

Background: Dual specificity phosphatases (DUSPs) have emerged as important regulators of MAPK signaling and responses, including immune responses.
Results: Dusp16-deficient gene trap mice were generated and show a phenotype of perinatal lethality and altered cytokine responses to TLR stimulation.
Conclusion: Dusp16 has non-redundant functions in the innate immune system.
Significance: This is the first description of a mouse model of Dusp16 deficiency.
Keywords: Cytokines/Interferon, Dendritic Cells, Endotoxin, Gene Knock-out, Innate Immunity, Macrophages, MAP Kinases (MAPKs), Dual Specificity Phosphatase, Gene Trap, Perinatal Lethality
Abstract
MAPK activity is negatively regulated by members of the dual specificity phosphatase (Dusp) family, which differ in expression, substrate specificity, and subcellular localization. Here, we investigated the function of Dusp16/MKP-7 in the innate immune system. The Dusp16 isoforms A1 and B1 were inducibly expressed in macrophages and dendritic cells following Toll-like receptor stimulation. A gene trap approach was used to generate Dusp16-deficient mice. Homozygous Dusp16tp/tp mice developed without gross abnormalities but died perinatally. Fetal liver cells from Dusp16tp/tp embryos efficiently reconstituted the lymphoid and myeloid compartments with Dusp16-deficient hematopoietic cells. However, GM-CSF-induced proliferation of bone marrow progenitors in vitro was impaired in the absence of Dusp16. In vivo challenge with Escherichia coli LPS triggered higher production of IL-12p40 in mice with a Dusp16-deficient immune system. In vitro, Dusp16-deficient macrophages, but not dendritic cells, selectively overexpressed a subset of TLR-induced genes, including the cytokine IL-12. Dusp16-deficient fibroblasts showed enhanced activation of p38 and JNK MAPKs. In macrophages, pharmacological inhibition and siRNA knockdown of JNK1/2 normalized IL-12p40 secretion. Production of IL-10 and its inhibitory effect on IL-12 production were unaltered in Dusp16tp/tp macrophages. Altogether, the Dusp16 gene trap mouse model identifies an essential role in perinatal survival and reveals selective control of differentiation and cytokine production of myeloid cells by the MAPK phosphatase Dusp16.
Introduction
Recognition of microbial danger molecules by TLRs2 rapidly triggers activating signaling pathways in innate immune cells. The MAPK family members ERK1/2, p38, and JNK are all activated in response to TLR ligands through phosphorylation of the Thr-X-Tyr motif and subsequently make essential contributions to TLR-induced gene expression, the induction of antimicrobial effector molecules, and the induction of adaptive immune responses. On the other hand, MAPK-dependent inflammatory gene expression has the potential to cause acute hyperinflammation and chronic immunopathology. Therefore, TLR-induced MAPK activation has to be tightly regulated to balance the need for efficient defense against pathogens and the avoidance of deleterious inflammatory side effects.
Indeed, when macrophages are stimulated, MAPK activation is typically transient with a peak within the 1st h after stimulation and a return to base-line levels within several hours. As the total protein levels of ERK, p38, and JNK are generally not affected in response to TLR stimulation, the return to basal levels of MAPK phosphorylation is brought about primarily by MAPK phosphatases belonging to the family of dual specificity phosphatases (Dusps) that comprises 10 active members in mouse and man (1). Dusp MAPK phosphatases contain a MAPK-binding domain that determines their selectivity for different substrate MAPKs. In addition, the expression of different Dusp genes is regulated in various cell types and in response to different stimuli, and the individual Dusp proteins localize to different subcellular compartments. Inducibly expressed Dusps are expected to mediate negative feedback inhibition of MAPK-dependent TLR-induced gene expression. In fact, for several Dusp genes, a non-redundant function in regulation of innate immune cell activation has been demonstrated: Dusp1/MKP-1 is strongly induced by TLR signals and down-regulates p38 activation, thereby controlling the expression of a subset of TLR-induced inflammatory genes (2–5). Abnormally high cytokine release in Dusp1−/− mice in response to LPS and in several bacterial infections results in tissue immunopathology and/or increased lethality (6–9). Dusp2−/− macrophages and mast cells display hyperphosphorylation of ERK most likely due to uninhibited cross-talk between different MAPK pathways, and this is associated with decreased cytokine production (10). Dusp4/MKP-2 was recently shown to regulate the macrophage response to the intracellular pathogen Leishmania mexicana (11). Dusp10/MKP-5 was the first family member for which a knock-out mouse phenotype with complex effects on JNK activation, cytokine production from macrophages, and increased IFNγ levels in CD4 T cells was described (12).
Similar to Dusp1 and Dusp2, expression of Dusp16/MKP-7 mRNA is inducible in macrophages by TLR stimulation (13). Several transcript variants of Dusp16 have been described that encode the MAPK-binding and catalytic domains of the protein but differ in the length of the C-terminal domain. Of these, two isoforms were shown to be translated: isoform A1 encodes a larger 677-amino acid protein containing PEST sequences that are absent in the shorter B1 isoform (13). Dusp16 preferentially dephosphorylates JNK and p38 MAPK (14) and may serve as a shuttle protein (15) or anchor activated MAPK in the cytoplasm (16). Dusp16 protein stability is controlled by ERK-dependent phosphorylation (16–18). Recently, expression of Dusp16 in Th2, but not Th1, cells was described, and a function of Dusp16 in Th2 differentiation was suggested by the phenotype of transgenic mice expressing dominant-negative Dusp16 under the control of the lck promoter in CD4 T cells (19). However, despite the expression of Dusp16 in TLR-activated myeloid cells, to date, no genetic model for the investigation of Dusp16 in innate immunity has been available.
Here, we generated and analyzed a Dusp16-deficient gene trap mouse line carrying an insertion of a neomycin-β-gal cassette in the Dusp16 locus. Homozygous Dusp16−/− mice died neonatally without displaying any gross anatomical abnormality. Using fetal liver cell-derived radiation chimeras, an impaired proliferation of bone marrow progenitors in response to GM-CSF but not M-CSF was found. Furthermore, we observed the selective overproduction of a subset of TLR-induced cytokines, most notably of IL-12, by macrophages lacking Dusp16. Selective control of IL-12 expression by Dusp16 was also found in Dusp16 chimeric mice challenged with LPS in vivo. Together, our data reveal a dual role for Dusp16 in the differentiation and effector function of myeloid cells.
EXPERIMENTAL PROCEDURES
Generation of Dusp16 Gene Trap Mutant Mice
The 129P2 ES cell line AE0704 was obtained from the International Gene Trap Consortium through the Sanger Institute Gene Trap Resource. The integration of the β-galactosidase-neomycin transferase (β-Geo) cassette in the fourth intron of the Dusp16 gene was confirmed by RT-PCR using the primers RL330 (5′-GCAGGTGGCTTTGCTGAGTTCT-3′; binding in exon 4 of Dusp16 transcript ENSMUST00000100857) and RL345 (5′-AGTATCGGCCTCAGGAAGATCG-3′; specific for the β-Geo coding sequence in the gene trap vector pGT0lxr). The ES cells were microinjected into C57BL/6 blastocysts, and male chimeras were mated to C57BL/6 females for germ line transmission. Dusp16tp/+ mice were backcrossed on the C57BL/6 background for 10 generations. Dusp16tp/+ mice were intercrossed to obtain homozygous Dusp16tp/tp mice. The gene trap integration site was mapped using long range PCR with a set of primers covering the fourth intron of the Dusp16 gene. The primers RL528 (5′-CGCTCTTACCAAAGGGCAAACCC-3′) and RL497 (5′-CAAGGCCAGCCATTCTCTCAGGT-3′) were used for sequencing the integration site. RL528 was combined with primers RL536 (5′-GACCTGTGCATAACTGGCCCTACTAC-3′) and RL535 (5′-CCATCTCATGGCAGAGGAGTGACT-3) for genotyping mice by PCR from tail DNA. Exclusive integration of the gene trap in the Dusp16 locus was also confirmed by Southern blot analysis of genomic DNA that was digested with EcoRI or KpnI and probed with PCR-generated probes hybridizing to the fourth intron of Dusp16 (RL1011, 5′-AGTTTAAGTTGGACCAGGAAGGTAGACA-3′, and RL1012, 5′-GGAGCTATATTTAGAGGCAGTTGTGAGC-3′, yielding a 663-bp fragment) and β-Geo gene trap sequences (RL492, 5′-CATCCCGCATCTGACCACCA-3′, and RL493, 5′-GGGATAGTTTTCTTGCGGCCCTAA-3′, yielding a 513-bp fragment), respectively.
Analysis of Newborn Mice
Timed matings of Dusp16tp/+ mice were set up, and pregnant females were killed at different embryonic days (between E11.5 and E18.5) to determine the genotype of the embryos by PCR. To evaluate the postnatal mortality of Dusp16tp mice, mice giving birth were closely monitored, and the newborns were weighed and photographed. Dead or dying pups were removed together with one or two randomly chosen littermates for histopathological analysis. Pups were decapitated, and the body and head were fixed in 4% PBS-buffered formalin overnight and embedded in paraffin. 4-μm sections were stained with hematoxylin and eosin (H&E) and periodic acid-Schiff and evaluated by light microscopy using a Zeiss Axioplan microscope.
Radiation Chimeras
Plug-positive females from timed matings of Dusp16tp/+ mice were sacrificed between E12.5 and E15.5. Embryo genotyping for Dusp16 and determination of the sex was carried out by PCR. Fetal liver cell suspensions were incubated for a few hours in medium containing IL-3, IL-6, and stem cell factor until intravenous injection of Dusp16+/+ and Dusp16tp/tp cells into lethally irradiated (9 or 10 grays) CD45.1 heterozygous mice. The cells from one fetal liver were used to reconstitute two irradiated CD45.1 heterozygous mice. Reconstitution efficiency was tested after 4–8 weeks by flow cytometry of peripheral blood cells using the syngeneic markers CD45.1 (identifying recipient-derived cells) and CD45.2. To ensure replacement of tissue-resident macrophages and DCs by donor-derived cells, experiments with chimeric mice were performed after a minimum of 10–12 weeks postreconstitution.
Flow Cytometry Analysis of Lymphoid and Myeloid Cell Lineages
Single cell suspensions of spleen, mesenteric lymph nodes, and thymus were prepared using 70-μm nylon cell strainers. Cells were resuspended in cold PBS with 2% FCS and incubated with antibodies to CD16/CD32 for blockade of Fc receptors. Appropriate dilutions of FACS antibodies were added after 15 min, and the cells were incubated for an additional 20 min at 4 °C in the dark. The following antibodies were used: CD45.1-FITC, CD45.2-allophycocyanin, CD4-phycoerythrin, CD8-eFluor, CD3-PerCP, B220-FITC, CD11c-PeCy7, CD11b-eFluor, and F4/80-FITC. For analysis of intracellular IL-12p40, brefeldin A was added to macrophages 1 h after LPS stimulation to block secretion of the cytokine. After 6 h, cells were fixed with 2% paraformaldehyde, permeabilized with a saponin-containing buffer, and stained with phycoerythrin-labeled anti-IL-12p40 antibody. Cells were analyzed on a FACSCanto II flow cytometer using unstained and single stained controls to adjust compensation settings.
Murine Embryonic Fibroblast Lines
Embryonic fibroblasts were isolated at E13.5 from Dusp16+/+ and Dusp16tp/tp embryos. Briefly, eviscerated embryo bodies were digested with trypsin to prepare single cell suspensions, which were then grown in complete DMEM containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C and in 5% CO2. Primary cells were subcultured every 3 days until they reached crisis and became established immortalized cell lines. Cells were treated with hydrogen peroxide (Sigma) diluted in full medium for the indicated time points. Cells were lysed in MKK lysis buffer (0.27 m sucrose, 1 mm EDTA, 1 mm EGTA, 10 mm Tris, 1% Triton X-100) containing protease inhibitors and phosphatase inhibitors (Roche Applied Science).
Macrophage and DC Cultures
Bone marrow cells were flushed out of mouse femur and tibia. Following erythrocyte lysis with NH4Cl, standard macrophage differentiation cultures were initiated in Petri dishes using 10% L-cell-conditioned medium as the source of M-CSF in complete DMEM (containing 10% FBS, β-mercaptoethanol, and antibiotics). Non-adherent cells were counted on day 1 and replated at a density of 6 × 105/ml in Petri dishes in 10% L-cell-conditioned medium, and macrophages were harvested and counted again on day 7 (20). For generation of DCs, we followed an established protocol based on Lutz et al. (21). Briefly, bone marrow cells were incubated at a density of 5 × 105/ml in Petri dishes using complete RPMI 1640 medium with 10% supernatant of Ag8653 melanoma cells stably transfected to express GM-CSF. On days 3 and 6 of culture, an additional 10 ml of growth medium were added followed by harvesting and counting of DCs on day 8. Instead of bone marrow cells from radiation chimeras, in some experiments, fetal liver cells from E12.5–E15.5 embryos were differentiated to macrophages or DCs using the same experimental protocols. Macrophages were stimulated with LPS and harvested at the indicated time points for stepwise preparation of cytoplasmic and nuclear extracts using the NE-PER kit (Thermo Scientific) following the manufacturer's instructions.
siRNA Knockdown in Macrophages
For efficient knockdown of gene expression in primary macrophages, a recently published protocol was used (22). In brief, macrophages were differentiated from C57BL/6 bone marrow cells in M-CSF-containing medium in hydrophobic Teflon bags for 7 days. 2 × 106 macrophages were electroporated in 100 μl of Opti-MEM with 0.4 nmol of Dharmacon ON-TARGETplus SMARTpool siRNA. After electroporation, cells were distributed onto 2 wells of a 12-well plate and incubated for 30 min in DMEM alone followed by addition of FCS and antibiotics. 24 h after electroporation, macrophages were used for stimulation with LPS. Supernatants were collected 8 h later, and cells were harvested for analysis of RNA or protein.
Pharmacological Inhibition of MAPK Signaling
The MEK1 inhibitor PD184352 (Selleck Chemicals), the p38 inhibitor SB203580 (Calbiochem), and the JNK inhibitors SP600125 (Calbiochem) and JNK inhibitor VIII (JNK-VIII) (Calbiochem) were all solubilized in DMSO.
Western Blot Analysis of MAPK Activation
Protein lysates were separated by SDS-PAGE and blotted onto Protran membranes. Antibodies against phospho-p38, p38, phospho-JNK1/2, JNK1/2, phospho-ERK1/2, and ERK1/2 were purchased from Cell Signaling Technology. The antibody against α-tubulin was purchased from Sigma. Membranes were blocked and antibodies were diluted in 5% BSA or 10% milk.
LPS Challenge Experiments
Mice were injected intraperitoneally with a sublethal dose of 10 μg/g of body weight LPS from Escherichia coli O55:B5 (Sigma-Aldrich, catalogue number L2880) in 200 μl of PBS. At the indicated times, mice were sacrificed, blood was collected, and tissues were harvested and homogenized in Trifast reagent (Peqlab, Erlangen, Germany) for RNA preparation.
Expression Analysis
RNA was prepared from homogenates of cells and tissues in Trifast reagent as detailed by the manufacturer. Reverse transcription was carried out with the High Capacity cDNA RT kit (Applied Biosystems). For quantitative real time PCR analysis, the Roche Universal Probe Library system was used on an ABI 7900 Sequence Detection System. Sequences for primers and probes are available on request. Threshold cycle values for genes of interest were normalized to Hprt as housekeeping gene, and -fold changes relative to untreated samples were calculated using the ΔΔCT method. In the case of the Dusp16 A1 isoform mRNA, no specific primer/probe combination could be identified in the Universal Probe Library; therefore, a conventional TaqMan probe was designed and ordered from Metabion (Martinsried, Germany). For microarray-based transcriptome analysis of LPS-induced gene expression in macrophages, RNA from two biological replicates was pooled at a concentration of 50 ng/μl, labeled, and processed for hybridization to Affymetrix Mouse Gene ST 1.0 GeneChips according to the manufacturer's protocol. Cel files were normalized using Robust Multi-array Average (Affymetrix Expression Console) to obtain signal intensities.
Cytokine and Chemokine ELISAs
Concentrations of IL-6, IL-10, IL-12p40, IL-12p70, and IL-23p19 were measured in cell culture supernatants and mouse sera (Duoset, R&D Systems) at the appropriate dilutions following the manufacturer's instructions.
Statistical Analysis
Student's t test was used to determine statistical significance. Throughout the figures, * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.
RESULTS
Dusp16 Expression Is Induced by TLR Ligands in Vitro and in Vivo
Several Dusp16 transcripts have been described to be generated by alternative splicing, but their expression in tissues and isolated cells has not yet been analyzed quantitatively. We designed primers and probes to specifically measure the expression of the A1 and B1 isoforms in tissues and macrophages using real time quantitative PCR (Fig. 1A). Expression of both isoforms was detectable in a broad range of tissues with higher levels of the A1 isoform in all tissues examined (Fig. 1B). In macrophages, both A1 and B1 mRNAs were inducible by TLR ligands to a similar extent. LPS and CpG caused strong up-regulation of Dusp16 with expression levels peaking between 2 and 10 h; in contrast, the effect of the TLR3 ligand poly(I-C) was much weaker, suggesting that Myd88-dependent pathways are responsible for TLR-induced Dusp16 expression (Fig. 1C). A stronger induction was observed in macrophages compared with DCs; however, this difference was attributable to higher basal expression of Dusp16 in DCs (Fig. 1D). In the spleens, livers, and lungs of mice injected with LPS, expression of Dusp16 A1 and B1 mRNAs was both induced severalfold (Fig. 1E). Together, these data confirm and extend the previously described induction of Dusp16 by TLR ligands in innate immune cells (13) and suggest that the isoforms A1 and B1 are generated by alternative splicing at a steady ratio favoring A1 over B1.
FIGURE 1.

TLR-triggered expression of Dusp16 isoforms A1 and B1 in vitro and in vivo. A, primers and probes used for qRT-PCR of mRNA of Dusp16 isoforms A1 and B1. Depicted are exons 6 and 7 of transcripts NM_130447 (A1) and NM_001048054 (B1); the binding sites of the primers and of the TaqMan probe are indicated. B, expression of both isoforms in tissues of C57BL/6 mice. ΔCT (dCt) values were normalized to Hprt. Shown are mean ± S.D. (error bars) of three mice. C, kinetics of expression in BMMs after stimulation with TLR ligands. LPS (10 ng/ml), CpG 1826 (0.5 μm), poly(I-C) (10 μg/ml). -Fold changes were calibrated to untreated control macrophages. Shown are mean ± S.D. (error bars) of quadruplicate samples of a representative experiment. D, dose response of Dusp16 A1 and B1 mRNA expression in BMMs (closed symbols) and bone marrow-derived DCs (BMDC) (open symbols) 12 h after stimulation with LPS (ng/ml) or CpG 1826 (μm). -Fold induction was calculated relative to untreated BMM samples. Shown are mean and S.D. (error bars) of quadruplicate determinations (two biological replicates, two technical replicates). E, Dusp16 expression in the spleen after intraperitoneal (i.p.) LPS injection (10 μg/g of body weight). Shown are mean ± S.D. (error bars) of four mice per group.
Generation of Dusp16-deficient Mice with a Gene Trap Approach
To generate a mouse model of Dusp16 deficiency, we used an ES cell line with a β-Geo gene trap insertion in the Dusp16 locus (Fig. 2A). We mapped the integration site of the gene trap cassette to the fourth intron of the major Dusp16 transcript ENSMUST00000100857 (Ensembl), allowing genotyping by PCR. The unique integration of the cassette at this site was confirmed by Southern blot (Fig. 2A). The splice acceptor site provided by the β-Geo cassette leads to the generation of a chimeric Dusp16-β-Geo transcript that lacks Dusp16 exon 7, which encodes the catalytic Dusp domain and the large C-terminal part of the protein containing nuclear localization signal, nuclear export signal, and PEST sequences. As no homozygous Dusp16trap mice were obtained at weaning (see below), we examined the embryonic Dusp16 expression in the different genotypes. Analysis of Dusp16 mRNA expression by Northern blot of homozygous Dusp16trap embryos showed the expected absence of the endogenous Dusp16 mRNA signal and the presence of the fusion transcript detected by a β-Geo-specific probe (Fig. 2B). By qRT-PCR using the A1- and B1-specific probes and primers, the nearly complete absence of both splice variants in homozygous embryos was confirmed. In contrast, primers binding to exons 3 and 4 gave a largely unaltered signal, suggesting that transcription from the trapped Dusp16 locus generates the expected Dusp16-β-Geo mRNA (Fig. 2, A and C). The enzymatic activity of the protein encoded by this transcript was used to visualize the pattern of Dusp16 expression in embryos at E11.5 and E14.5, which showed a strong signal in the brain but also from other tissues (Fig. 2D).
FIGURE 2.

Generation of Dusp16 gene trap mice and perinatal lethality. A, schematic overview of Dusp16 locus with β-Geo trap in intron 4 (left panel). The KpnI and EcoRI sites and the position of the probes are indicated. Southern blots of KpnI- and EcoRI-digested DNA from embryos obtained after heterozygote matings are shown in the right panel. In the KpnI blot, the 5.4-kb band stems from the gene trap allele, and the 18-kb band stems from WT. In the EcoRI blot, the 7.8-kb band stems from the gene trap allele, and the high molecular weight signal is due to nonspecific hybridization of the β-Geo probe to undigested DNA. Results of PCR genotyping are indicated. B, Northern blot analysis of Dusp16 mRNA expression in Dusp16 gene trap embryos. Total RNA prepared from E10.5 embryos was analyzed by Northern blot using probes for Dusp16 (3′ of intron 4) and the β-Geo cassette. Ribosomal RNA bands in ethidium bromide (Etbr)-stained gel indicate equal loading. C, qRT-PCR of embryo RNA analyzed with primers specific for A1, B1 (see Fig. 1A), or total Dusp16 (binding in exons 3 and 4, therefore also amplifying the Dusp16-β-Geo mRNA). Shown are mean and S.D. (error bars) of two embryos per genotype. D, X-Gal staining of whole embryos taken at E11.5 and E14.5. E, frequency of Dusp16 genotypes at different embryonic and postnatal ages. The total number of mice was 134 (E10–E12), 496 (E13–E15), 155 (E16–E18), 53 (P0), and 500 (weaning). F, neonatal lethality of Dusp16tp/tp mice. The total number of mice for each genotype in the analysis was nine (+/+), 12 (tp/+), and 11 (tp/tp). G, reduced birth weight of Dusp16tp/tp mice. Shown are mean and S.D. (error bars) and p value of Student's t test (** indicates p < 0.01 and *** indicates p < 0.001). The difference between Dusp16trap/+ and Dusp16+/+ was not significant.
Dusp16-deficient Mice Die in the Neonatal Period
Genotyping of a large number of litters from matings of Dusp16tp/+ mice at different stages of backcrossing to C57BL/6 failed to yield any homozygous Dusp16tp/tp offspring when analyzed at weaning. To determine at which developmental stage the Dusp16 deficiency caused lethality, we genotyped mice at different embryonic ages and closely followed the pups after birth. In utero, the frequency of all genotypes was as expected at all ages examined (Fig. 2E). Homozygous mice were born at the expected frequency but then died without exception within the first 16 h (Fig. 2F). In contrast, wild-type and heterozygous pups showed a comparable, much lower perinatal mortality (Fig. 2F). Macroscopically, the Dusp16-deficient pups showed no obvious abnormality but were smaller and weighed significantly less (Fig. 2G). Histopathological analysis of the pups did not reveal any abnormality in the cardiovascular system, lungs, liver, and kidney. Thus, the reason for the neonatal lethality of Dusp16-deficient mice is at present unclear but probably not caused by any gross developmental defects. The occurrence of death in Dusp16tp/tp pups within the first 16 after birth would be consistent with an inability to adapt either metabolism and/or energy homeostasis to postnatal stress conditions (23).
Dusp16-deficient Hematopoietic Stem Cells Reconstitute Lymphocyte and Myeloid Cell Compartments
To generate mice with a Dusp16-deficient immune system, we reconstituted lethally irradiated syngeneic CD45.1+ mice with Dusp16tp/tp and wild-type fetal liver cells. Phenotyping of splenocytes and lymph node cells from reconstituted mice by flow cytometry showed comparable frequencies and total numbers of CD4+ and CD8+ T cells as well as B220+ B lymphocytes (Fig. 3A). T cell development in the thymus was not affected by Dusp16 deficiency in hematopoietic cells as the distribution of CD4 and CD8 single and double positive cells was similar between mice reconstituted with wild-type and Dusp16tp/tp fetal liver cells (Fig. 3A). Staining of myeloid cells from peritoneal lavage and bronchoalveolar lavage fluid showed that 3 months after transfer >95% of resident macrophages were derived from CD45.1-negative donor cells, again without impact of Dusp16 deficiency (Fig. 3A). Furthermore, the percentage of Ly6G+ granulocytes, CD11b+F4/80+ macrophages, and CD11c+B220+ plasmacytoid DCs in the spleen was comparable between WT and Dusp16tp/tp-reconstituted mice (Fig. 3, B and C).
FIGURE 3.

Reconstitution of lymphoid and myeloid immune cells by Dusp16-deficient fetal liver cells in radiation chimeras. A, FACS phenotyping of cells isolated from the thymus, mesenteric lymph nodes (mes LN), spleen, peritoneal lavage, and bronchoalveolar lavage (BAL) of radiation chimeras 12 weeks after reconstitution with Dusp16+/+ (closed symbols) or Dusp16tp/tp (open symbols) fetal liver cells. The fraction of CD45.1+ recipient-derived cells indicates the degree of chimerism. Shown are mean and S.D. (error bars) of n = 9–10 mice. Differences were not significant between genotypes. B, gating strategy for identification of phagocytic and antigen-presenting cells in the spleen of chimeras. C, frequencies of myeloid cells in the spleen of radiation chimeras. Shown are mean and S.D. (error bars) of n = 4 mice from one representative experiment of two with six mice per genotype total. D, cell yield of macrophage and DC differentiation cultures. The number of cells obtained was normalized to the mean of Dusp16+/+ bone marrow cells. Shown are mean and S.D. (error bars) from pooled data from three experiments comprising a total of 8–11 mice. E, proliferation of bone marrow cells in response to M-CSF and GM-CSF. [3H]Thymidine incorporation was measured in quadruplicates on day 3 of culture. Shown are mean and S.D. (error bars) of n = 4–5 from one representative experiment of at least three. SSC, side scatter.
Reduced Proliferative Response of Dusp16-deficient Bone Marrow Cells to GM-CSF
However, we noted that the fraction of CD11c+CD11b+ myeloid DCs was moderately reduced in Dusp16tp/tp-reconstituted spleens (Fig. 3C). In vitro, the M-CSF-driven proliferation of macrophage progenitors from the bone marrow of reconstituted mice was not influenced by the Dusp16 genotype (Fig. 3, D and E). In contrast, when Dusp16tp/tp bone marrow cells were cultured with GM-CSF to differentiate DCs, we noticed a significantly reduced cell yield at days 7–8 of culture when DCs were harvested (Fig. 3D). Consistent with impaired proliferation, [3H]thymidine incorporation was significantly reduced in response to GM-CSF, but not to M-CSF, in Dusp16tp/tp bone marrow cells (Fig. 3E). Altogether, although Dusp16 appears not to be required for homeostasis of the immune system under steady state conditions in vivo, the GM-CSF-driven proliferation of bone marrow progenitors was impaired in the absence of Dusp16 in vitro, and the fraction of CD11c+CD11b+ myeloid DCs in the spleen was reduced.
Intraperitoneal LPS Challenge in Dusp16 Radiation Chimeras
Next, we used the Dusp16 radiation chimeras to investigate whether the hematopoietic expression of this MAPK phosphatase shapes the cytokine response following intraperitoneal injection of LPS. Following injection of 10 μg/g of bodyweight LPS, mice were monitored clinically for signs of sepsis, and the levels of IL-6, IL-10, IL-12p40, and IL-23p19 were measured in the serum at several time points (Fig. 4A). There was no difference between wild type and Dusp16tp/tp-reconstituted chimeras in the development of clinical sepsis signs (immobility, rough fur, and conjunctivitis), and none of the mice died at this sublethal LPS dose. Analysis of serum cytokine levels showed that 2 h after LPS IL-12p40 and IL-6 levels were comparable between mice. However, mice reconstituted with Dusp16tp/tp fetal liver cells had higher serum levels of IL-12p40 at the 6- and 12-h time points, whereas IL-6 was higher only after 12 h (Fig. 4A). In contrast, significantly lower peak concentrations of IL-10 and IL-23p19 were observed 2 h after LPS injection in the sera of chimeras deficient in Dusp16 in the hematopoietic compartment. Thus, lack of hematopoietic Dusp16 in radiation chimeras led to a selective overproduction of IL-12p40 in response to LPS.
FIGURE 4.

LPS challenge of Dusp16 radiation chimeras. A, serum cytokine levels after intraperitoneal injection of 10 μg/g body weight LPS. Data were pooled from three experiments with different timings. Shown are mean and S.E. (error bars) of n = 4–12 mice per data point. B, qRT-PCR analysis of Dusp16 A1 and B1 mRNA expression following LPS intraperitoneally (i.p.). Data were normalized to Hprt and calibrated to the “WT, 0-h” condition. Shown are mean and S.D. (error bars) of n = 5 per data point (except untreated for which n = 2). * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.
Quantitative RT-PCR for Dusp16 A1 and B1 isoforms from the tissues of LPS-injected radiation chimeras allowed us to assess the contribution of radiosensitive hematopoietic cells to basal and LPS-induced Dusp16 expression because the Dusp16tp transcript is not detected by the A1- and B1-specific primer/probe combinations (Fig. 4B). Consistent with high reconstitution rates observed by FACS (Fig. 3A), both isoforms were expressed 10–100-fold less in the lymphoid organs bone marrow and spleen of Dusp16tp/tp-reconstituted mice. In contrast, the induction of Dusp16 A1 and B1 isoforms in lung and liver by LPS was much less affected by the genotype of the reconstituting fetal liver cells, indicating that in these organs radioresistant, non-hematopoietic cells constitute the bulk of basal and LPS-induced Dusp16 expression.
Dusp16-deficient Macrophages, but Not Dendritic Cells, Selectively Overproduce IL-12 in Response to TLR Stimulation
To more closely examine the impact of Dusp16 on TLR4-induced cytokine responses, we next used macrophages and DCs differentiated from bone marrow cells of radiation chimeras in M-CSF- and GM-CSF-containing media, respectively. Stimulation of bone marrow-derived macrophages deficient in Dusp16 with LPS resulted in a strongly enhanced release of IL-12p40, IL-12p70, and IL-23p19 into the supernatant, whereas the production of IL-6 and TNF was more or less comparable with WT BMMs (Fig. 5A). Intriguingly, overproduction of IL-12p40, IL-12p70, and IL-23p19 in the absence of Dusp16 was specifically observed in macrophages but not in Dusp16tp/tp bone marrow-derived DCs, which produced amounts comparable with their WT counterparts (Fig. 5B). Of note, Dusp16tp/+ macrophages derived from bone marrow cells of radiation chimeras produced amounts of IL-12p40 similar to Dusp16+/+ macrophages, indicating that expression of the Dusp16tp transcript per se does not alter cytokine production (Fig. 5C). Increased IL-12p40 protein was also detected in Dusp16-deficient macrophages, but not DCs, by intracellular cytokine staining after LPS stimulation (Fig. 5D).
FIGURE 5.

Dusp16-deficient macrophages overproduce IL-12. A and B, cytokine secretion was measured 24 h after stimulation of BMMs (A) and bone marrow-derived DCs (BMDC) (B) in the supernatants. Shown are mean and S.E. (error bars) of pooled data from three to five experiments using bone marrow from independent radiation chimeric mice. C, IL-12p40 production by WT, Dusp16+/tp, and Dusp16tp/tp BMMs after stimulation with LPS and CpG. D, IL-12p40 intracellular cytokine staining in BMMs. Shown are mean and S.D. (error bars) of n = 2–3 from a representative of two experiments. Med, medium. E, increased expression of IL-12p40, CD69, Nos2, and Cxcl11 by BMMs from Dusp16tp/tp chimeras. Macrophages were stimulated with LPS (100 ng/ml) for the indicated times. qRT-PCR analysis was normalized to the mean of all WT medium samples (Media). Shown are mean and S.D. (error bars) of n = 4 from a representative of two experiments. Error bars are often smaller than the symbols. The reduced Dusp16 A1 and B1 mRNA levels from Dusp16tp/tp-reconstituted mice indicate the degree of chimerism in the bone marrow. Asterisks indicate significant differences between WT and Dusp16tp/tp determined by two-sided, unpaired Student's t test. * indicates p < 0.05, ** indicates p < 0.01, and *** indicates p < 0.001.
To determine whether IL-12 expression is controlled at the mRNA level and to identify additional Dusp16-regulated LPS target genes, Affymetrix microarray profiling was performed (not shown). Several LPS-induced genes were found to be expressed at higher levels in the absence of Dusp16 and were validated by qRT-PCR, confirming IL-12p40 regulation at the mRNA level and identifying Cxcl11, Cd69, and Nos2 as Dusp16-regulated transcripts (Fig. 5E).
Kinetics of MAPK Phosphorylation in Dusp16tp/tp Embryonic Fibroblasts and Macrophages
Because Dusp16 is a MAPK phosphatase, the impact of its loss in Dusp16tp/tp cells on MAPK phosphorylation was addressed next. We first used mouse embryonic fibroblast lines generated from Dusp16+/+ and Dusp16tp/tp embryos to determine the kinetics of H2O2-induced MAPK activation (Fig. 6A). In the absence of Dusp16, enhanced phosphorylation of JNK and p38 were observed especially at the 30-min and 1-h time points. However, this difference was transient, and phosphorylation of all three MAPKs returned to basal levels 2 h after stimulation. Next, we assessed the LPS-induced phosphorylation of MAPK in Dusp16-deficient bone marrow-derived macrophages. Overall, the pattern of MAPK phosphorylation was similar between genotypes with a weak increase in JNK1/2 phosphorylation at the later 2-h time point. (Fig. 6B). To determine whether increased JNK activation in Dusp16-deficient macrophages may play a role in increased IL-12p40 production, we used pharmacological inhibitors to test the requirement for the individual MAPKs (Fig. 6C). Although inhibition of p38 MAPK with SB23580 led to increased IL-12p40 production, the MEK1 inhibitor PD184352 had no significant effect. In contrast, inhibition of JNK activity with SP600125 significantly impaired IL-12p40 release from Dusp16tp/tp and to a lesser degree from WT macrophages. A similar effect was observed with JNK-VIII, an unrelated pharmacological inhibitor of JNK1/2, that specifically impaired the production of IL-12p40, but not of IL-10, from WT and Dusp16tp/tp macrophages (Fig. 6D). In addition, we performed siRNA knockdown to test whether JNK1/2 is involved in the increased IL-12p40 production by Dusp16-deficient macrophages. Combined knockdown of the JNK1/2-encoding mRNAs Mapk8 and Mapk9 led to a significant reduction in IL-12p40 levels in macrophage culture supernatants (Fig. 6, E and F).
FIGURE 6.

Kinetics of MAPK phosphorylation in Dusp16tp/tp fibroblasts and macrophages. A, mouse embryonic fibroblast lines generated from Dusp16+/+ and Dusp16tp/tp embryonic fibroblasts were stimulated with 300 μm H2O2 for the indicated times, and lysates were prepared and analyzed by immunoblot for phosphorylated and total MAPK proteins. Samples from WT and Dusp16tp/tp mouse embryonic fibroblasts were separated together on one gel except for total p38 for which two gels were run but blotted together onto one membrane. Shown is a representative of two experiments with independent mouse embryonic fibroblast lines for each genotype. B, WT (+/+) and Dusp16-deficient (tp/tp) BMMs differentiated from two individual chimeric mice per genotype were plated at 106/well in 12-well plates, rested overnight, and stimulated with 100 ng/ml LPS for 0 min, 15 min, 45 min, and 2 h. Total cells were prepared and analyzed by Western blotting. Immunoblot images for phosphorylated MAPK and the Grb2 loading control are shown. A and B, the positions of molecular weight markers are indicated on the left side of the blots; arrowheads indicate bands corresponding to the p46 and p54 isoforms of JNK1/2. An asterisk indicates a smaller sized, nonspecific band obtained with the phospho-JNK1/2 (pJNK1/2) antibody. C, effect of inhibition of MEK1 (PD184352, 10 μm), p38 (SB203580, 5 μm), and JNK (SP600125, 10 μm) on IL-12p40 production. WT and Dusp16tp/tp BMMs were preincubated with inhibitors or solvent controls for 1 h followed by stimulation with 100 ng/ml LPS. Supernatants were harvested for IL-12p40 ELISA after overnight incubation. Percent IL-12p40 secretion was calculated relative to the DMSO solvent control for each inhibitor separately. Shown are mean and S.D. (error bars) of sextuplicate stimulations from a representative of two independent experiments performed. D, effect of JNK inhibitor JNK-VIII on IL-12p40 and IL-10 production. BMMs were pretreated with JNK-VIII and stimulated with LPS as indicated. Cytokine levels were calculated relative to LPS only for WT and Dusp16tp/tp BMMs separately. Shown are mean and S.E. (error bars) of pooled data from two independent experiments using macrophages from five individual mice per genotype. Asterisks indicate significant differences compared with DMSO solvent controls. E and F, siRNA knockdown of JNK1/2 impairs IL-12p40 production. WT and Dusp16tp/tp BMMs were transfected with a combination of siRNAs targeting Mapk8 and Mapk9 (encoding for JNK1 and JNK2, respectively) or with control non-silencing siRNA (ns). After a 24-h resting period, LPS was added for 8 h. E, analysis of JNK1/2 protein by Western blot. F, IL-12p40 production was measured by ELISA and normalized to the levels produced by BMMs transfected with control siRNA. Shown are mean and S.E. (error bars) of pooled data from three experiments (n = 6 mice for WT and Dusp16tp/tp each). Knockdown efficiency was 63–76% for Mapk9 and 42–61% for Mapk8 mRNA. Asterisks indicate significant differences compared with control siRNA. * indicates p < 0.05 and *** indicates p < 0.001. pERK1/2, phospho-ERK1/2; pp38, phospho-p38.
IL-10 Expression and Inhibitory Capacity Are Not Affected in Dusp16tp/tp Macrophages
IL-12 production in response to TLR stimulation is under tight control of the immunoregulatory cytokine IL-10 (24). Therefore, we considered the possibility that the overproduction of IL-12p40 in Dusp16tp/tp macrophages may be secondary to diminished IL-10 production. However, as shown in Fig. 7A, Dusp16tp/tp macrophages secreted comparable amounts of IL-10 after stimulation with LPS. In addition, we confirmed that IL-10 is an effective negative regulator of LPS-induced IL-12p40 production in Dusp16tp/tp macrophages (Fig. 7B).
FIGURE 7.
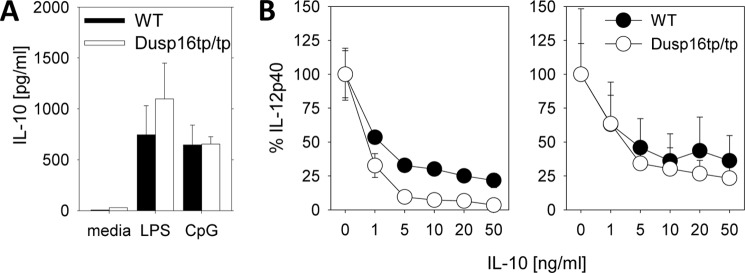
IL-10 expression and inhibitory effect in Dusp16tp/tp macrophages. A, LPS-induced production of IL-10 from BMMs was measured by ELISA in the supernatants 24 h after stimulation. Shown are mean and S.D. (error bars) of quadruplicate samples representative of three experiments. B, recombinant IL-10 was added to BMMs derived from radiation chimeras at the indicated concentrations followed by stimulation with LPS (10 ng/ml, left; 100 ng/ml, right). IL-12p40 levels in the supernatant were determined after overnight culture and are depicted relative to the amount measured in the absence of IL-10. Shown are mean and S.D. (error bars) derived from three independent mice per genotype, each stimulated in duplicate wells.
Dusp16tp/tp Macrophages Overexpress the Transcription Factor IRF-1
As overexpression of IL-12p40 is observed at the protein as well mRNA level, increased expression or activity of transcription factors controlling Il12b transcription may underlie the phenotype of Dusp16tp/tp macrophages. In this context, the interferon regulatory factor IRF-1 is of interest because it binds to the IL-12p40 promoter (25), and its down-regulation has been recently implicated in the diminished IL-12p40 expression in Dusp1/MKP-1-deficient macrophages (26). In Dusp16tp/tp macrophages, the mRNA expression of IRF-1 was significantly increased under basal and especially LPS-stimulated conditions, paralleling the overexpression of IL-12p40 (Fig. 8). However, siRNA-mediated knockdown of Irf1 expression failed to show any inhibitory effect on the levels of IL-12p40 expression despite efficient reduction of IRF-1 protein levels (Fig. 8C). As the Krüppel-like factors KLF10 and KLF11 have recently been implicated in the down-regulation of LPS-induced IL-12p40 expression (27), we asked whether their expression may be affected in Dusp16-dficient macrophages. However, as determined by qRT-PCR, no clear difference between Dusp16+/+ and Dusp16tp/tp macrophages was observed (Fig. 8B), indicating that these transcriptional repressors are not involved in Dusp16-mediated control of IL-12p40 expression.
FIGURE 8.
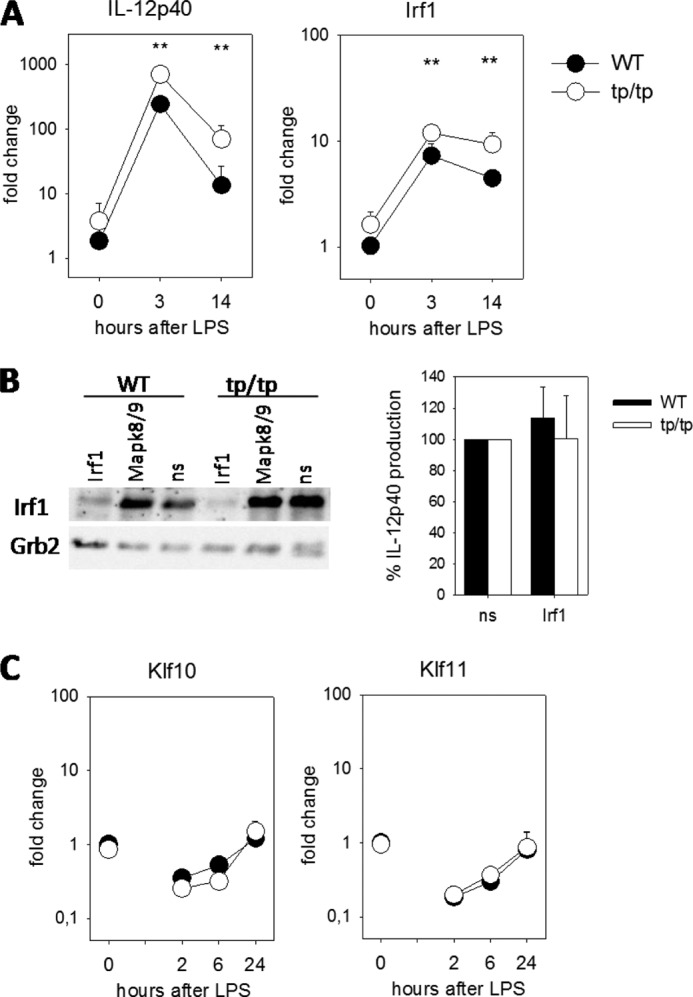
Increased expression of IRF-1 in Dusp16tp/tp macrophages correlates with overexpression of IL-12p40. A, mRNA expression of IL-12p40 and IRF-1 in BMMs was measured by qRT-PCR after stimulation with LPS for 3 and 14 h. Data were normalized to Hprt and calibrated to the “WT, untreated” condition. Shown are mean and S.D. (error bars) of quadruplicate samples (two biological replicates and two PCR replicates) from a representative of three independent experiments. B, siRNA knockdown of IRF-1. Macrophages were electroporated and stimulated as indicated in the legend to Fig. 6E. Left, IRF-1 protein levels as determined by Western blot 8 h after LPS. Right, mean and S.E. (error bars) of normalized IL-12p40 production relative to non-silencing siRNA (ns) of pooled data from three experiments (n = 6 mice for WT and Dusp16tp/tp each). Knockdown efficiency of Irf1 mRNA was 60–78%. C, qRT-PCR of Klf10 and Klf11 expression. Stimulation of macrophages and processing of samples were as in A but using different time points. ** indicates p < 0.01.
DISCUSSION
The results obtained using the Dusp16 gene trap mouse model described here suggest an essential function of this MAPK phosphatase in adaptation to early postnatal life and a selective regulatory role in the expression of several TLR-induced genes in macrophages. Although we confirmed the integration of the gene trap in the fourth intron of the Dusp16 locus and the absence of full-length Dusp16 A1 and B1 mRNA transcripts, the expression of the Dusp16-β-Geo transcript from the trapped allele can lead to the translation of a fusion protein containing the Dusp16 MAPK-binding domain that may act as a dominant-negative protein on MAPK signaling. Therefore, effects independent of the loss of functional Dusp16 can at present not be excluded. However, heterozygous Dusp16tp/+ mice showed no perinatal lethality or weight reduction as observed in Dusp16tp/tp newborns; in addition, Dusp16tp/+ macrophages responded comparably with Dusp16+/+ controls to TLR stimulation, indicating that expression of the gene trap Dusp16 allele is not sufficient to explain the phenotype observed in the Dusp16tp/tp mice and macrophages.
The cause of neonatal death in Dusp16tp/tp mice remains to be defined. Our histopathological analysis did not reveal any gross structural defects or malformations in the cardiovascular and respiratory systems, liver, or kidney. Many physiological processes and organ systems are involved in the successful adaptation to postnatal stress and may be affected in Dusp16tp/tp newborns (23). It will be important to measure the metabolic adaptation of Dusp16tp/tp newborns to birth in terms of energy homeostasis and fluid and electrolyte balance. Given the altered response of Dusp16tp/tp macrophages to TLR stimulation, one possible explanation could be that a dysregulated response of the innate immune system upon encountering microbial ligands during and shortly after birth causes early death of the mice. However, we consider this unlikely because the rapid neonatal death of the mice seems too fast for an inability to control inflammatory responses as e.g. seen in the knock-out mice with disrupted Socs1 (28), Zfp36 (29), or Il10 (30) gene, all of which are born normally and survive the 1st week of life. In addition, the absence of a hyperinflammatory response in the radiation chimeras reconstituted with Dusp16tp/tp fetal liver cells argues against such a scenario.
Our results suggest that Dusp16 has a dual function in the innate immune system. On the one hand, it shapes the output of cytokines and other TLR-induced gene products by macrophages, most notably leading to increased production of IL-12. On the other hand, it regulates the responsiveness of bone marrow progenitor cells to the growth factor GM-CSF. Whether the decreased number of CD11b+CD11c+ DCs in the spleens of Dusp16tp/tp-reconstituted mice is an in vivo correlate of the impaired DC generation we observed in response to GM-CSF in vitro is currently unknown. However, the importance of GM-CSF for DC homeostasis is increasingly realized (31). GM-CSF signaling comprises activation of ERK1/2 MAPK, which is required for differentiation and survival of human monocytes to DCs (32, 33), whereas p38 activation appears to negatively impact DC differentiation (32, 34). Altered MAPK activation in Dusp16-deficient DCs, therefore, may interfere with differentiation in response to GM-CSF but could also cause reduced cell yield by increasing apoptotic cell death in the cultures. To dissect these possibilities will be a goal of future experiments.
The selectively increased production of IL-12 by macrophages in Dusp16tp/tp mice in vitro and in vivo suggests a rather specific role for Dusp16 in shaping TLR-induced macrophage responses. Of note, none of the Dusp knock-out phenotypes reported to date showed a similar specific enhancement of IL-12 production. Dusp1−/− mice overproduce IL-6 and IL-10 but have decreased IL-12p40 (35, 36). In Dusp4-deficient mice, one study reported reduced inflammatory gene expression during sepsis (37), whereas another group found elevated production of IL-12p40, IL-6, and TNF in response to LPS (11). The mechanism by which Dusp16 controls IL-12p40 expression is linked to the activation of JNK, although we observed only a quite weak increase in JNK1/2 phosphorylation in Dusp16-deficient macrophages (Fig. 6B). First, pharmacological inhibition of JNK activity with SP600125 or JNK-VIII, but not of the other MAPK, diminished IL-12p40 production; in addition, siRNA knockdown of JNK1/2 expression attenuated IL-12p40 production. The relatively weak effect of Dusp16 on JNK1/2 activation may indicate that other Dusps expressed in macrophages can compensate partially for Dusp16. Rather than to impair total cellular JNK1/2 phosphorylation levels, Dusp16 may act in a compartment-specific manner e.g. by shuttling or anchoring JNK in the cytoplasm or the nucleus (15, 16). In addition, Dusp16 may exert JNK-dependent effects by acting as a scaffold, connecting certain substrate proteins with the MAPK (38). Dusp16 control of IL-12 expression may involve transcriptional regulators directly binding to the Il12b promoter/enhancer or may be indirect through secondary effects. Altered IL-10 production was an obvious candidate for the latter class because it is a potent endogenous feedback inhibitor of IL-12p40 production (24, 39). In addition, we have previously found that IL-10 synergized with LPS in enhancing Dusp16 expression (3). However, Dusp16 appears not to be required for IL-10-mediated inhibition of IL-12p40 expression, and the exaggerated IL-12 production in Dusp16-deficient macrophages is not linked to changes in IL-10 expression (Fig. 7). Therefore, Dusp16 controls IL-12 expression by a mechanism that is independent of IL-10. Thus, exaggerated IL-12p40 is likely due to increased activity of transcriptional regulators in the absence of Dusp16. Candidate transcription factors to investigate here include NFκB (40) and AP-1 family members and CCAAT/enhancer-binding protein β (41). Expression of the recently identified negative regulators of IL-12p40 production, KFL10 and KFL11 (27), was not affected in Dusp16-deficient macrophages, making a role for these transcription factors unlikely. IRF-1 is another transcription factor known to contribute critically to IL-12p40 expression (25, 42). Our finding that Irf1 expression levels in Dusp16tp/tp macrophages are significantly elevated (Fig. 8) suggested a possible explanation for the overproduction of IL-12p40. Interestingly, the impaired IL-12p40 expression in Dusp1-deficient macrophages was recently shown to correlate with lower levels of IRF-1 (26). Thus, IRF-1 may be a key factor in the modulation of cytokine production by Dusps. However, siRNA-mediated knockdown of Irf1 expression did not impair IL-12p40 production in our system. Future experiments in macrophages from mice lacking Irf1 on a Dusp16tp background will be required to definitely determine the role of IRF-1.
Dusp16-mediated regulation of IL-12 production has the potential to control Th1 differentiation during infection but also following vaccination with TLR-triggering adjuvants. The recent demonstration of high Dusp16 expression in Th2- but not Th1-differentiated CD4+ T cells (19) provides evidence for a dual function of this MAPK phosphatase in the regulation of adaptive immunity both at the antigen-presenting cell and T cell level. Of note, Dusp16 has recently been linked to intracellular survival of Mycobacterium tuberculosis because the mycobacterial effector protein Eis increases lysine acetylation of Dusp16, which is associated with a block in JNK activation, reactive oxygen species production, and phagosome maturation (43, 44). Taken together, Dusp16 extends the family of immunologically relevant Dusp MAPK phosphatases based on the phenotype of genetic mouse models (Dusp1, Dusp2, Dusp4, and Dusp10). Mice deficient in different Dusp genes show distinct and sometimes unexpected phenotypes, indicating that individual family members fulfill specific functions in regulation and specification of MAPK-mediated innate immune responses that are related to substrate specificity, inducible expression, and subcellular localization (45).
Acknowledgments
We thank Dr. Christian Bogdan for discussion. The skilled technical support of Harald Dietrich, Anne Urbat, and Katrin Jozefowski is gratefully acknowledged. We are grateful to Susanne Weiss for performing blastocyst injections of AE0704 ES cells. We thank Jeanette Hein and Manfred Kirsch for animal husbandry.
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB 643, TP A10 (to R. L.) and a Bayerische Forschungsstiftung fellowship (to M. N.). Work in the Keyse laboratory was supported by Cancer Research UK Program Grant CR-UK Stress Response Laboratory C8227/A12053.
- TLR
- Toll-like receptor
- DC
- dendritic cell
- Dusp
- dual specificity phosphatase
- β-Geo
- β-galactosidase-neomycin transferase
- E
- embryonic day
- JNK-VIII
- JNK inhibitor VIII
- qRT-PCR
- quantitative RT-PCR
- BMM
- bone marrow-derived macrophage.
REFERENCES
- 1. Dickinson R. J., Keyse S. M. (2006) Diverse physiological functions for dual-specificity MAP kinase phosphatases. J. Cell Sci. 119, 4607–4615 [DOI] [PubMed] [Google Scholar]
- 2. Chi H., Barry S. P., Roth R. J., Wu J. J., Jones E. A., Bennett A. M., Flavell R. A. (2006) Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hammer M., Mages J., Dietrich H., Schmitz F., Striebel F., Murray P. J., Wagner H., Lang R. (2005) Control of dual-specificity phosphatase-1 expression in activated macrophages by IL-10. Eur. J. Immunol. 35, 2991–3001 [DOI] [PubMed] [Google Scholar]
- 4. Salojin K. V., Owusu I. B., Millerchip K. A., Potter M., Platt K. A., Oravecz T. (2006) Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J. Immunol. 176, 1899–1907 [DOI] [PubMed] [Google Scholar]
- 5. Zhao Q., Wang X., Nelin L. D., Yao Y., Matta R., Manson M. E., Baliga R. S., Meng X., Smith C. V., Bauer J. A., Chang C. H., Liu Y. (2006) MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J. Exp. Med. 203, 131–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frazier W. J., Wang X., Wancket L. M., Li X. A., Meng X., Nelin L. D., Cato A. C., Liu Y. (2009) Increased inflammation, impaired bacterial clearance, and metabolic disruption after Gram-negative sepsis in Mkp-1-deficient mice. J. Immunol. 183, 7411–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hammer M., Echtenachter B., Weighardt H., Jozefowski K., Rose-John S., Männel D. N., Holzmann B., Lang R. (2010) Increased inflammation and lethality of Dusp1−/− mice in polymicrobial peritonitis models. Immunology 131, 395–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rodriguez N., Dietrich H., Mossbrugger I., Weintz G., Scheller J., Hammer M., Quintanilla-Martinez L., Rose-John S., Miethke T., Lang R. (2010) Increased inflammation and impaired resistance to Chlamydophila pneumoniae infection in Dusp1(−/−) mice: critical role of IL-6. J. Leukoc. Biol. 88, 579–587 [DOI] [PubMed] [Google Scholar]
- 9. Wang X., Meng X., Kuhlman J. R., Nelin L. D., Nicol K. K., English B. K., Liu Y. (2007) Knockout of Mkp-1 enhances the host inflammatory responses to Gram-positive bacteria. J. Immunol. 178, 5312–5320 [DOI] [PubMed] [Google Scholar]
- 10. Jeffrey K. L., Brummer T., Rolph M. S., Liu S. M., Callejas N. A., Grumont R. J., Gillieron C., Mackay F., Grey S., Camps M., Rommel C., Gerondakis S. D., Mackay C. R. (2006) Positive regulation of immune cell function and inflammatory responses by phosphatase PAC-1. Nat. Immunol. 7, 274–283 [DOI] [PubMed] [Google Scholar]
- 11. Al-Mutairi M. S., Cadalbert L. C., McGachy H. A., Shweash M., Schroeder J., Kurnik M., Sloss C. M., Bryant C. E., Alexander J., Plevin R. (2010) MAP kinase phosphatase-2 plays a critical role in response to infection by Leishmania mexicana. PLoS Pathog. 6, e1001192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang Y., Blattman J. N., Kennedy N. J., Duong J., Nguyen T., Wang Y., Davis R. J., Greenberg P. D., Flavell R. A., Dong C. (2004) Regulation of innate and adaptive immune responses by MAP kinase phosphatase 5. Nature 430, 793–797 [DOI] [PubMed] [Google Scholar]
- 13. Matsuguchi T., Musikacharoen T., Johnson T. R., Kraft A. S., Yoshikai Y. (2001) A novel mitogen-activated protein kinase phosphatase is an important negative regulator of lipopolysaccharide-mediated c-Jun N-terminal kinase activation in mouse macrophage cell lines. Mol. Cell. Biol. 21, 6999–7009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanoue T., Yamamoto T., Maeda R., Nishida E. (2001) A Novel MAPK phosphatase MKP-7 acts preferentially on JNK/SAPK and p38 α and β MAPKs. J. Biol. Chem. 276, 26629–26639 [DOI] [PubMed] [Google Scholar]
- 15. Masuda K., Shima H., Watanabe M., Kikuchi K. (2001) MKP-7, a novel mitogen-activated protein kinase phosphatase, functions as a shuttle protein. J. Biol. Chem. 276, 39002–39011 [DOI] [PubMed] [Google Scholar]
- 16. Masuda K., Katagiri C., Nomura M., Sato M., Kakumoto K., Akagi T., Kikuchi K., Tanuma N., Shima H. (2010) MKP-7, a JNK phosphatase, blocks ERK-dependent gene activation by anchoring phosphorylated ERK in the cytoplasm. Biochem. Biophys. Res. Commun. 393, 201–206 [DOI] [PubMed] [Google Scholar]
- 17. Katagiri C., Masuda K., Urano T., Yamashita K., Araki Y., Kikuchi K., Shima H. (2005) Phosphorylation of Ser-446 determines stability of MKP-7. J. Biol. Chem. 280, 14716–14722 [DOI] [PubMed] [Google Scholar]
- 18. Masuda K., Shima H., Katagiri C., Kikuchi K. (2003) Activation of ERK induces phosphorylation of MAPK phosphatase-7, a JNK specific phosphatase, at Ser-446. J. Biol. Chem. 278, 32448–32456 [DOI] [PubMed] [Google Scholar]
- 19. Musikacharoen T., Bandow K., Kakimoto K., Kusuyama J., Onishi T., Yoshikai Y., Matsuguchi T. (2011) Functional involvement of dual specificity phosphatase 16 (DUSP16), a c-Jun N-terminal kinase-specific phosphatase, in the regulation of T helper cell differentiation. J. Biol. Chem. 286, 24896–24905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lang R., Rutschman R. L., Greaves D. R., Murray P. J. (2002) Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J. Immunol. 168, 3402–3411 [DOI] [PubMed] [Google Scholar]
- 21. Lutz M. B., Kukutsch N., Ogilvie A. L., Rössner S., Koch F., Romani N., Schuler G. (1999) An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J. Immunol. Methods 223, 77–92 [DOI] [PubMed] [Google Scholar]
- 22. Wiese M., Castiglione K., Hensel M., Schleicher U., Bogdan C., Jantsch J. (2010) Small interfering RNA (siRNA) delivery into murine bone marrow-derived macrophages by electroporation. J. Immunol. Methods 353, 102–110 [DOI] [PubMed] [Google Scholar]
- 23. Turgeon B., Meloche S. (2009) Interpreting neonatal lethal phenotypes in mouse mutants: insights into gene function and human diseases. Physiol. Rev. 89, 1–26 [DOI] [PubMed] [Google Scholar]
- 24. Zhou L., Nazarian A. A., Smale S. T. (2004) Interleukin-10 inhibits interleukin-12 p40 gene transcription by targeting a late event in the activation pathway. Mol. Cell. Biol. 24, 2385–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maruyama S., Sumita K., Shen H., Kanoh M., Xu X., Sato M., Matsumoto M., Shinomiya H., Asano Y. (2003) Identification of IFN regulatory factor-1 binding site in IL-12 p40 gene promoter. J. Immunol. 170, 997–1001 [DOI] [PubMed] [Google Scholar]
- 26. Korhonen R., Huotari N., Hömmö T., Leppänen T., Moilanen E. (2012) The expression of interleukin-12 is increased by MAP kinase phosphatase-1 through a mechanism related to interferon regulatory factor 1. Mol. Immunol. 51, 219–226 [DOI] [PubMed] [Google Scholar]
- 27. Zhang W., Wang X., Xia X., Liu X., Suo S., Guo J., Li M., Cao W., Cai Z., Hui Z., Subramaniam M., Spelsberg T. C., Wang J., Wang L. (2013) Klf10 inhibits IL-12p40 production in macrophage colony-stimulating factor-induced mouse bone marrow-derived macrophages. Eur. J. Immunol. 43, 258–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Marine J. C., Topham D. J., McKay C., Wang D., Parganas E., Stravopodis D., Yoshimura A., Ihle J. N. (1999) SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell 98, 609–616 [DOI] [PubMed] [Google Scholar]
- 29. Carballo E., Lai W. S., Blackshear P. J. (1998) Feedback inhibition of macrophage tumor necrosis factor-α production by tristetraprolin. Science 281, 1001–1005 [DOI] [PubMed] [Google Scholar]
- 30. Kühn R., Löhler J., Rennick D., Rajewsky K., Müller W. (1993) Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 [DOI] [PubMed] [Google Scholar]
- 31. Greter M., Helft J., Chow A., Hashimoto D., Mortha A., Agudo-Cantero J., Bogunovic M., Gautier E. L., Miller J., Leboeuf M., Lu G., Aloman C., Brown B. D., Pollard J. W., Xiong H., Randolph G. J., Chipuk J. E., Frenette P. S., Merad M. (2012) GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity 36, 1031–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van de Laar L., Coffer P. J., Woltman A. M. (2012) Regulation of dendritic cell development by GM-CSF: molecular control and implications for immune homeostasis and therapy. Blood 119, 3383–3393 [DOI] [PubMed] [Google Scholar]
- 33. Xie J., Qian J., Yang J., Wang S., Freeman M. E., 3rd, Yi Q. (2005) Critical roles of Raf/MEK/ERK and PI3K/AKT signaling and inactivation of p38 MAP kinase in the differentiation and survival of monocyte-derived immature dendritic cells. Exp. Hematol. 33, 564–572 [DOI] [PubMed] [Google Scholar]
- 34. Geest C. R., Coffer P. J. (2009) MAPK signaling pathways in the regulation of hematopoiesis. J. Leukoc. Biol. 86, 237–250 [DOI] [PubMed] [Google Scholar]
- 35. Hammer M., Mages J., Dietrich H., Servatius A., Howells N., Cato A. C., Lang R. (2006) Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J. Exp. Med. 203, 15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang J., Wang H., Song Z., Lin X., Zhang C. (2011) Involvement of MAPK phosphatase-1 in dexamethasone-induced chemoresistance in lung cancer. J. Chemother. 23, 221–226 [DOI] [PubMed] [Google Scholar]
- 37. Cornell T. T., Rodenhouse P., Cai Q., Sun L., Shanley T. P. (2010) Mitogen-activated protein kinase phosphatase 2 regulates the inflammatory response in sepsis. Infect. Immun. 78, 2868–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Willoughby E. A., Collins M. K. (2005) Dynamic interaction between the dual specificity phosphatase MKP7 and the JNK3 scaffold protein β-arrestin 2. J. Biol. Chem. 280, 25651–25658 [DOI] [PubMed] [Google Scholar]
- 39. Murray P. J., Smale S. T. (2012) Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat. Immunol. 13, 916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanjabi S., Hoffmann A., Liou H. C., Baltimore D., Smale S. T. (2000) Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc. Natl. Acad. Sci. U.S.A. 97, 12705–12710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Plevy S. E., Gemberling J. H., Hsu S., Dorner A. J., Smale S. T. (1997) Multiple control elements mediate activation of the murine and human interleukin 12 p40 promoters: evidence of functional synergy between C/EBP and Rel proteins. Mol. Cell. Biol. 17, 4572–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goodridge H. S., Harnett W., Liew F. Y., Harnett M. M. (2003) Differential regulation of interleukin-12 p40 and p35 induction via Erk mitogen-activated protein kinase-dependent and -independent mechanisms and the implications for bioactive IL-12 and IL-23 responses. Immunology 109, 415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim K. H., An D. R., Song J., Yoon J. Y., Kim H. S., Yoon H. J., Im H. N., Kim J., Kim D. J., Lee S. J., Lee H. M., Kim H. J., Jo E. K., Lee J. Y., Suh S. W. (2012) Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc. Natl. Acad. Sci. U.S.A. 109, 7729–7734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shin D. M., Jeon B. Y., Lee H. M., Jin H. S., Yuk J. M., Song C. H., Lee S. H., Lee Z. W., Cho S. N., Kim J. M., Friedman R. L., Jo E. K. (2010) Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 6, e1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lang R., Hammer M., Mages J. (2006) DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J. Immunol. 177, 7497–7504 [DOI] [PubMed] [Google Scholar]