

Background: Kv7 potassium channels are regulated by protein kinase C (PKC) and may assemble as Kv7.4/Kv7.5-heteromers.
Results: Kv7.4/Kv7.5-heteromers are endogenously expressed in artery myocytes; both subunits are differentially regulated by PKC.
Conclusion: Regulation of Kv7 channels by PKC depends on its subunit composition.
Significance: Insights into the mechanisms controlling Kv7 currents are important to understand how membrane potential is regulated.
Keywords: Patch Clamp Electrophysiology, Potassium Channels, Protein Phosphorylation, Signal Transduction, Vascular Smooth Muscle Cells, KCNQ Channel, Mesenteric Artery, Vasopressin
Abstract
The Kv7 family (Kv7.1–7.5) of voltage-activated potassium channels contributes to the maintenance of resting membrane potential in excitable cells. Previously, we provided pharmacological and electrophysiological evidence that Kv7.4 and Kv7.5 form predominantly heteromeric channels and that Kv7 activity is regulated by protein kinase C (PKC) in response to vasoconstrictors in vascular smooth muscle cells. Direct evidence for Kv7.4/7.5 heteromer formation, however, is lacking. Furthermore, it remains to be determined whether both subunits are regulated by PKC. Utilizing proximity ligation assays to visualize single molecule interactions, we now show that Kv7.4/Kv.7.5 heteromers are endogenously expressed in vascular smooth muscle cells. Introduction of dominant-negative Kv7.4 and Kv7.5 subunits in mesenteric artery myocytes reduced endogenous Kv7 currents by 84 and 76%, respectively. Expression of an inducible protein kinase Cα (PKCα) translocation system revealed that PKCα activation is sufficient to suppress endogenous Kv7 currents in A7r5 rat aortic and mesenteric artery smooth muscle cells. Arginine vasopressin (100 and 500 pm) and the PKC activator phorbol 12-myristate 13-acetate (1 nm) each inhibited human (h) Kv7.5 and hKv7.4/7.5, but not hKv7.4 channels expressed in A7r5 cells. A decrease in hKv7.5 and hKv7.4/7.5 current densities was associated with an increase in PKC-dependent phosphorylation of the channel proteins. These findings provide further evidence for a differential regulation of Kv7.4 and Kv7.5 channel subunits by PKC-dependent phosphorylation and new mechanistic insights into the role of heteromeric subunit assembly for regulation of vascular Kv7 channels.
Introduction
Kv7 voltage-activated potassium channels are expressed in many excitable cell types, including neurons, cardiac, skeletal, vascular, and visceral myocytes (1–15) where they function to stabilize resting membrane potential and restrict cellular excitability (6, 16). The five members of the Kv7 family (Kv7.1–Kv7.5) are encoded by five mammalian genes (KCNQ1–5), and the individual gene products assemble as homomeric or heteromeric tetramers to form functional channels (17).
The composition of Kv7 channels differs among cell types and species. KCNQ1, KCNQ4, and KCNQ5 genes are expressed in the murine, rat, and human vasculature (10, 13, 18, 19), with the KCNQ4 transcripts being most abundant in mouse and rat, and an approximately equal expression of KCNQ1, KCNQ3, KCNQ4, and KCNQ5 in human arteries (13). Pharmacological screening of functional channels in vascular smooth muscle cells suggested that Kv7.4 and/or Kv7.5 are the predominant functional isoforms (10, 11, 13, 18). This conclusion was drawn based on the expression pattern and the ability of retigabine and flupirtine, selective Kv7.2–7.5 channel activators, to enhance Kv7 currents in freshly isolated vascular myocytes and to relax constricted arteries (10, 11, 13, 18).
Vasoconstrictor agonists binding to Gq/11-coupled receptors have been reported to suppress the activity of Kv7 channels in vascular smooth muscle cells, leading to membrane depolarization and enhanced Ca2+ influx via L-type voltage-sensitive Ca2+ channels (11, 12, 20, 21). We have previously shown that physiologically relevant concentrations of vasoconstrictors reduced Kv7 channel activity in vascular smooth muscle cells through a PKC-dependent mechanism (11, 12, 21). Furthermore, based on pharmacological and electrophysiological approaches, we proposed that Kv7.4 and Kv7.5 form predominantly heteromeric channels in vascular smooth muscle cells (22). However, biochemical evidence for the formation and regulation of endogenous Kv7.4/7.5 heteromers is lacking. It remains to be determined whether the Kv7.4 and Kv7.5 subunits in vascular smooth muscle cells are phosphorylated or if they are equally sensitive to PKC-dependent regulation by vasoconstrictor agonists. Our previous studies revealed that the vasoconstrictor hormone arginine vasopressin (AVP)2 induces translocation of PKCα from the cytosol to the plasma membrane in A7r5 rat aortic smooth muscle cells (23). Here we provide evidence that endogenous Kv7.4 and Kv7.5 subunits form heteromeric channels and that activation of PKCα is sufficient to reduce endogenous Kv7 channel activity in both A7r5 cells and rat mesenteric artery smooth muscle cells (MASMCs). We further demonstrate that both Kv7.4 and Kv7.5 channel proteins can undergo PKC-dependent phosphorylation, which depends on the multimeric state of the Kv7 channels. The ability to be phosphorylated corresponds to the sensitivity of the Kv7 channels to AVP in the rank order: Kv7.5 > Kv7.4/7.5 ≫ Kv7.4.
EXPERIMENTAL PROCEDURES
Adenoviral Constructs
The adenoviruses to express human KCNQ4 (Adv-hKCNQ4) and human KCNQ5 (Adv-hKCNQ5, with a FLAG epitope on the amino terminus) were created previously using the AdEasyTM Adenoviral Vector System (Stratagene) (22). The adenovirus to express the rapamycin-induced PKCα translocation system (PKCα-FKBP; PKCα conjugated to enhanced green fluorescent protein and the FK506-binding protein) (24) was a generous gift from Dr. Luis F. Santana (University of Washington, Seattle, WA). This vector also expresses a FKBP12-rapamycin-binding element. Upon rapamycin treatment, the FKBP12-rapamycin-binding element recruits the PKCα-FKBP to the plasma membrane, which results in the activation of PKCα (24).
Single amino acid substitutions at residues located in the pore region (P-loop) of Kv7.4 and Kv7.5 (G285S in Kv7.4 and G278S in Kv7.5) channels have been found to exert dominant-negative (DN) effects on Kv7 channel conductance (7, 9, 25). Dominant-negative hKCNQ4(G285S) was a generous gift from Dr. Mark S. Shapiro (University of Texas Health Sciences Center, San Antonio, TX). The QuikChange Site-directed Mutagenesis Kit (Stratagene) was used according to the manufacturer's protocol to generate the dominant-negative hKCNQ5(G278S) mutant (9). The mutation was confirmed by DNA sequencing (ACGT, Inc.). Bicistronic adenoviruses Adv-hKCNQ4(G285S) and Adv-hKCNQ5(G278S), created using the AdEasyTM Adenoviral Vector System (Stratagene) according to manufacturer's instructions, were amplified, purified on CsCl gradients, and stored at −80 °C until use.
Cell Culture
A7r5 cells were cultured as described previously (26). For overexpression studies, subcultured A7r5 cells were infected with Adv-hKCNQ4 or Adv-hKCNQ5 or both at a multiplicity of infection of 100, plated at 30% confluence, replated 3 days later at a 1:2 dilution and used for immunoprecipitation experiments or electrophysiological measurements 10–12 days after infection. For PKCα translocation studies, A7r5 cells were infected with PKCα-FKBP adenovirus or GFP virus with a multiplicity of infection of 100 and used 3 days later for electrophysiological measurements and fluorescence imaging. Cells expressing the exogenous proteins were identified based on detection of GFP fluorescence.
Expression of Dominant-negative Adv-hKCNQ4(G285S) and Adv-hKCNQ5(G278S) Mutants and PKCα-FKBP in Mesenteric Arteries and Isolation of Myocytes
All animal studies were approved by the Loyola University Chicago Institutional Animal Care and Use Committee and conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, Washington, D. C.). Adult male Sprague-Dawley rats were anesthetized with isoflurane and segments of the small intestinal mesentery were surgically removed as described previously (27).
The methods for infection of MASMCs in intact arterial segments were adapted from a protocol kindly provided by Dr. Dee Van Riper (Albany Medical College, Albany, NY). Virus-containing medium was prepared as follows: 90 μl of adenoviral stock (1 × 1010 to 8 × 1010 particles/ml based on absorbance at 260 nm) was combined with 10 μl of antennapedia peptide (H2N-RQIAIWFQNRRMKWAA-OH, New England Peptide LLC, 0.1 mm) for 30 min; 900 μl of serum-free DMEM (supplemented with 4.5 g/liter of glucose, 110 mg/liter of pyruvate, and 584 mg/liter of l-glutamine) was added to the adenovirus/antennapedia mixture. The antennapedia peptide has been shown to increase the infection rate (28). Mesenteric arteries were cleared of adventitia and adipose tissue. A specially designed chamber containing one glass pipette connected to a pressure column was used to remove the endothelium and infect the arterial segments. One end of each arterial segment was cannulated on a glass pipette and tied in place using nylon suture. Luminal blood was removed and endothelium denudation was performed as described previously (29). Once denuded, the arterial lumen was filled with DMEM containing one of the adenoviruses, tied off using nylon suture, and pressurized to 80 mm Hg. Each arterial segment was incubated at 37 °C for 1 h under 80 mm Hg pressure, transferred to a 35-mm dish containing 2 ml of DMEM/F-12 media supplemented with insulin/transferrin/selenium (Sigma), and placed in a cell culture incubator (37 °C, 5% CO2) for 24 h. Efficiency of infection was assessed by the expression of GFP fluorescence in smooth muscle cells of intact arteries (Fig. 3A). MASMCs from infected arteries were isolated as described previously (11). Based on the presence of detectable GFP fluorescence in MASMCs isolated from infected arteries, ∼30–50% of the myocytes in infected arteries expressed the exogenous constructs. Freshly isolated MASMCs were kept on ice until use. For electrophysiological recordings, the cell suspension was added to a recording chamber with a 25-mm glass coverslip base and allowed to adhere for at least 15 min at room temperature.
FIGURE 3.
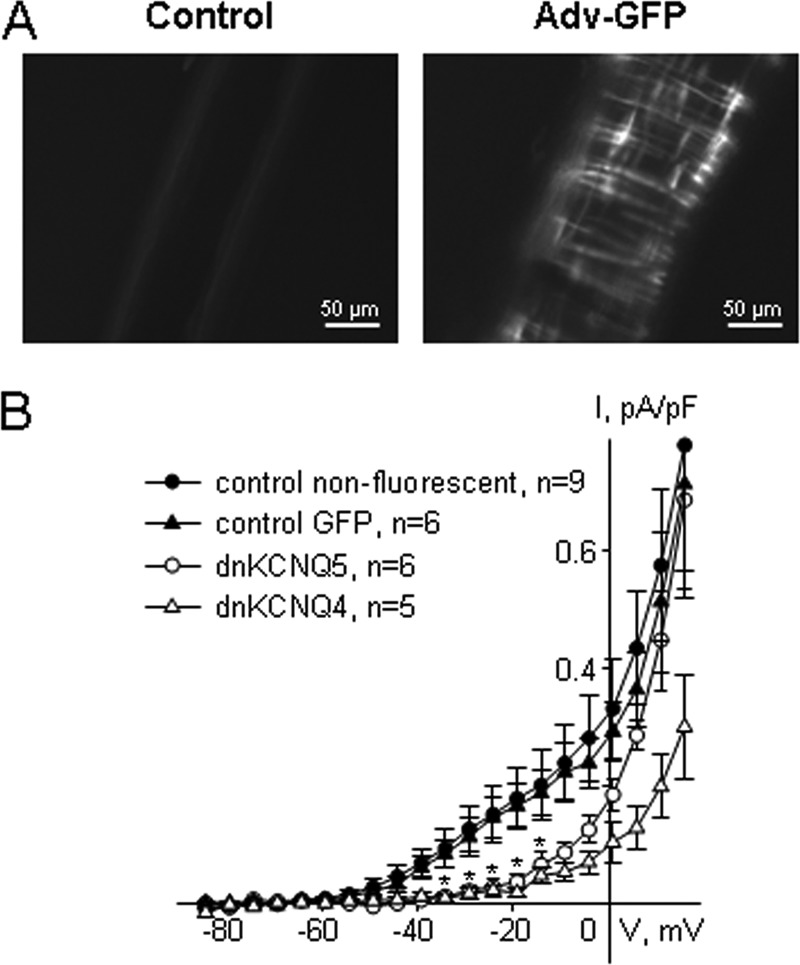
Dominant-negative hKCNQ4(G285S) and hKCNQ5(G278S) overexpressed in MASMCs suppress endogenous Kv7 currents. A, images of an uninfected mesenteric artery (left) or an artery infected with GFP-expressing adenovirus (right) after a 24-h incubation following the infection. B, mean Kv7 current amplitudes measured in non-fluorescent myocytes from MAs infected with either Adv-hKCNQ4(G285S) or with Adv-hKCNQ5(G278S) (filled circle, n = 9); in GFP fluorescent myocytes from the MAs infected with empty GFP-expressing adenovirus (filled triangles, n = 6); in GFP fluorescent myocytes from the MAs infected with Adv-hKCNQ5(G278S) (dnKCNQ5; open circle, n = 6); and in GFP fluorescent myocytes from the MAs infected with Adv-hKCNQ4(G285S) (dnKCNQ4; open triangles, n = 5). * indicates a significant difference for dnKCNQ4 and dnKCNQ5 from control GFP, p < 0.05, two-way ANOVA.
Patch Clamp
The whole cell perforated patch configuration was used to measure membrane currents in smooth muscle cells under voltage-clamp conditions. All experiments were performed at room temperature with continuous perfusion of bath solution as described previously (11, 12, 29). The standard bath solution for A7r5 cells contained (in mm): 5 KCl, 130 NaCl, 10 HEPES, 2 CaCl2, 1.2 MgCl2, 5 glucose, pH 7.3. Standard internal (pipette) solution for A7r5 cells contained (in mm): 110 potassium gluconate, 30 KCl, 5 HEPES, 1 K2EGTA, pH 7.2. Osmolality was adjusted to 298 mosmol/liter with d-glucose. The standard bath solution for MASMCs contained (in mm): 140 NaCl, 5.36 KCl, 1.2 MgCl2, 2 CaCl2, 10 HEPES, 10 d-glucose, pH 7.3, 298 mosmol/liter. Standard internal (pipette) solution for MASMCs contained (in mm): 135 KCl, 5 NaCl, 10 HEPES, 0.05 K2EGTA, 1 MgCl2, 20 d-glucose, pH 7.2, 298 mosmol/liter. To isolate Kv7 currents, 100 μm GdCl3 (sufficient to block L- and T-type Ca2+ channels, non-selective cation channels, and to shift activation of 4-AP-sensitive Kv channels to more positive voltages) was added to external solutions (12).
Voltage-clamp command voltages were generated using an Axopatch 200B amplifier under control of PCLAMP8 software. 120 μg/ml of Amphotericin B in the internal solution was used for membrane patch perforation. Series resistances after amphotericin perforation were 8–15 MΩ and were compensated by 60% in cells overexpressing Kv7 channels. Whole-cell currents were digitized at 2 kHz and filtered at 1 kHz. The last 2000 points recorded during each voltage step (corresponding to 1000 ms recording time) were averaged and normalized by cell capacitance to obtain end pulse steady-state K+ current. Stable currents were recorded for at least 15 min prior to drug application.
Overexpressed hKv7 currents were recorded using a 5-s voltage step protocol from a −74 mV holding voltage to test voltages ranging from −114 to −4 mV followed by a 1-s step to −30 mV. To analyze the voltage dependence of channel activation the conductance (G) was calculated from steady-state K+ currents according to the equation: G = I/(V − Erev), where I is the steady-state current, V is the step potential, and Erev is the reversal potential (30). Erev for potassium was calculated to be −86 mV using the Nernst equation. Conductance plots were fitted to a Boltzmann distribution: G(V) = Gmax/[1 + exp(V0.5 −V)/s], where G is conductance, Gmax is a maximal conductance, V0.5 is the voltage of half-maximal activation, and s is the slope factor. To measure endogenous currents in A7r5 cells, a 5-s voltage step protocol was used (from a −74 mV holding voltage to test voltages ranging from −94 mV to +36 mV). Kv7 currents in MASMCs were recorded by application of 5-s voltage steps from a −4 mV holding voltage to test voltages ranging from −84 to +16 mV. Time courses of rapamycin effects on Kv7 currents were recorded at −20 mV holding voltage.
Imaging
PKCα translocation in PKCα-FKBP-expressing A7r5 cells and MASMCs was monitored with an Olympus 1X71 inverted epifluorescence microscope and Hamamatsu Orca 12-bit digital camera. Gray scale images were captured at 480 nm excitation and 535 nm emission wavelengths. Regions of interest in the periphery (membrane) and center (cytosol) of GFP fluorescent cells were defined using Simple PCI software (Compix Inc.). Average gray level fluorescence intensity of each region of interest was recorded at a rate of 0.1 Hz, while concurrently recording Kv7 currents by patch clamp. The ratio of gray levels from the region of interest at the membrane to that of the cytosol was determined at each time point.
Duolink Proximity Ligation Assay (PLA)
Duolink PLA assays (Olink Bioscience) were performed as per the manufacturer's instructions. MASMCs were allowed to adhere to 8-well tissue culture slides (Permanox,® Nunc), and then fixed for 15 min with 2% paraformaldehyde in phosphate-buffered saline (PBS). The cells were permeabilized with 0.5% Triton X-100 in PBS for 20 min at room temperature and then blocked with 3% bovine serum albumin in PBS for 2 h. Primary mouse anti-KCNQ4 antibodies (Abcam, ab84820) at 1:200 dilution; rabbit anti-KCNQ5 (Millipore-Chemicon, AB5599) at 1:200 dilution; rabbit anti-KCNQ5 (Abcam, ab96707) at 1:100 dilution; rabbit anti-TRPC6 (Alomone Labs, ACC-017) at 1:200 dilution were applied at 37 °C for 2 h in a humidifying chamber. As an antibody control, slides were incubated with PBS without primary antibody. To detect individual target proteins, a single primary antibody was used and the slides were washed and incubated (1 h, 37 °C) with secondary antibodies conjugated with plus and minus Duolink II PLA probes (1:5). The plus and minus PLA probes (antibodies with attached DNA strands) serve as a template for hybridization of circularization oligonucleotides (31). After washing, slides were incubated with ligation-ligase solution to join the adjacent oligonucleotides by ligation into circular DNA molecules (30 min, 37 °C). This was followed by incubation with an amplification-polymerase solution (2 h, 37 °C), as per the manufacturer's protocol. Under these conditions, the circular DNA molecules were amplified by rolling-circle amplification primed by one of the proximity probes, thus creating a concatemeric amplification product that remained covalently attached to the proximity probe (31). The rolling-circle amplification product was subsequently detected by hybridization of fluorescently labeled oligonucleotides. Slides were mounted with a minimal volume of Duolink II Mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) for 15–30 min and PLA signals (Duolink In Situ Detection Reagents Red (λexcitation/emission 598/634 nm)) were identified as fluorescent dots under a Zeiss Axiovert 200M microscope with an EC Plan-Neofluor ×100/1.30 oil objective and a Zeiss Axio CamMRc5 camera. A series of images (∼8–10/cell) was acquired with z axis scanning (1-μm z axis interval) and a wavelet fusion algorithm was used to combine those images into one single composite z-stack image (Zeiss AxioVision Rel.4.8.2 software). Z-stacked images were merged with DAPI and phase-contrast images using Adobe Photoshop CS6 software. Images were captured and processed identically across all experimental groups.
To determine subunit interactions, a pair of primary antibodies was used, one raised in mouse and the other in rabbit. Slides were then incubated with secondary anti-rabbit/mouse antibodies conjugated with plus/minus Duolink II PLA probes (1:5). PLA signals were detected as described above. Quantification of receptor interactions (as PLA signals per cell) was performed using Duolink Image Tool software (Sigma). Images containing both signal and nuclei channels were merged and imported in TIFF formats. Merged images were checked visually and verified for analytical quality. A total of 21 fields (7 fields from each of 3 different cell preparations) was analyzed for each interaction.
Immunoprecipitation
A7r5 cells were infected with Adv-hKCNQ4 or Adv-hKCNQ5 or both and subcultured on 100-mm dishes for 11–12 days. Infected A7r5 cells were serum-deprived overnight. On the day of the experiment, A7r5 cells were incubated for 3 h at room temperature in modified Krebs buffer (in mm): 5.9 KCl, 135 NaCl, 11.6 HEPES, 1.5 CaCl2, 1.2 MgCl2, 11.5 glucose, pH 7.3, and either left untreated or treated for 30 min with 100 pm AVP, 500 pm AVP, or 1 nm PMA. For a subset of experiments, A7r5 cells infected with Adv-hKCNQ5 were incubated for 2 h at room temperature in modified Krebs buffer, pretreated with 5 μm Ro-31-8220 for 1 h, and treated for 30 min with 500 pm AVP plus 5 μm Ro-31-8220. Immediately after treatments, A7r5 cells were lysed with ice-cold lysis buffer containing (in mm): 100 mm NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 30 mm sodium pyrophosphate, 5 mm β-glycerophosphate, 20 mm HEPES, pH 7.4. For immunoprecipitation, lysates containing 800 μg of protein were incubated overnight at 4 °C with 4 μg of anti-FLAG rabbit polyclonal antibody (Sigma F7425), 10 μg of anti-phospho-(Ser) PKC substrate antibody (Cell Signaling Technology, number 2261), or 5 μg of anti-KCNQ4 antibody (Santa Cruz Biotechnology, H-130). Lysates incubated without primary antibody or with 10 μg of rabbit IgG were used as controls. After overnight incubation, 20 μl of Dynabeads® Protein G (Invitrogen) were added to antibody-containing lysates for 2 h. The immune complexes were washed 3 times for 5 min, eluted with 1× Laemmli sample buffer (5% SDS, 10% glycerol, 50 mm Tris, pH 6.8, 0.01% bromphenol blue, 10% β-mercaptoethanol, 100 mm dithiothreitol), heated for 5 min at 97 °C, and separated by SDS-PAGE on 10% acrylamide gels. Proteins were transferred to PVDF membranes, blocked with 5% nonfat milk, and probed with anti-FLAG antibody (rabbit polyclonal 1:1000, Sigma F7425), anti-phospho-(Ser) PKC substrate antibody (rabbit polyclonal 1:1000, Cell Signaling Technology, number 2261), or anti-KCNQ4 antibody (mouse monoclonal 1:1000, Abcam AB84820) overnight at 4 °C. For loading controls, 50 μg of protein from lysates for each experiment were run on separate 10% acrylamide gels, probed with rabbit anti-FLAG antibody for hKv7.5 expression, with mouse anti-KCNQ4 antibody for hKv7.4 expression and with both antibodies for hKv7.4 and hKv7.5 expressed together. Membranes were washed 6 times for 5 min at room temperature and incubated with anti-rabbit IgG or anti-mouse IgG HRP-conjugated secondary antibodies for 1 h at room temperature. Membranes were washed and proteins were visualized by enhanced chemiluminescence (ECL kit, Pierce). ImageJ software (32) was used for densitometry.
Statistics
Data are expressed as mean ± S.E. SigmaStat (Systat Software, Inc.) was used for all statistical analyses. Paired Student's t test was used for comparisons of parameters measured before and after treatments. Comparisons among multiple treatment groups were evaluated by analysis of variance (ANOVA) followed by a Holm-Sidak post hoc test or ANOVA on Ranks followed by a multiple comparisons versus control group (Dunn method). Differences associated with p values ≤0.05 were considered statistically significant.
Materials
Cell culture media were from MediaTech (Herndon, VA). Collagenase, elastase, [Arg8]vasopressin, and ITS were from Sigma. XE991 dihydrochloride was from Ascent Scientific (Princeton, NJ). Amphotericin B, PMA, and Ro-31-8220 were from Calbiochem. Rapamycin was from LC Labs (Woburn, MA). Dynabeads® Protein G were from Invitrogen. The Duolink PLA kit was from Olink Bioscience (Uppsala, Sweden).
RESULTS
Detection of Heteromeric Kv7.4/7.5 Channels in MASMCs
To visualize individual Kv7.4 and Kv7.5 channels and the heteromeric assembly of endogenous Kv7.4/7.5 channels in mesenteric artery myocytes we utilized the Duolink in situ PLA. As a negative control, we also visualized TRPC6 non-selective cation channels, which are known to be expressed in MASMCs (33), but, as an ion channel of a different class, would not be expected to physically interact with Kv7 channel subunits. Kv7.4, Kv7.5, and TRPC6 could each be visualized individually in MASMCs (Fig. 1). Omission of the primary antibody abolished the PLA signal (Fig. 1, bottom row). To detect an interaction between Kv7.4 and Kv7.5, MASMCs were incubated with both mouse anti-KCNQ4 and rabbit anti-KCNQ5 antibodies. All MASMCs examined (n = 21 cells from 3 different preparations) displayed punctal fluorescence signals indicating close proximity (<40 nm) between Kv7.4 and Kv7.5 subunits (Fig. 2, A and B). Similar results were obtained using a different rabbit anti-KCNQ5 antibody (Abcam ab96707, data not shown). PLA signals were not detected when the primary anti-KCNQ5 antibody was omitted (Fig. 2A, bottom row). When anti-KCNQ4 was paired with anti-TRPC6, very few PLA signals were detected (an average of 2.6 ± 0.6 signals/cell), significantly fewer than with the Kv7.4/Kv7.5 antibody pair (37.7 ± 6.6 signals/cell, p < 0.001) (Fig. 2, A and B).
FIGURE 1.

Detection of endogenous Kv7.4, Kv7.5, and TRP6 channels in MASMCs based on PLA. Images of MASMCs stained with mouse anti-KCNQ4 antibody (Kv7.4), rabbit anti-KCNQ5 antibody (Kv7.5), or rabbit anti-TRPC6 antibody (TRPC6). Ab ctrl, omission of the primary antibody. phase, phase contrast. PLA, PLA fluorescence signal. PLA/DAPI, merged PLA/DAPI signals. Merge, merged phase contrast/PLA/DAPI signals. Images are representative of 3 independent experiments. Scale bar: 10 μm.
FIGURE 2.

Detection of interactions between endogenous Kv7.4 and Kv7.5 in MASMCs based on PLA. A, images of MASMCs stained with a combination of mouse anti-KCNQ4 antibody and rabbit anti-KCNQ5 antibody (Kv7.4-Kv7.5, top panel) or with a combination of mouse anti-KCNQ4 antibody and rabbit anti-TRPC6 antibody (Kv7.4-TRPC6, middle panel). Ab ctrl, omission of one secondary antibody after incubation with mouse anti-KCNQ4 and rabbit anti-KCNQ5 antibodies. phase, phase contrast. PLA, PLA fluorescence signal. PLA/DAPI, merged PLA/DAPI signals. Merge, merged phase-contrast/PLA/DAPI signals. Images are representative of 3 independent experiments. Scale bar: 10 μm. B, quantification of the number of PLA signals per cell for Kv7.4-Kv7.5 (black bar, n = 21) and Kv7.4-TRPC6 (white bar, n = 21) from 3 independent cell preparations (7 randomly selected non-overlapping vision fields per experiment). *, p < 0.001, Mann-Whitney Rank Sum Test.
Expression of Dominant-negative Kv7.4 or Kv7.5 Reduces Native Kv7 Currents in MASMCs
To investigate the involvement of both Kv7.4 and Kv7.5 subunits in the function of endogenous Kv7 channels in mesenteric artery (MA) myocytes, we used bicistronic adenoviruses to express dominant-negative human Kv7.4 (hKCNQ4(G285S)) or dominant-negative human Kv7.5 (hKCNQ5(G278S)) subunits together with GFP in segments of MAs with disrupted endothelium. Introduction of the adenoviruses into denuded MA segments induced expression of the dominant-negative subunits in smooth muscle cells within 24 h. Infection was confirmed by GFP fluorescence in the smooth muscle cell layer oriented perpendicularly to the length of the arteries (Fig. 3A, right panel). No GFP fluorescence was detected in the smooth muscle cell layer in non-infected arteries (Fig. 3A, left panel). Introduction of hKCNQ5(G278S) into MASMCs reduced Kv7 current densities by 76 ± 4 and 74 ± 4% (in the voltage range from −30 to −10 mV) relative to current densities measured from two different controls (1: non-fluorescent myocytes, and 2: GFP-fluorescent myocytes from MAs infected with empty GFP-expressing adenovirus). Introduction of hKCNQ4(G285S) also reduced Kv7 current densities in the same voltage range by 84 ± 3 and 83 ± 3%, relative to current densities from the same pair of controls (Fig. 3B). It is important to note that at voltages positive to −10 mV, significant contributions of potassium currents other than Kv7 currents are usually detected in MASMCs (11). In the present study, the only statistically significant difference in current densities among non-fluorescent, GFP-expressing, hKCNQ4(G285S)-expressing, and hKCNQ5(G278S)-expressing myocytes in the voltage range from −10 to +15 mV was between hKCNQ4(G285S)-expressing and non-fluorescent controls at +15 mV (p < 0.05, two-way ANOVA).
Activation of PKCα Is Sufficient for Suppression of Native Kv7 Currents in A7r5 Cells and MASMCs
Our previous findings suggest an involvement of PKC in suppression of endogenous Kv7 currents in MASMCs, which express both KCNQ4 and KCNQ5 (11, 22), and in the A7r5 smooth muscle cell line, which expresses only KCNQ5 (12, 20). To test whether PKC activation is sufficient to suppress native Kv7 currents, we utilized a rapamycin-induced PKCα translocation system (24). Application of rapamycin promotes cytosol-to-membrane translocation of PKCα-FKBP sufficient for PKCα activation (24). GFP fluorescent A7r5 cells infected with PKCα-FKBP adenovirus were chosen for electrophysiological recording of endogenous Kv7.5 current with concurrent detection of PKCα-FKBP translocation. Application of rapamycin (100 nm) induced 89 ± 5% suppression of sustained Kv7.5 current recorded at −20 mV holding voltage (p < 0.01, n = 3, paired t test; Fig. 4, A and B). In some cases a reduction of the Kv7 current preceded a detectable change in PKCα distribution, measured as the ratio of membrane to cytosol fluorescence intensity (Fig. 4, C and D). As a control, A7r5 cells were infected with empty GFP-expressing adenovirus; application of 100 nm rapamycin had no effect on Kv7.5 current amplitude and there was no detectable GFP translocation in GFP-expressing A7r5 cells (data not shown).
FIGURE 4.

Suppression of endogenous Kv7 currents in A7r5 cells and MASMCs upon activation of PKCα using a rapamycin-induced PKCα translocation system. A, representative time course of Kv7 current inhibition in an A7r5 cell upon application of rapamycin (100 nm). B, mean I-V curves of steady-state Kv7 currents recorded in A7r5 cells with a voltage step protocol before (control, filled circles) and after treatment with 100 nm rapamycin for 12 min (open circles) (n = 3). C, representative time course of PKCα translocation recorded simultaneously with current inhibition presented on panel A in an A7r5 cell upon application of rapamycin (100 nm). D, representative images of an A7r5 cell used for recordings for panels A and C before application of rapamycin (100 nm, left panel) and at the end of application of rapamycin for 12 min (right panel). E, representative time course of Kv7 current inhibition in a MASMC upon application of rapamycin (100 nm). F, mean I-V curves of steady-state Kv7 currents recorded in MASMCs with a voltage step protocol before (control, filled circles), after treatment with 100 nm rapamycin for 12 min (open circles) and after treatment with 10 μm XE991 (filled triangles). Currents were normalized by the current recorded at −20 mV before application of rapamycin. *, significant difference from control, p < 0.05, Student's paired t test, n = 4). G, representative time course of PKCα translocation recorded in an MASMC upon application of rapamycin (100 nm). H, representative images of the MASMC used for recordings for panel G before application of rapamycin (100 nm, left panel) and at the end of application of rapamycin for 10 min (right panel).
We also examined whether translocation of PKCα from the cytosol to the membrane is sufficient to suppress the endogenous Kv7 current in MASMCs infected with PKCα-FKBP adenovirus. Application of rapamycin (100 nm) induced a 50 ± 8% suppression of sustained Kv7 current recorded at −20 mV holding voltage in GFP-fluorescent myocytes (p < 0.01, n = 4, paired t test; Fig. 4, E and F) in parallel with translocation of PKCα-FKBP to the plasma membrane (Fig. 4, G and H).
Differential Regulation of Kv7.4 and Kv7.5 Currents by AVP
To test whether both Kv7.4 and Kv7.5 subunits are involved in the PKC-dependent modulation of vascular Kv7 channel activity, we used A7r5 cells as an expression system to introduce exogenous hKv7.4 and hKv7.5, individually or together. Adenoviral vectors were used to overexpress hKv7.4, hKv7.5, or both, as described previously (22). The overexpressed channels produce robust currents that are easily distinguished from the native currents, which have ∼60–100-fold smaller current densities (compare y axis scales in Fig. 4B with those in Fig. 5, A, C, and E). In A7r5 cells overexpressing hKv7.5 channels, AVP, at two different concentrations (100 and 500 pm), significantly suppressed the hKv7.5 current (by 66 ± 7% (n = 9) and 80 ± 6% (n = 6), respectively, at −20 mV; Fig. 5A). Direct activation of PKC by PMA (1 nm) also significantly suppressed the hKv7.5 current (by 86 ± 4% at −20 mV, n = 6; Fig. 5A). A Boltzmann fit of the activation curves of exogenous hKv7.5 channels revealed that application of both AVP (100 and 500 pm) and PMA (1 nm) significantly reduced maximal conductance (Gmax) without changes in the voltage of half-maximal activation (V0.5). The activation curves of hKv7.5 channels had a steeper slope in the presence of AVP and PMA (Table 1).
FIGURE 5.

hKv7.5 and hKv7.4 exogenously expressed in A7r5 cells as homomeric and heteromeric channels are differentially regulated by AVP. A, C, and E, I-V curves of steady-state Kv7 currents through homomeric hKv7.5 (A), homomeric hKv7.4 (C), and heteromeric hKv7.4/7.5 (E) recorded in A7r5 cells before (control, filled circles) and after treatments with 100 pm AVP for 10 min (open circles), 500 pm AVP for 10 min (open triangles), and 1 nm PMA for 10 min (open squares). B, D, and F, averaged fractional conductance plots in control (filled circles), and after treatments with 100 pm AVP for 10 min (open circles), 500 pm AVP for 10 min (open triangles), and 1 nm PMA for 10 min (open squares) in A7r5 cells expressing hKv7.5 (B, n = 6–12), hK7.4 (D, n = 4–6), or both hKv7.4 and hKv7.5 (F, n = 5) fitted to the Boltzmann equation.
TABLE 1.
Activation parameters of hKv7.5 channels
| hKv7.5 | Control | 100 pm AVP | 500 pm AVP | 1 nm PMA |
|---|---|---|---|---|
| Gmax, pS/pF | 0.31 ± 0.05 | 0.10 ± 0.03a | 0.07 ± 0.03a | 0.04 ± 0.01a |
| V0.5, mV | −42.2 ± 2.3 | −41.6 ± 3.0 | −44.0 ± 2.8 | −49.8 ± 6.1 |
| s, mV | 12.9 ± 0.7 | 8.5 ± 0.4a | 7.0 ± 0.7a | 7.5 ± 0.6a |
a Significant difference from control, one-way ANOVA, p < 0.001, n = 9–12.
In A7r5 cells overexpressing hKv7.4 channels, neither AVP (100 or 500 pm) nor PMA (1 nm) significantly reduced the Kv7 current. The only statistically significant change to the parameters of the activation curve was an ∼5 mV positive shift in V0.5 in the presence of 500 pm AVP (Table 2; Fig. 5, C and D).
TABLE 2.
Activation parameters of hKv7.4 channels
| hKv7.4 | Control | 100 pm AVP | 500 pm AVP | 1 nm PMA |
|---|---|---|---|---|
| Gmax, pS/pF | 0.64 ± 0.19 | 0.58 ± 0.17 | 0.54 ± 0.15 | 0.51 ± 0.23 |
| V0.5, mV | −32.4 ± 1.9 | −30.9 ± 2.1 | −27.8 ± 2.2a | −26.2 ± 1.6 |
| s, mV | 11.3 ± 0.3 | 10.8 ± 0.5 | 9.8 ± 0.4 | 9.4 ± 0.5 |
a Significant difference from control, paired t test, p < 0.05, n = 5.
When both hKv7.4 and hKv7.5 channels were overexpressed together, the resulting currents were suppressed by both AVP and PMA. Application of 100 pm AVP resulted in a 52 ± 10% suppression of current at −20 mV (n = 6). Subsequent application of 500 pm AVP induced 72 ± 8% suppression at the same voltage (n = 6). PMA (1 nm), added after washout of AVP, induced 85 ± 4% current suppression (n = 6, Fig. 5E). Boltzmann fits of the activation curves of heteromeric hKv7.4/7.5 channels revealed that application of both AVP (100 and 500 pm) and PMA (1 nm) significantly reduced Gmax. A trend toward a positive shift of the activation curves reached significance only for 500 pm AVP, whereas a steeper slope of the activation curves of hKv7.4/7.5 channels was observed only in the presence of 1 nm PMA (Table 3).
TABLE 3.
Activation parameters of hKv7.4/7.5 channels
| hKv7.4/7.5 | Control | 100 pm AVP | 500 pm AVP | 1 nm PMA |
|---|---|---|---|---|
| Gmax, pS/pF | 0.62 ± 0.15 | 0.33 ± 0.12a | 0.21 ± 0.08a | 0.10 ± 0.03a |
| V0.5, mV | −42.0 ± 2.0 | −37.3 ± 1.8 | −35.4 ± 1.7b | −37.7 ± 2.4 |
| s, mV | 13.4 ± 0.9 | 10.5 ± 0.9 | 9.9 ± 0.6 | 6.9 ± 1.9a |
a Significant difference from control, one-way ANOVA, p < 0.05, n = 5.
b Significant difference from control, paired t test, p < 0.05, n = 5.
PKC-dependent Phosphorylation of Kv7.4 and Kv7.5 Channel Proteins
PKC-dependent phosphorylation of Kv7 channel subunits in response to AVP and PMA treatments was assessed by immunoprecipitation of the channel proteins either with an anti-FLAG antibody (for FLAG-tagged hKv7.5 and heteromeric hKv7.4/7.5 channels) or with an anti-KCNQ4 antibody. The FLAG-tagged Kv7.5 protein appears as a double band at ∼104 kDa in Western blots, whereas Kv7.4 appears as a double band at ∼60 kDa. The anti-FLAG and anti-Kv7.4 bands (see Fig. 7) are absent in non-infected A7r5 cells (results not shown). Phosphorylation of Kv7.4 and Kv7.5 was detected by immunoblotting with an anti-phosphoserine PKC substrate antibody. Treatment of A7r5 cells overexpressing Kv7.5 with 100 or 500 pm AVP (30 min) increased phosphorylation of FLAG-tagged hKv7.5 channel protein by 3.9 ± 1.1- and 4.7 ± 1.8-fold, respectively. Treatment with 1 nm PMA (30 min) induced a 5.0 ± 1.0-fold increase in Kv7.5 channel phosphorylation (Fig. 6, A and D). Pretreatment of A7r5 cells overexpressing hKv7.5 with the PKC inhibitor Ro-31-8220 (5 μm for 1 h) prevented the increase in phosphorylation upon treatment with 500 pm AVP without changes in the basal level of hKv7.5 channel phosphorylation (Fig. 6, B and E). Although Kv7.5 channels were increasingly phosphorylated on serine residues in response to treatments with AVP (100 and 500 pm) and PMA (1 nm), basal phosphorylation of Kv7.4 channels was not changed upon AVP or PMA treatments (Fig. 6, C and F).
FIGURE 7.

AVP and PMA increase phosphorylation of both hKv7.5 and hKv7.4 proteins within heteromeric channels. A, representative Western blots (WB) of Kv7.4/7.5 channels immunoprecipitated (IP) with anti-FLAG antibody and probed with anti-phospho-(Ser) PKC substrate antibody (left), the same membrane re-probed with anti-FLAG (middle) and anti-KCNQ4 antibody (right). A7r5 cells overexpressing both hKv7.4 and hKv7.5 were left untreated or treated with AVP (100 pm or 500 pm) or PMA (1 nm) for 30 min as indicated below the panels. B and C, densitometric analysis of phosphorylation of hKv7.5 (B) and hKv7.4 (C), relative to untreated control, upon treatment with 100 and 500 pm AVP and 1 nm PMA (n = 8, *, significant difference from control, p < 0.05, ANOVA on Ranks).
FIGURE 6.

AVP and PMA stimulate PKC-dependent phosphorylation of hKv7.5 but not hKv7.4 channels. A, representative Western blots (WB) of hKv7.5 channels immunoprecipitated with anti-FLAG antibody and blotted with anti-phospho-(Ser) PKC substrate antibody (top). A7r5 cells overexpressing hKv7.5 were left untreated or treated with AVP (100 pm or 500 pm) or PMA (1 nm) for 30 min as indicated below the panel. Whole cell lysates (WCL, 50 μg of total protein) were run separately and probed with anti-FLAG antibody. B, representative Western blots of Kv7.5 channels immunoprecipitated (IP) with anti-FLAG antibody and blotted with anti-phospho-(Ser) PKC substrate antibody (top panel). A7r5 cells overexpressing hKv7.5 were left untreated or were treated with 500 pm AVP for 30 min with or without pretreatment with Ro-31-8220 (5 μm) as indicated below the panel. Whole cell lysates (50 μg of total protein) were run separately and probed with anti-FLAG antibody. C, representative Western blots of hKv7.4 channels immunoprecipitated with anti-KCNQ4 antibody and blotted with anti-phospho-(Ser) PKC substrate antibody (top). A7r5 cells overexpressing hKv7.4 were treated as described for panel A. Whole cell lysates (50 μg of total protein) were run separately and probed with anti-KCNQ4 antibody. D, mean densitometric analyses of Western blots to evaluate phosphorylation of Kv7.5 channels. Anti-phospho-(Ser) PKC substrate blots were normalized to input signal from WCL blots for the same samples. Treatments with 100 and 500 pm AVP, and 1 nm PMA were compared with untreated controls (n = 7, * significant difference from control, p < 0.01, one-way ANOVA). E, densitometric analysis of Kv7.5 channel phosphorylation, relative to untreated control, upon treatment with 500 pm AVP with or without pretreatment with Ro-31-8220 (5 μm) (n = 3, *, significant difference from indicated columns, p < 0.001, one-way ANOVA). F, densitometric analysis of the level of hKv7.4 channel phosphorylation, relative to untreated control, upon treatment with 100 and 500 pm AVP, and 1 nm PMA (n = 7).
Because hKv7.4 and hKv7.5 form predominantly heteromeric channels when expressed together (22), we chose to co-immunoprecipitate hKv7.4 and hKv7.5 channel subunits and examine the phosphorylation state of both proteins. A7r5 cells overexpressing both hKv7.4 and hKv7.5 were left untreated or were treated with AVP (100 or 500 pm) or PMA (1 nm) for 30 min. FLAG-tagged hKv7.5 channel subunits and associated proteins were co-immunoprecipitated with anti-FLAG antibodies. Interestingly, immunoblotting with anti-phosphoserine PKC substrate antibody revealed an increase in phosphorylation of both co-immunoprecipitated Kv7.4 and Kv7.5 channel subunits in response to treatments with AVP and PMA (Fig. 7).
DISCUSSION
Kv7 channels form as tetrameric assemblies of Kv7.1-Kv7.5α subunits. Each of the five Kv7 family members can form homotetramers, but some heterotetrameric combinations have also been described (17). For example, Kv7.2/7.3 and Kv7.5/7.3 heteromeric channels underlie neuronal M-currents (6, 8, 9). Recently, Kv7.4/Kv7.5 heterotetramers were demonstrated in an expression system based on Förster resonance energy transfer (FRET) and also based on co-expression of dominant-negative mutants of KCNQ4(G285S) and KCNQ5(G278S) with corresponding wild type KCNQ5 and KCNQ4 subunits (25, 34). In the present study we also utilized a dominant-negative approach to reveal suppression of native Kv7 currents. Introducing either KCNQ4(G285S) or KCNQ5(G278S) dominant-negative subunits in MA myocytes significantly reduced densities of native Kv7 currents, leading us to conclude that Kv7.4 and/or Kv7.5 subunits are essential components of the native channel complexes (Fig. 3B). Introduction of either KCNQ4(G285S) or KCNQ5(G278S) also induced a positive shift in the voltage of activation, supporting the proposed role of KCNQ-encoded channels in regulation of resting membrane voltage in smooth muscle myocytes.
The notion that both Kv7.4 and Kv7.5 subunits are found in the same channels (i.e. that they form heteromeric channels) was reinforced by a strong proximity signal for Kv7.4 and Kv7.5 (but not Kv7.4 and TRPC6) using the Duolink PLA assay, which detects protein-protein interactions only when the interacting partners are within 40 nm of one another (31, 35). Although the possibility remains that Kv7.5 homomeric channels are within 40 nm of Kv7.4 homomeric channels, previous electrophysiological and pharmacological studies support predominant formation of heteromeric channels rather than a mixture of homomeric channels (22). The present results provide the first direct biochemical evidence for the existence of functional Kv7.4/7.5 heteromers natively expressed in mammalian cells.
Gq/11-coupled receptor activation leads to suppression of vascular smooth muscle Kv7 currents, although the mechanisms involved in suppression of Kv7 channel activity in vascular myocytes remain to be elucidated. Several mechanisms previously described for agonist-induced suppression of neuronal M-currents could potentially operate in vascular myocytes (e.g. depletion of plasma membrane phosphatidylinositol 4,5-bisphosphate (36–40), or elevation of cytosolic [Ca2+]) (16, 41). Our previous findings, however, strongly support involvement of PKC, a less commonly proposed regulator of M-currents (42, 43), in the suppression of endogenous Kv7 currents by physiologically relevant concentrations of AVP. PKC inhibitors calphostin C and Ro-31-8220 prevented AVP-induced constriction of MAs and abolished AVP-induced suppression of Kv7 currents in MASMCs and A7r5 cells, suggesting that AVP acts in a PKC-dependent manner (11, 12, 27). Furthermore, direct activation of PKC with 1 nm PMA was sufficient to suppress Kv7 currents in both MASMCs and A7r5 cells (11, 12).
Treatment of A7r5 cells with 100 pm AVP was previously found to induce translocation of one classical PKC isoform (PKCα) and two novel PKC isoforms (PKCδ, and PKCϵ) from the cytosol to the plasma membrane (23). In the present study, we used an inducible PKCα translocation system to monitor Kv7 currents during activation of PKCα. Translocation of PKCα to the plasma membrane was sufficient to induce suppression of the Kv7 currents, although the extent of current suppression differed between MASMCs and A7r5 cells. This difference cannot be explained by variations of the amount of PKCα constructs expressed in the two types of smooth muscle cells because MASMCs and A7r5 cells were infected for the same amount of time and cells with comparable levels of GFP fluorescence were selected for the experiments. Moreover, the exogenously expressed PKCα proteins would be expected to vastly exceed the number of endogenous Kv7 channels. One plausible explanation for our observations, however, could be that differences in Kv7 channel composition led to a difference in the extent of the Kv7 current suppression in A7r5 cells and MASMCs.
Currently available information suggests that natively expressed Kv7 channels in A7r5 cells are composed exclusively of homomeric Kv7.5 subunits (12, 22), whereas MASMCs predominantly express heteromeric Kv7.4/7.5 channels (Figs. 2 and 3) (22). Although no specific information is available about the influence of Kv7.4 and Kv7.5 subunit composition on regulation of homomeric versus heteromeric channel function, protein kinases have been reported to exert differential regulatory actions on different Kv7 channel subtypes. For example, overexpressed Src tyrosine kinase was found to distinguish among neuronal Kv7 subtypes (44). Overexpression of Src suppressed currents through homomeric Kv7.3, Kv7.4, Kv7.5, and heteromeric Kv7.2/7.3 channels. These effects were attributed to increased tyrosine phosphorylation of the channel proteins. Notably, Src overexpression did not increase tyrosine phosphorylation or suppress currents through homomeric Kv7.1 or Kv7.2 channels (44). Unlike the other channel subtypes, Kv7.4 channels were constitutively phosphorylated on tyrosine and this was not detectably changed by Src co-expression (44), similar to our finding that basal phosphorylation of hKv7.4 homomers on serine residues was not increased by AVP or PMA treatment.
Although we failed to detect an AVP-induced increase in PKC-dependent phosphorylation or regulation of exogenously expressed homomeric hKv7.4 channels in A7r5 vascular smooth muscle cells, homomeric hKv7.5 and heteromeric hKv7.4/7.5 channels were regulated. One may speculate that Kv7.5 channel subunits are required for recruitment of PKC to the channel complex, thereby ensuring its proximity to both Kv7.4 and Kv7.5 subunits in the heteromeric channel complex, which then enables each of these subunits to be phosphorylated.
The expression of Kv7.4 and Kv7.5 channels in arterial myocytes may change under pathological conditions. For example, recent studies by Jepps et al. (45) and Khanamiri et al. (46) revealed decreased Kv7.4 expression, but not Kv7.5 expression, in arterial myocytes derived from hypertensive rat models compared with normotensive controls. Such changes would be expected to change the subunit stoichiometry or the proportion of homomeric versus heteromeric Kv7.4/7.5 channels, which, based on our present findings, might in turn alter the PKC-dependent phosphorylation and regulation of the channels in response to vasoconstrictor hormones.
The specific serine residues in the Kv7.5 and Kv7.4 proteins that become phosphorylated still remain to be elucidated. The anti-phosphoserine antibody that was used recognizes classical PKC substrates phosphorylated on serine residues that contain the following motif: -K/R-X-S-FILVY-K/R-. The only serines in Kv7.5 that conform exactly to this motif are Ser-385 and Ser-441, located on the cytosolic C-terminal domain. Based on proposed amino acid sequence alignments with other human Kv7 channel proteins (9), neither Ser-385 nor Ser-441 in Kv7.5 corresponds to the previously identified serine residues proposed as likely PKC phosphorylation sites in rat Kv7.2 (Ser-534, Ser-541, Ser-563, and Ser-570 (42, 47)). However, Ser-441 in human Kv7.5 is aligned with Ser-494 in human Kv7.4 (this site also conforms with the phosphoserine PKC substrate antibody recognition motif). Bal and colleagues (48) implicated threonine at position 553 (Thr-553) in A-kinase anchoring protein-mediated PKC-dependent phosphorylation of Kv7.4 channels. Thus, it is likely that multiple phosphorylation sites contribute to the physiological regulation of Kv7.4/7.5 channel function.
Although AVP, at subnanomolar concentrations, was ineffective in suppressing Kv7.4 currents in A7r5 cells, it is important to note that Kv7.4 currents were robustly suppressed by saturating concentrations of a muscarinic receptor agonist when they were expressed in CHO cells (48) and in Xenopus oocytes (47). Nakajo and Kubo (47) also found that Kv7.2/7.3 channels exhibited different responses to PMA when expressed in Xenopus oocytes compared with HEK293 cells (47). These discrepancies highlight the importance of studying function and regulation of channels in their native environment using physiologically relevant agonist concentrations to more accurately predict their roles in physiological responses.
In summary, the current study provides novel molecular and biochemical evidence that endogenous Kv7 channels in MASMCs form as Kv7.4/7.5 heteromeric channels. The regulation of these heteromeric channels by the vasoconstrictor hormone arginine vasopressin is dependent on PKC activation and likely involves PKCα-dependent phosphorylation of the channel proteins on serine residues; the extent of channel phosphorylation and current suppression depends on the subunit composition of the channels.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 HL089564 (to K. L. B.) and American Heart Association Pre-doctoral Fellowship 0715618Z (to A. R. M.).
- AVP
- arginine vasopressin
- MASMC
- mesenteric artery smooth muscle cell
- ANOVA
- analysis of variance
- FKBP
- FK506-binding protein
- FRB
- FKBP12-rapamycin-binding element
- MA
- mesenteric artery
- PLA
- proximity ligation assay
- PMA
- phorbol 12-myristate 13-acetate
- DN
- dominant-negative.
REFERENCES
- 1. Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996) K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384, 78–80 [DOI] [PubMed] [Google Scholar]
- 2. Biervert C., Schroeder B. C., Kubisch C., Berkovic S. F., Propping P., Jentsch T. J., Steinlein O. K. (1998) A potassium channel mutation in neonatal human epilepsy. Science 279, 403–406 [DOI] [PubMed] [Google Scholar]
- 3. Charlier C., Singh N. A., Ryan S. G., Lewis T. B., Reus B. E., Leach R. J., Leppert M. (1998) A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat. Genet. 18, 53–55 [DOI] [PubMed] [Google Scholar]
- 4. Schroeder B. C., Kubisch C., Stein V., Jentsch T. J. (1998) Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396, 687–690 [DOI] [PubMed] [Google Scholar]
- 5. Singh N. A., Charlier C., Stauffer D., DuPont B. R., Leach R. J., Melis R., Ronen G. M., Bjerre I., Quattlebaum T., Murphy J. V., McHarg M. L., Gagnon D., Rosales T. O., Peiffer A., Anderson V. E., Leppert M. (1998) A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 18, 25–29 [DOI] [PubMed] [Google Scholar]
- 6. Wang H. S., Pan Z., Shi W., Brown B. S., Wymore R. S., Cohen I. S., Dixon J. E., McKinnon D. (1998) KCNQ2 and KCNQ3 potassium channel subunits. Molecular correlates of the M-channel. Science 282, 1890–1893 [DOI] [PubMed] [Google Scholar]
- 7. Kubisch C., Schroeder B. C., Friedrich T., Lütjohann B., El-Amraoui A., Marlin S., Petit C., Jentsch T. J. (1999) KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–446 [DOI] [PubMed] [Google Scholar]
- 8. Lerche C., Scherer C. R., Seebohm G., Derst C., Wei A. D., Busch A. E., Steinmeyer K. (2000) Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J. Biol. Chem. 275, 22395–22400 [DOI] [PubMed] [Google Scholar]
- 9. Schroeder B. C., Hechenberger M., Weinreich F., Kubisch C., Jentsch T. J. (2000) KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J. Biol. Chem. 275, 24089–24095 [DOI] [PubMed] [Google Scholar]
- 10. Yeung S. Y., Pucovský V., Moffatt J. D., Saldanha L., Schwake M., Ohya S., Greenwood I. A. (2007) Molecular expression and pharmacological identification of a role for K(v)7 channels in murine vascular reactivity. Br. J. Pharmacol. 151, 758–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mackie A. R., Brueggemann L. I., Henderson K. K., Shiels A. J., Cribbs L. L., Scrogin K. E., Byron K. L. (2008) Vascular KCNQ potassium channels as novel targets for the control of mesenteric artery constriction by vasopressin, based on studies in single cells, pressurized arteries, and in vivo measurements of mesenteric vascular resistance. J. Pharmacol. Exp. Ther. 325, 475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brueggemann L. I., Moran C. J., Barakat J. A., Yeh J. Z., Cribbs L. L., Byron K. L. (2007) Vasopressin stimulates action potential firing by protein kinase C-dependent inhibition of KCNQ5 in A7r5 rat aortic smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 292, H1352-H1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ng F. L., Davis A. J., Jepps T. A., Harhun M. I., Yeung S. Y., Wan A., Reddy M., Melville D., Nardi A., Khong T. K., Greenwood I. A. (2011) Expression and function of the K+ channel KCNQ genes in human arteries. Br. J. Pharmacol. 162, 42–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCallum L. A., Pierce S. L., England S. K., Greenwood I. A., Tribe R. M. (2011) The contribution of Kv7 channels to pregnant mouse and human myometrial contractility. J. Cell Mol. Med. 15, 577–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jepps T. A., Greenwood I. A., Moffatt J. D., Sanders K. M., Ohya S. (2009) Molecular and functional characterization of Kv7 K+ channel in murine gastrointestinal smooth muscles. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G107-G115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hernandez C. C., Zaika O., Tolstykh G. P., Shapiro M. S. (2008) Regulation of neural KCNQ channels. Signalling pathways, structural motifs and functional implications. J. Physiol. 586, 1811–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwake M., Jentsch T. J., Friedrich T. (2003) A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 4, 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Joshi S., Sedivy V., Hodyc D., Herget J., Gurney A. M. (2009) KCNQ modulators reveal a key role for KCNQ potassium channels in regulating the tone of rat pulmonary artery smooth muscle. J. Pharmacol. Exp. Ther. 329, 368–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chadha P. S., Zunke F., Zhu H.-L., Davis A. J., Jepps T. A., Olesen S. P., Cole W. C., Moffatt J. D., Greenwood I. A. (2012) Reduced KCNQ4-encoded voltage-dependent potassium channel activity underlies impaired β-adrenoceptor-mediated relaxation of renal arteries in hypertension. Hypertension 59, 877–884 [DOI] [PubMed] [Google Scholar]
- 20. Mani B. K., Brueggemann L. I., Cribbs L. L., Byron K. L. (2009) Opposite regulation of KCNQ5 and TRPC6 channels contributes to vasopressin-stimulated calcium spiking responses in A7r5 vascular smooth muscle cells. Cell Calcium 45, 400–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mani B. K., O'Dowd J., Kumar L., Brueggemann L. I., Ross M., Byron K. L. (2013) Vascular KCNQ (Kv7) potassium channels as common signaling intermediates and therapeutic targets in cerebral vasospasm. J. Cardiovasc. Pharmacol. 61, 51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brueggemann L. I., Mackie A. R., Martin J. L., Cribbs L. L., Byron K. L. (2011) Diclofenac distinguishes among homomeric and heteromeric potassium channels composed of KCNQ4 and KCNQ5 subunits. Mol. Pharmacol. 79, 10–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fan J., Byron K. L. (2000) Ca2+ signalling in rat vascular smooth muscle cells. A role for protein kinase C at physiological vasoconstrictor concentrations of vasopressin. J. Physiol. 524, 821–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Navedo M. F., Cheng E. P., Yuan C., Votaw S., Molkentin J. D., Scott J. D., Santana L. F. (2010) Increased coupled gating of l-type Ca2+ channels during hypertension and Timothy syndrome. Circ. Res. 106, 748–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bal M., Zhang J., Zaika O., Hernandez C. C., Shapiro M. S. (2008) Homomeric and heteromeric assembly of KCNQ (Kv7) K+ channels assayed by total internal reflection fluorescence/fluorescence resonance energy transfer and patch clamp analysis. J. Biol. Chem. 283, 30668–30676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Byron K. L., Taylor C. W. (1993) Spontaneous Ca2+ spiking in a vascular smooth muscle cell line is independent of the release of intracellular Ca2+ stores. J. Biol. Chem. 268, 6945–6952 [PubMed] [Google Scholar]
- 27. Henderson K. K., Byron K. L. (2007) Vasopressin-induced vasoconstriction. Two concentration-dependent signaling pathways. J. Appl. Physiol. 102, 1402–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gratton J. P., Yu J., Griffith J. W., Babbitt R. W., Scotland R. S., Hickey R., Giordano F. J., Sessa W. C. (2003) Cell-permeable peptides improve cellular uptake and therapeutic gene delivery of replication-deficient viruses in cells and in vivo. Nat. Med. 9, 357–362 [DOI] [PubMed] [Google Scholar]
- 29. Brueggemann L. I., Mackie A. R., Mani B. K., Cribbs L. L., Byron K. L. (2009) Differential effects of selective cyclooxygenase-2 inhibitors on vascular smooth muscle ion channels may account for differences in cardiovascular risk profiles. Mol. Pharmacol. 76, 1053–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wickenden A. D., Zou A., Wagoner P. K., Jegla T. (2001) Characterization of KCNQ5/Q3 potassium channels expressed in mammalian cells. Br. J. Pharmacol. 132, 381–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Söderberg O., Leuchowius K. J., Gullberg M., Jarvius M., Weibrecht I., Larsson L. G., Landegren U. (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232 [DOI] [PubMed] [Google Scholar]
- 32. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ. 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brueggemann L. I., Markun D. R., Henderson K. K., Cribbs L. L., Byron K. L. (2006) Pharmacological and electrophysiological characterization of store-operated currents and capacitative Ca2+ entry in vascular smooth muscle cells. J. Pharmacol. Exp. Ther. 317, 488–499 [DOI] [PubMed] [Google Scholar]
- 34. Spitzmaul G., Tolosa L., Winkelman B. H., Heidenreich M., Frens M. A., Chabbert C., de Zeeuw C. I., Jentsch T. J. (2013) Vestibular role of KCNQ4 and KCNQ5 K+ channels revealed by mouse models. J. Biol. Chem. 288, 9334–9344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 36. Delmas P., Brown D. A. (2005) Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat. Rev. Neurosci. 6, 850–862 [DOI] [PubMed] [Google Scholar]
- 37. Zhang H., Craciun L. C., Mirshahi T., Rohács T., Lopes C. M., Jin T., Logothetis D. E. (2003) PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37, 963–975 [DOI] [PubMed] [Google Scholar]
- 38. Suh B. C., Hille B. (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35, 507–520 [DOI] [PubMed] [Google Scholar]
- 39. Li Y., Gamper N., Hilgemann D. W., Shapiro M. S. (2005) Regulation of Kv7 (KCNQ) K+ channel open probability by phosphatidylinositol 4,5-bisphosphate. J. Neurosci. 25, 9825–9835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zaika O., Lara L. S., Gamper N., Hilgemann D. W., Jaffe D. B., Shapiro M. S. (2006) Angiotensin II regulates neuronal excitability via phosphatidylinositol 4,5-bisphosphate-dependent modulation of Kv7 (M-type) K+ channels. J. Physiol. 575, 49–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gamper N., Li Y., Shapiro M. S. (2005) Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol. Biol. Cell 16, 3538–3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hoshi N., Zhang J. S., Omaki M., Takeuchi T., Yokoyama S., Wanaverbecq N., Langeberg L. K., Yoneda Y., Scott J. D., Brown D. A., Higashida H. (2003) AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat. Neurosci. 6, 564–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tunquist B. J., Hoshi N., Guire E. S., Zhang F., Mullendorff K., Langeberg L. K., Raber J., Scott J. D. (2008) Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Nat. Acad Sci. U.S.A. 105, 12557–12562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gamper N., Stockand J. D., Shapiro M. S. (2003) Subunit-specific modulation of KCNQ potassium channels by Src tyrosine kinase. J. Neurosci. 23, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jepps T. A., Chadha P. S., Davis A. J., Harhun M. I., Cockerill G. W., Olesen S. P., Hansen R. S., Greenwood I. A. (2011) Down-regulation of Kv7.4 channel activity in primary and secondary hypertension. Circulation 124, 602–611 [DOI] [PubMed] [Google Scholar]
- 46. Khanamiri S., Soltysinska E., Jepps T. A., Bentzen B. H., Chadha P. S., Schmitt N., Greenwood I. A., Olesen S. P. (2013) Contribution of Kv7 channels to basal coronary flow and active response to ischemia. Hypertension 62, 1090–1097 [DOI] [PubMed] [Google Scholar]
- 47. Nakajo K., Kubo Y. (2005) Protein kinase C shifts the voltage dependence of KCNQ/M channels expressed in Xenopus oocytes. J. Physiol. 569, 59–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bal M., Zhang J., Hernandez C. C., Zaika O., Shapiro M. S. (2010) Ca2+/calmodulin disrupts AKAP79/150 interactions with KCNQ (M-Type) K+ channels. J. Neurosci. 30, 2311–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]