 [DOI] [PubMed] [Google Scholar]
- 24. Larimer F. W., Chain P., Hauser L., Lamerdin J., Malfatti S., Do L., Land M. L., Pelletier D. A., Beatty J. T., Lang A. S., Tabita F. R., Gibson J. L., Hanson T. E., Bobst C., Torres J. L., Peres C., Harrison F. H., Gibson J., Harwood C. S. (2004) Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 22, 55–61 [DOI] [PubMed] [Google Scholar]
- 25. Sauer U. (2006) Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vincenzini M., Marchini A., Ena A., De Philippis R. (1997) H2 and poly-β-hydroxybutyrate, two alternative chemicals from purple non sulfur bacteria. Biotechnol. Lett. 19, 759–762 [Google Scholar]
- 27. Mukhopadhyay M., Patra A., Paul A. K. (2005) Production of poly(3-hydroxybutyrate) and poly(3-hydroxybutyrate-co-3-hydroxyvalerate) by Rhodopseudomonas palustris SP5212. World J. Microbiol. Biotechnol. 21, 765–769 [Google Scholar]
- 28. De Philippis R. (1992) Factors affecting poly-β-hydroxybutyrate accumulation in cyanobacteria and in purple non-sulfur bacteria. FEMS Microbiol. Lett. 103, 187–194 [Google Scholar]
- 29. Anderson A. J., Dawes E. A. (1990) Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol. Rev. 54, 450–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Melnicki M. R., Eroglu E., Melis A. (2009) Changes in hydrogen production and polymer accumulation upon sulfur-deprivation in purple photosynthetic bacteria. Int. J. Hydrog. Energy 34, 6157–6170 [Google Scholar]
- 31. Cronan J. (2002) Phospholipid modifications in bacteria. Curr. Opin. Microbiol. 5, 202–205 [DOI] [PubMed] [Google Scholar]
- 32. Nam T.-W., Ziegelhoffer E. C., Lemke R. A. S., Donohue T. J. (2013) Proteins needed to activate a transcriptional response to the reactive oxygen species singlet oxygen. mBio 4, 10.1128/mBio.00541-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer E., Sauer U. (2003) A novel metabolic cycle catalyzes glucose oxidation and anaplerosis in hungry Escherichia coli. J. Biol. Chem. 278, 46446–46451 [DOI] [PubMed] [Google Scholar]
- 34. Wendisch V. F., de Graaf A. A., Sahm H., Eikmanns B. J. (2000) Quantitative determination of metabolic fluxes during coutilization of two carbon sources: comparative analyses with Corynebacterium glutamicum during growth on acetate and/or glucose. J. Bacteriol. 182, 3088–3096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao J., Shimizu K. (2003) Metabolic flux analysis of Escherichia coli K12 grown on 13C-labeled acetate and glucose using GC-MS and powerful flux calculation method. J. Biotechnol. 101, 101–117 [DOI] [PubMed] [Google Scholar]
- 36. Tang Y. J., Chakraborty R., Martín H. G., Chu J., Hazen T. C., Keasling J. D. (2007) Flux analysis of central metabolic pathways in Geobacter metallireducens during reduction of soluble Fe(III)-nitrilotriacetic acid. Appl. Environ. Microbiol. 73, 3859–3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rühl M., Le Coq D., Aymerich S., Sauer U. (2012) 13C-flux analysis reveals NADPH-balancing transhydrogenation cycles in stationary phase of nitrogen-starving Bacillus subtilis. J. Biol. Chem. 287, 27959–27970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ishii N., Nakahigashi K., Baba T., Robert M., Soga T., Kanai A., Hirasawa T., Naba M., Hirai K., Hoque A., Ho P. Y., Kakazu Y., Sugawara K., Igarashi S., Harada S., Masuda T., Sugiyama N., Togashi T., Hasegawa M., Takai Y., Yugi K., Arakawa K., Iwata N., Toya Y., Nakayama Y., Nishioka T., Shimizu K., Mori H., Tomita M. (2007) Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science 316, 593–597 [DOI] [PubMed] [Google Scholar]
- 39. Kranz R., Gabbert K., Locke T., Madigan M. (1997) Polyhydroxyalkanoate production in Rhodobacter capsulatus: genes, mutants, expression, and physiology. Appl. Environ. Microbiol. 63, 3003–3009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lebedeva N. V., Malinina N. V., Ivanovskiı̆ R. N. (2002) [A comparative study of the isocitrate dehydrogenases of Chlorobium limicola forma Chlorobium thiosulfatophilum and Rhodopseudomonas palustris]. Mikrobiologiia 71, 762–767 [PubMed] [Google Scholar]
- 41. Tahama H., Shinoyama H., Ando A., Fujii T. (1990) Purification and characterization of isocitrate lyase from Rhodopseudomonas sp. No. 7. Agricultural Biological Chemistry 54, 3177–3183 [Google Scholar]
- 42. Singh S. K., Matsuno K., LaPorte D. C., Banaszak L. J. (2001) Crystal structure of Bacillus subtilis isocitrate dehydrogenase at 1.55 A. Insights into the nature of substrate specificity exhibited by Escherichia coli isocitrate dehydrogenase kinase/phosphatase. J. Biol. Chem. 276, 26154–26163 [DOI] [PubMed] [Google Scholar]
- 43. Jones S. E., Lennon J. T. (2010) Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. U.S.A. 107, 5881–5886 [DOI] [PMC free article] [PubMed] [Google Scholar]