

Background: Insig proteins are required for feedback regulation of cholesterol synthesis in cultured cells and liver. Insig may play a similar role in intestine.
Results: Insig deficiency in enterocytes leads to constitutively elevated cholesterol synthesis in intestine.
Conclusion: Intestinal regulation of cholesterol homeostasis requires Insig.
Significance: This work provides insight into how enterocytes coordinately balance de novo cholesterol synthesis and absorption.
Keywords: Cholesterol Regulation, Dyslipidemia, Gene Knockout, Intestine, Lipid Absorption, Lipid Synthesis, Ezetimibe, HMG-CoA Reductase, Insig, SREBP
Abstract
Enterocytes are the only cell type that must balance the de novo synthesis and absorption of cholesterol, although the coordinate regulation of these processes is not well understood. Our previous studies demonstrated that enterocytes respond to the pharmacological blockade of cholesterol absorption by ramping up de novo sterol synthesis through activation of sterol regulatory element-binding protein-2 (SREBP-2). Here, we genetically disrupt both Insig1 and Insig2 in the intestine, two closely related proteins that are required for the feedback inhibition of SREBP and HMG-CoA reductase (HMGR). This double knock-out was achieved by generating mice with an intestine-specific deletion of Insig1 using Villin-Cre in combination with a germ line deletion of Insig2. Deficiency of both Insigs in enterocytes resulted in constitutive activation of SREBP and HMGR, leading to an 11-fold increase in sterol synthesis in the small intestine and producing lipidosis of the intestinal crypts. The intestine-derived cholesterol accumulated in plasma and liver, leading to secondary feedback inhibition of hepatic SREBP2 activity. Pharmacological blockade of cholesterol absorption was unable to further induce the already elevated activities of SREBP-2 or HMGR in Insig-deficient enterocytes. These studies confirm the essential role of Insig proteins in the sterol homeostasis of enterocytes.
Introduction
Insig1 and Insig2 are two closely-related endoplasmic reticulum (ER)3 membrane proteins essential for feedback inhibition of cholesterol synthesis in cultured cells and in the liver. Insig proteins mediate this regulation by virtue of their sterol-dependent interaction with two other ER membrane proteins: Scap, an escort protein required for the proteolytic processing and activation of sterol regulatory element-binding proteins (SREBPs), and HMG-CoA reductase (HMGR), the enzyme that catalyzes the rate-determining step of the cholesterol biosynthetic pathway. SREBPs are a family of membrane-bound transcription factors that activate transcription of all genes required for the synthesis of cholesterol and fatty acids (1). When cells are depleted of sterols, SREBPs are transported by Scap from ER to Golgi, where they are processed proteolytically to yield active nuclear fragments (nSREBPs). Sterol-induced binding of Insig to Scap leads to retention of the Scap-SREBP complex in ER, thereby preventing the proteolytic processing of SREBPs to their active nuclear forms (2, 3). Sterol-induced binding of Insigs to HMGR leads to its ubiquitination and proteasomal degradation (4, 5). By virtue of these dual activities, Insigs potently suppress sterol synthesis in conditions when cells have an adequate supply of sterols.
The two Insig isoforms, Insig1 and Insig2, have overlapping functions in terms of Scap and HMGR binding (6, 7). In vivo studies of Insig function have been limited by both functional redundancy and neonatal lethality. Mice with single ablation of either Insig1 or Insig2 did not have obvious phenotypes (8). Mice with germ line ablation of both Insig1 and Insig2 failed to survive the first day after birth; these mice had severe craniofacial malformations with failure of mid-facial fusion and exencephaly, reminiscent of developmental anomalies noted in Smith-Lemli-Opitz syndrome and other inborn errors of cholesterol synthesis (9).
Given that the liver is quantitatively the most important organ for cholesterol synthesis in rodents, the initial focus of studies on Insig function in adult animals was restricted to the liver (8). To overcome the issue of neonatal lethality, we produced mice with germ line disruption of Insig2 and postnatal interferon-inducible MX1-Cre-mediated deletion of Insig1 in the liver. These Insig1/2-double deficient livers exhibited constitutive SREBP processing and a blunted ability to degrade HMGR, leading to overproduction of sterols and fatty acids synthesis and producing fatty liver. These studies confirmed the essential role of Insigs in preventing lipid accumulation in liver.
The small intestine is quantitatively second to liver in terms of de novo sterol synthesis in rodents and is the most important in other species, like rabbit and guinea pig (10–12), with most of the synthetic capacity localized within the intestinal epithelium. The relative importance of the intestine in whole-body sterol synthesis in humans is not well understood. In one study using [14C]acetate incorporation in human small bowel biopsies, intestinal sterol synthesis rates were 1–4-fold higher than hepatic rates (13). In all mammals in whom measurements have been made, the intestine and liver are the sites whose sterol synthesis is most subject to regulation by any condition that alters net sterol balance and are the sites most important for contributing cholesterol to the plasma (12).
As central players in the regulation of whole-body sterol flux, enterocytes are unusual in that they have three sources of cholesterol. Their unique function is to absorb cholesterol from the gut lumen (which is composed of cholesterol from the diet, biliary secretions, and sloughing of mucosal cells). In addition, they share the two ubiquitous sources of cellular cholesterol (i.e. the uptake of LDL cholesterol from plasma and de novo sterol synthesis from acetate). In other studied cell types, Insig proteins balance utilization of the latter two sources by suppressing LDL uptake and de novo synthesis when cells are sterol-replete. How Insigs function in enterocytes is unknown. Our previous studies indicated that, when deprived of cholesterol from the gut lumen by virtue of administration of ezetimibe, which blocks cholesterol absorption by inhibiting the apical cholesterol transporter NPC1L1 (Niemann-Pick C1-like 1), enterocytes increased de novo synthesis by activating SREBP-2 processing (14). We also observed a marked overaccumulation of HMGR protein, out of proportion to the modest SREBP-2-mediated increase of its cognate mRNA. We hypothesized that the ezetimibe-induced derepression of SREBP-2 processing and accumulation of HMGR enzyme was mediated via deactivation of Insig proteins.
Before packaging in nascent chylomicrons, dietary cholesterol is trafficked to the enterocyte's ER membranes, where it is esterified by acyl-CoA:cholesterol acyltransferase (15). Insig, Scap, and nascent SREBP also reside in ER membranes. Studies in cultured fibroblasts have demonstrated that sterol sensing by Scap responds in a cooperative manner such that SREBP transport is blocked when ER cholesterol exceeds 5% of total ER lipid (16). Insig depletion raises the feedback threshold for cholesterol such that SREBPs are constitutively cleaved even when sterol levels are high. If sterol sensing operates in the enterocyte as it does in other cell types, the depletion of ER membrane cholesterol by the blockade of cholesterol absorption should derepress SREBP transport by blocking interactions of Insig with Scap.
In the current studies, we ask whether Insigs function in the enterocyte in the same fashion as other cells and whether this regulation has physiologic relevance. We generated mice with an intestine-specific deletion of Insig1 using Villin-Cre in combination with a germ line deletion of Insig2. These mice are referred to as Vil-Insig− mice. Enterocytes from these mice have increased levels of nSREBPs and elevated mRNA levels for lipogenic SREBP target genes. A marked accumulation of HMGR enzyme in excess of its mRNA increase is also observed. As a consequence, intestinal de novo sterol synthesis is elevated, causing cholesterol to accumulate in the intestine, liver, and plasma. These results indicate that Insig proteins are essential for the feedback inhibition of cholesterol synthesis in the intestine.
EXPERIMENTAL PROCEDURES
Animals and Diets
Mice carrying floxed Insig1 and null Insig2 alleles (Insig1f/f;Insig2−/−) (8) were bred to Villin-Cre (Vil-Cre) transgenic mice (004782, The Jackson Laboratory) (17) to generate Insig1f/f;Insig2−/−;Vil-Cre mice that lack Insig1 and Insig2 in the intestines. For brevity, these mice were designated as Vil-Insig− mice. Mice were genotyped by PCR using crude lysates of ear punches (102-T, Viagen Biotech) with primers previously described (8). Mice were housed in colony cages with a 12-h light/12-h dark cycle and fed a standard chow diet containing 6% fat (Teklad Mouse/Rat Diet 7002, Harlan Teklad). In some studies, mice were fed with a fat-free diet (901683, MP Biomedicals) or a ground chow diet containing ezetimibe (SRP04000e, Sequoia Research Products). All mice were euthanized in the fed state at the end of the dark cycle. All animal experiments were performed with the approval of the Institutional Animal Care and Research Advisory Committee of the University of Texas Southwestern Medical Center.
Metabolic Parameters
Mice were euthanized with isoflurane, blood was obtained from the inferior vena cava in EDTA-coated tubes, and plasma was separated and stored at −80 °C. Tissue triglyceride and cholesterol concentrations were measured as described using commercial kits (18). Plasma cholesterol and triglyceride concentrations were measured using the Vitros 250 Chemistry Analyzer (Ortho Clinical Diagnostics). Plasma lipoproteins were separated by FPLC and triglycerides, and cholesterol concentrations in each fraction were measured as described (19).
Real-time RT-PCR
Total RNA was prepared from mouse tissues using RNA STAT-60 kit (TEL-TEST Inc.), and quantitative real-time RT-PCR was performed as described (14, 20, 21).
Isolated Intestine Epithelial Cell Preparation
An intestinal epithelial cell preparation was performed as described (14). Cells from this isolation are composed primarily of villus cells and are termed enterocytes for simplicity. In some experiments, the entire small intestine was used, whereas in other experiments, the small and large intestine were anatomically divided as described in the legends to Figs. 1 and 3.
FIGURE 1.
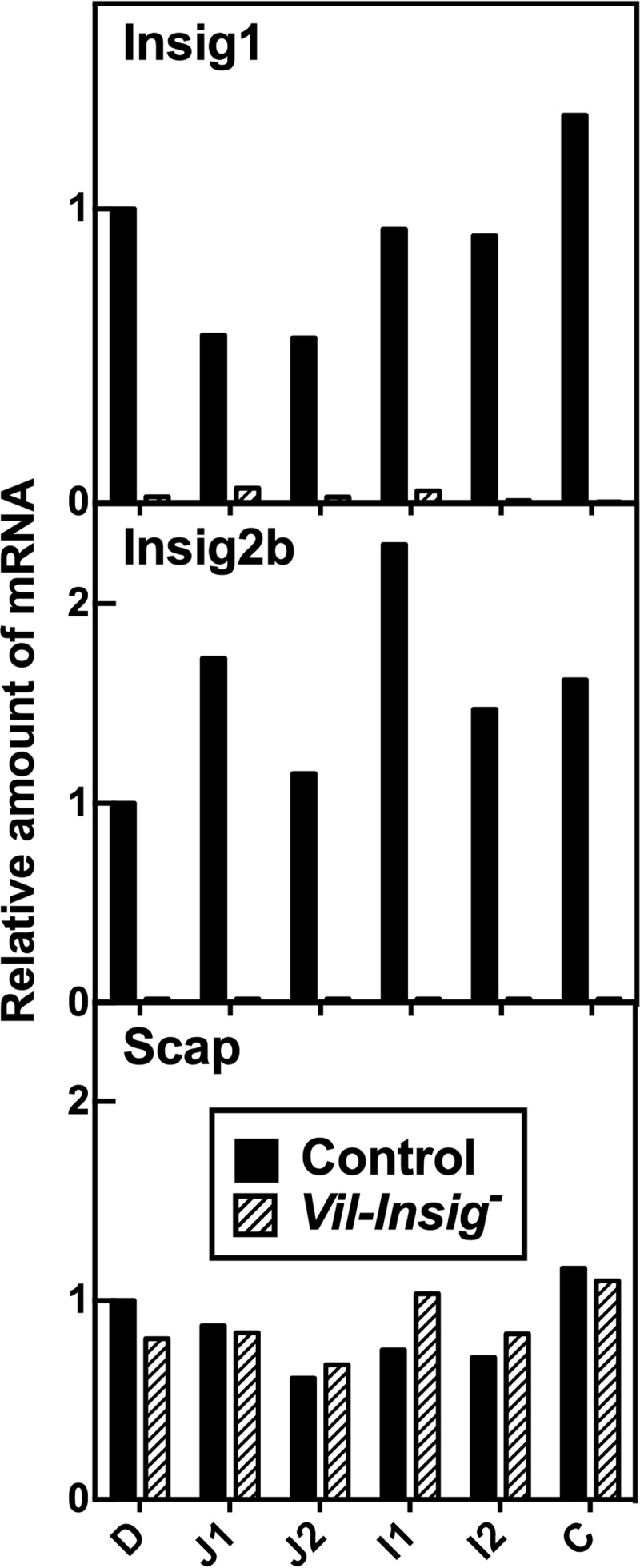
Disruption of Insig1 and Insig2 along the longitudinal axis of intestine. Equal amounts of RNA from isolated enterocytes from intestine segments of control and Vil-Insig− mice (female, 11–15 weeks of age, 4 mice/group) were pooled and subjected to quantitative RT-PCR mice using cyclophilin as the invariant control. Each value represents the amount of mRNA relative to that in the duodenum of the control mice, which is arbitrarily defined as 1.0. Intestines were anatomically divided as follows. Duodenum (D) denotes the segment from the distal end of the pylorus to 1 cm past the insertion of the common bile duct. The remaining small bowel was further divided into four equal sections: jejunum 1 and 2 and ileum 1 and 2 (J1, J2, I1, and I2). The colon (C) was analyzed without further division.
FIGURE 3.

Increased lipid synthesis in intestines of Vil-Insig− mice. Control and Vil-Insig− mice (male, 17–19 weeks of age, 7 mice/group) were injected intraperitoneally with 3H-labeled water (50 mCi in 0.2 ml of saline). One hour later, tissues were removed and processed for isolation of digitonin-precipitable sterols (A and C) and fatty acids (B and D). The small intestine was divided into three equal length segments termed proximal (prox.), middle (mid.), and distal (dist.). Each bar represents mean ± S.E. (error bars) of data from seven mice. The sterol and fatty acid synthetic rates were calculated as μmol of 3H radioactivity incorporated/h per g (A and B) or per organ (C and D). *, p < 0.01, the level of statistical significance (two-tailed Student's t test) between control and Vil-Insig− mice.
Immunoblot Analyses of Intestine and Liver Extracts
Whole-cell extracts were prepared individually from mice (∼150 mg wet weight of enterocytes or 100 mg of liver per mouse). Tissues or cell pellets were disrupted by Polytron homogenization in 1.5 ml of lysis buffer containing 1% SDS, 1% Triton X-100, 1% Nonidet P-40, 50 mm Tris-HCl, pH 7.4, and 5 mm EDTA with protease inhibitors as described (14), and insoluble material was separated by centrifugation at 4800 × g for 10 min and discarded. Extracts were pooled as described in the figure legend and subjected to SDS-PAGE and immunoblot analysis as described (18, 22–24).
Measurement of in Vivo Cholesterol and Fatty Acid Synthesis Rates
Rates of cholesterol and fatty acid synthesis were measured in mice using 3H-labeled water as described previously (25). The mice were fed ad libitum with chow diets and then fasted for 4 h prior to injection of 3H-labeled water. The rates of cholesterol and fatty acid synthesis were calculated as μmol of 3H-labeled water incorporated into fatty acids or digitonin-precipitable sterols/h/g of tissue.
Histology
Jejuna were fixed in 4% paraformaldehyde in PBS overnight at 4 °C. The fixed tissues were cryoprotected with sequential equilibrations of 10 and 18% sucrose for 12 h at 4 °C, embedded in OCT, cryosectioned at 8 μm, stained with Oil Red O, and counterstained with hematoxylin (26).
RESULTS
Mice with floxed Insig1 alleles and germ line deletion of Insig2 (Insig1f/f;Insig2−/−) were bred to Vil-Cre transgenic mice in which the Cre recombinase is driven from the gut-selective Villin promoter (27). The Vil-Cre-mediated deletion of Insig1, in combination with the germ line deletion of Insig2, rendered the resulting Insig1f/f;Insig2−/−;Vil-Cre mice deficient in both Insig1 and Insig2 in the intestines. For brevity, these mice were designated as Vil-Insig− mice. Mice derived from the same intercrosses with normal expression of both Insig1 and Insig2 (Insig1+/+;Insig2+/+ or Insig1+/+;Insig2+/+;Vil-Cre) were used as control mice. The Vil-Insig− mice were born at the expected ratio and were grossly indistinguishable from control littermates from birth to adulthood.
To verify that Insig expression was ablated in the intestines, we first measured mRNAs for the various Insig isoforms by real-time RT-PCR in enterocytes isolated from sequential segments of intestine along its longitudinal axis. We focused on isolated enterocytes rather than intact intestines for two reasons: first, Villin-cre is selectively active in epithelial cells of the villi and crypts of the intestine; second, enterocytes are the cells responsible for lipid synthesis and absorption in the intestine.
Fig. 1 shows the expression of the Insig mRNAs in enterocytes along the longitudinal axis of the intestine. In control mice, Insig1 and Insig2b mRNA vary slightly along the longitudinal axis. Insig2a, the liver-specific isoform of Insig2 (28), was undetectable in any segment of the intestine (data not shown). In Vil-Insig− mice, Insig1 mRNA levels were reduced by 91–99% in various segments of intestine, and Insig2b mRNA was not detectable. The expression of Scap was relatively constant across segments of intestine in both control and Vil-Insig− mice. Using polyclonal antibodies that are capable of detecting endogenous Insig proteins in mouse liver (18), we found that Insig1 and Insig2 proteins were not detectable in enterocyte extracts from either control or Vil-Insig− mice (data not shown), which suggests that the steady state abundance of Insig proteins is lower in the intestine than in the liver. This finding agrees with previous measurements of Insig1 and Insig2 mRNAs in various mouse tissues (2, 28).
In Fig. 2A, we measured protein levels of SREBP-1, SREBP-2, and HMGR by immunoblot analysis of homogenates of isolated enterocytes from control and Vil-Insig− mice. Nuclear SREBP-1 and SREBP-2 were markedly increased in the Vil-Insig− mice compared with control mice. Precursor SREBP-1 and SREBP-2 were modestly elevated in the Vil-Insig− mice as well. The relatively much larger increase in nuclear SREBP-1 and SREBP-2, compared with their precursors, suggests that the rate of SREBP processing is elevated in the Vil-Insig− mice. HMGR protein levels were increased massively in the Vil-Insig− mice compared with control mice, whereas its mRNA was increased by only 2.3-fold (Fig. 2B). The disproportionately larger increase in HMGR protein relative to its mRNA suggests that the degradation of HMGR protein is reduced in Vil-Insig− mice, consistent with the known effect of Insig on HMGR proteasomal degradation. Levels of Scap, NPC1L1, and calnexin, a control protein, were not significantly different between control and Vil-Insig− mice. These data show that Insig proteins are required for feedback inhibition of SREBPs and HMGR in enterocytes as they are in hepatocytes and other cells.
FIGURE 2.

Constitutive SREBP processing in Insig-deficient enterocytes. A, immunoblot analysis. Whole-cell extracts from enterocytes of control and Vil-Insig− mice (male, 14–21 weeks of age, 7 mice/group) were individually prepared and pooled. Aliquots of the pooled protein (60 μg for SREBP-1 and SREBP-2, 30 μg for all other proteins) were subjected to SDS-PAGE and immunoblot analysis. Precursor and nuclear forms of SREBPs are denoted as P and N, respectively. Asterisks, nonspecific bands. Calnexin was used as a loading control. B, relative mRNA levels in enterocytes of control and Vil-Insig− mice used in A. Total RNA from enterocytes was subjected to quantitative RT-PCR using cyclophilin as the invariant control. Each value represents the mean ± S.E. (error bars) of data from seven mice relative to that of control mice, which was arbitrarily defined as 1.0. *, p < 0.01, the level of statistical significance (two-tailed Student's t test) between control and Vil-Insig− mice.
Fig. 2B shows mRNA levels from enterocytes of control and Vil-Insig− mice. Relative to control mice, the mRNA levels of SREBP-1c, SREBP-2, and SREBP target genes in lipogenic pathways were increased by 2.0–130-fold in the enterocytes of Vil-Insig− mice, consistent with the increase in nSREBP-1 and nSREBP-2 (Fig. 2A). SREBP-2 mRNA level was increased by 2.0-fold, and the mRNAs of five SREBP-2 target genes involved in sterol synthesis or LDL uptake were increased by 2.3–12-fold. SREBP-1c mRNA level was increased by 6.4-fold, and mRNAs for two of its target genes involved in fatty acid synthesis were increased, fatty acid synthase (FAS) by 5.7-fold and SCD1 (stearoyl-CoA desaturase 1) by 130-fold. Expression of SREBP-1a was not affected in the Vil-Insig− intestines. The increase of SREBP-1c and SREBP-2 mRNA and precursor protein can be explained in that their transcription is stimulated by SREBP-binding sites present in their own promoters/enhancer regions acting in feed-forward fashion (29, 30). Of note, the genes for FAS, SCD1, and SREBP-1c are all stimulated by both SREBP and liver X receptors (LXRs) (31, 32), nuclear hormone receptors that are activated by oxysterols. The increase in these mRNAs may reflect the activation of LXRs in addition to increased SREBP processing. The mRNA for ATP-binding cassette transporter A1 (ABCA1), a cholesterol transporter whose expression is regulated by LXR but not SREBP (33, 34), was increased by 3.1-fold. However, other LXR-responsive genes were not affected in the Vil-Insig− mice. These include ABCG5 and ABCG8, the major cholesterol efflux transporters, and inducible degrader of the LDL receptor (IDOL), an E3 ligase that initiates degradation of the LDL receptor.
To test the effect of intestinal Insig deficiency on in vivo lipid synthesis, we next measured the incorporation of intraperitoneally injected 3H-labeled water into digitonin-precipitable sterols and fatty acids in control and Vil-Insig− mice (Fig. 3). Intestinal sterol synthesis, on a per gram of tissue basis, was increased in the Vil-Insig− mice by 10–14-fold in proximal, middle, and distal small intestine (Fig. 3A). Intestinal fatty acid synthesis was increased 2.2–3.2-fold (Fig. 3B). Sterol and fatty acid synthesis in the colon of Vil-Insig− mice were elevated by 2.8- and 2.2-fold, respectively.
In Fig. 3, C and D, we replotted the above data to compare the whole-organ lipid synthesis rates between control and Vil-Insig− mice. Whereas in control mice, the overall sterol synthesis rate in the intestine was approximately one-half that of the liver, in the Vil-Insig− mice, the small intestine was the predominant organ of sterol synthesis, with the synthetic rate being 7.4-fold that of the liver. Fatty acid synthesis in liver exceeded that of the intestine in both control and Vil-Insig− mice. Compared with control mice, hepatic sterol and fatty acid synthesis in the Vil-Insig− mice were reduced 32 and 13%, respectively, but neither difference reached statistical significance.
The data of Fig. 3 show that in the Vil-Insig− mice, the small intestine supersedes the liver as the major organ of sterol synthesis. To determine whether the exaggerated sterol synthesis in enterocytes affects tissue and plasma lipid levels, we measured cholesterol and triglyceride levels (Fig. 4). Total plasma cholesterol (Fig. 4A) was increased by 47 mg/dl in the Vil-Insig− mice (p < 0.01). This finding was reproduced in four independent studies; in pooled data from these representing 21 mice in each group, the mean increase in plasma cholesterol in the Vil-Insig− mice was 48 mg/dl. Fractionation of plasma lipoproteins by fast performance liquid chromatography (FPLC) revealed an increase in cholesterol in all lipoprotein fractions (Fig. 4C). Relative to that of control mice, cholesterol contents in intestine (Fig. 4E) and liver (Fig. 4G) tissues of Vil-Insig− mice were increased by 1.8- and 1.7-fold, respectively, with nearly all of the increase being in esterified cholesterol. Plasma (Fig. 4, B and D) and tissue (Fig. 4, F and H) triglyceride levels were unaffected.
FIGURE 4.

Increased tissue and plasma cholesterol in Vil-Insig− mice. A–D, plasma lipids and FPLC profiles from the same mice described in Fig. 2. For plasma cholesterol (A) and triglycerides (B), each bar represents the mean ± S.E. of values from seven mice. Equal aliquots of plasma from 7 mice/group were pooled and subjected to gel filtration by FPLC. Concentrations of cholesterol (C) and triglycerides (D) in each FPLC fraction were determined. Fractions containing VLDL/chylomicron (V-CM), LDL (L), and HDL (H) are indicated. E–H, lipid contents in isolated enterocytes and livers of control and Vil-Insig− mice (female, 17–20 weeks of age, 6 mice/group). Each bar represents mean ± S.E. (error bars) of data from six mice. E and G, tissue cholesterol levels. Total and free cholesterol are designated. F and H, tissue triglyceride levels. *, p < 0.01, the level of statistical significance (two-tailed Student's t test) between control and Vil-Insig− mice.
Enterocytes do not ordinarily accumulate large quantities of cholesterol because they incorporate it into chylomicrons that are secreted into the lymph (35). To identify the cell population(s) in which the excess lipids accumulated in the intestines of Vil-Insig− mice, we stained neutral lipids with Oil Red O in cryosections of jejuna from control and Vil-Insig− mice (Fig. 5). In the Vil-Insig− mice, the overall morphology of the jejunal villi and crypts appeared normal. Lipid droplets were seen to accumulate in the lower villus and crypt, being most evident at the crypt base.
FIGURE 5.
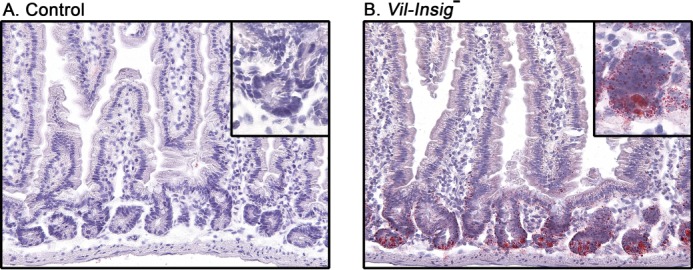
Lipid accumulation of in jejunal crypts of Vil-Insig− mice. Representative Oil Red O and hematoxylin-stained histologic sections of jejuna from control and Vil-Insig− mice are shown. Magnification, ×20. Inset magnification, ×40.
In Fig. 6, we carried out immunoblot and real-time RT-PCR analyses to determine whether the intestinal Insig deficiency affects the hepatic levels of SREBP proteins and their targeting genes. Compared with that of control mice, nSREBP-2 was reduced in livers of Vil-Insig− mice (Fig. 6A). As expected, the reduced nSREBP-2 was accompanied by reduced mRNAs of several SREBP-2 targets, including HMG-CoA synthase (HMGS), HMGR, farnesyl pyrophosphate synthase (FPPS), and PCSK9 (proprotein convertase subtilisin/kexin type 9). The amount of Insig1 mRNA, which is also controlled by SREBP-2, was reduced by 64% in livers of Vil-Insig− mice. This reduction is not a result of Cre-mediated disruption of Insig1 in the liver because Cre mRNA was not detectable in livers of Vil-Insig− mice (data not shown). Insig2a mRNA was not detectable in livers of Vil-Insig− mice due to the germ line Insig2 deletion. Because germ line deletion of Insig-2 alone did not have any significant effects on the hepatic levels of SREBP-1, SREBP-2, or HMGR or the expression of SREBP-stimulated genes (Ref. 8 and data not shown), we believe that the suppression of SREBP-2 processing in livers of Vil-Insig− mice is caused by the hepatic accumulation of cholesterol as a result of Insig-1 and Insig-2 deletion in the intestine rather than by Insig-2 deficiency in liver per se.
FIGURE 6.

Compensatory reduction of nuclear SREBP-2 in the livers of Vil-Insig− mice. The mice used here are the same as those described in the legend to Fig. 2. A, immunoblot analysis. Whole-cell extracts from livers of control and Vil-Insig− mice were individually prepared, pooled, and subjected to SDS-PAGE and immunoblot analysis. Precursor and nuclear forms of SREBPs are denoted as P and N, respectively. Asterisks, nonspecific bands. B, relative mRNA levels in livers of control and Vil-Insig− mice. Total RNA from liver was subjected to quantitative RT-PCR with ApoB as the invariant control. Each value represents the mean ± S.E. (error bars) of data from seven mice relative to that of control mice, which was arbitrarily defined as 1.0. *, p < 0.05; **p < 0.01, the level of statistical significance (two-tailed Student's t test) between control and Vil-Insig− mice.
Nuclear SREBP-1 was slightly elevated in livers of Vil-Insig− mice, and mRNAs for two of its target genes, FAS and SCD1, were increased by about 1.5-fold. The mRNAs for several LXR target genes, including ABCG5, AGCG8, and IDOL, were also up-regulated, suggesting that hepatic LXRs are being activated by the increased flux of cholesterol from the intestines of the Vil-Insig− mice. The hepatic mRNA levels for LXRα and ABCA1 were not different between control and Vil-Insig− mice.
Our previous studies showed that administration of the cholesterol absorption inhibitor ezetimibe triggered a dramatic activation of enterocyte SREBP-2 processing and increased abundance of HMGR. We hypothesized that ezetimibe blocks Insig action by lowering ER cholesterol. To test this hypothesis, we fed control and Vil-Insig− mice a diet containing 0.01% ezetimibe, a dose that blocks ∼90% of lumenal cholesterol absorption (36) (Fig. 7). We also fed mice with a fat-free diet, which eliminates exogenous cholesterol but allows enterocytes to access cholesterol from bile and from mucosal sloughing. In control mice, ezetimibe produced a large increase in nSREBP-2 and HMGR proteins, as expected (Fig. 7A, lanes 1 and 2), and the fat-free diet provoked a small increase in SREBP-2 without affecting HMGR (Fig. 7A, lane 3). These data suggest that whereas both dietary and intrinsic sources of lumenal cholesterol play a role in providing tonic repression of SREBP-2 and HMGR, the intrinsic sources seem to be more potent. In the Vil-Insig− mice, nSREBP-2 and HMGR levels overaccumulated, and neither ezetimibe nor the fat-free diet produced a further increase (Fig. 7A, lanes 4–6). Fig. 7B shows the levels of various mRNAs in enterocytes from the same groups of mice. mRNAs for SREBP-2 and three of its target mRNAs (Insig1, HMGR, and HMG-CoA synthase) were increased by ezetimibe in the control mice. Apart from Insig1, mRNA levels for these genes were increased in the Vil-Insig− mice irrespective of diet, in agreement with the levels of nSREBP-2 in Fig. 7A. Scap and Insig2b mRNA were not affected by dietary manipulation. The data of Fig. 7 indicate that Insigs are required for the feedback inhibition of enterocyte sterol synthesis by gut lumen-derived cholesterol.
FIGURE 7.

Ezetimibe fails to further activate SREBP-2 processing and increase HMGR protein in Insig-deficient enterocytes. Control and Vil-Insig− mice (female, 22–35 weeks of age, 4 mice/group) were fed ad libitum with a chow diet, a chow diet containing 0.01% ezetimibe (EZE), or a fat-free diet (FF) for 5 days prior to studies. A, immunoblot analysis of isolated enterocytes. Precursor and nuclear forms of SREBP-2 are denoted as P and N, respectively. Asterisks, nonspecific bands. B, relative mRNA levels in isolated enterocytes. Total RNA from enterocytes was subjected to quantitative RT-PCR using cyclophilin as the invariant control. Each bar represents the mean ± S.E. (error bars) of value from four mice relative to that of control mice, which was arbitrarily defined as 1.0.
DISCUSSION
The current results demonstrate that Insig proteins suppress cholesterol synthesis in enterocytes by inhibiting SREBP processing and promoting HMGR degradation in the presence of excess sterols. They further support that the entire SREBP pathway functions in the intestine as it does in cultured fibroblasts and livers.
In the absence of Insig-mediated suppression, enterocytes oversynthesize sterols, some of which exit the intestine and accumulate in plasma and liver. However, other destinations of the newly synthesized cholesterol remain unclear. Another pathway in the intestine that is gaining increasing attention is the transintestinal cholesterol excretion/efflux (TICE) process that contributes 30–70% of fecal neutral sterol excretion (reviewed in Refs. 37 and 38). Preliminary data suggest that fecal cholesterol contents of Vil-Insig− mice seem to be slightly higher than that of control mice under chow-fed conditions. Future quantitative whole-body cholesterol balance studies, such as the method developed by van der Vee et al. (39), will be necessary to elucidate the potential alterations of TICE in Vil-Insig− mice.
The effect of increasing intestinal cholesterol synthesis on the liver is reminiscent of the effect of feeding cholesterol-rich diets. Cholesterol originating from enterocytes accumulates in hepatocytes and suppresses sterol synthesis by reducing SREBP-2 processing. In addition, LXR-dependent genes are activated, increasing cholesterol excretion into the bile.
It was proposed years ago that increased intestinal cholesterol synthesis may contribute to dyslipidemia (40); elevated rates of intestinal sterol synthesis are noted in several rodent models of diabetes associated with dyslipidemia (41). In concert with the hypothesis that exaggerated intestinal sterol synthesis leads to hypercholesterolemia, we found a modest elevation in plasma cholesterol in the Vil-Insig− mice. One straightforward model would hold that increased enterocyte sterol synthesis directly increases secretion of chylomicron cholesterol; in support of this hypothesis, a previous report of thoracic lymph duct cannulation of diabetic rats showed increased secretion of chylomicron cholesterol in association with increased intestinal sterol synthesis (42). In the current studies, we saw no evidence for increased accumulation of cholesterol-rich, chylomicron-size particles in the Vil-Insig− mice at steady state. Detailed studies on the effect of modulating enterocyte SREBPs on chylomicron synthesis and secretion as well as other aspects of lipoprotein metabolism are ongoing and are beyond the scope of the current paper.
These findings highlight key similarities and differences in the physiologic regulation of SREBP-2 in liver and intestine, the two organs responsible for over 70% of sterol synthesis in mice (10). In the liver, SREBP-2 is highly expressed and actively processed so long as animals are fed a low cholesterol diet. High cholesterol diets repress SREBP-2 (18, 43). Hepatic Insig deficiency resulted in constitutive activation of SREBP-2 processing and a massive increase in HMGR protein, both of which were also refractory to the inhibitory effects of dietary cholesterol (8). In contrast, SREBP-2 processing and HMGR protein in enterocytes are low in animals even when they are maintained on a cholesterol-free diet (Fig. 7A). This is possibly due to the intestinal uptake of cholesterol from bile and from sloughed mucosal cells. High cholesterol diets fail to produce a further decline of the already suppressed levels of SREBP-2 processing protein in the intestine (data not shown). This suppression is relieved when Insigs are eliminated by recombination or when cholesterol absorption is blocked by ezetimibe. Insig deficiency activated intestinal SREBP-2 processing by roughly the same degree as did ezetimibe in control mice (Fig. 7A). Intestinal Insig deficiency also produced a massive increase of HMGR protein, a level magnitudes higher than that of ezetimibe-treated control mice. By the virtue of these dual actions, intestinal Insig deficiency resulted in a marked increase in sterol synthesis, with a whole-tissue biosynthetic rate of 4.6 μmol of [3H]H2O incorporated/h (Fig. 3C). This large capacity for sterol synthesis in the intestine is comparable with that in the Insig-deficient liver (6.4 μmol of [3H]H2O incorporated/h) (8).
It is unclear why enterocytes have evolved to maintain a high potential capacity for sterol synthesis, which is then kept tightly suppressed by Insig proteins. In the crypt-villus structure, sterol synthesis is highest at the crypt base and decreases with ascension to the villus tip (44, 45). Highly proliferative cells in the crypt require high levels of de novo lipid synthesis, which is suppressed as the cells differentiate and ascend the crypt in order to maintain cellular sterol homeostasis. It appears that rather than losing this synthetic capacity, upper villus enterocytes suppress it through the absorption of lumenal cholesterol. In the absence of Insig proteins, lumenal cholesterol is unable to suppress SREBPs, and consequently, upper villus enterocytes continue to synthesize cholesterol although they remain supplied from the gut lumen. Insigs are therefore key suppressors of sterol synthesis in the intestine.
These studies clarify the nature of the homeostatic regulation between the enterocyte's three sources of cholesterol: de novo synthesis, LDL uptake, and lumenal absorption. Our previous studies suggested that enterocytes maintain homeostasis when lumenal cholesterol becomes unavailable by ramping up de novo sterol synthesis and LDL uptake (14). To determine whether the large increase in sterol synthesis in Insig-deficient enterocytes causes a compensatory decrease in intestinal cholesterol absorption, we measured cholesterol absorption in control and Vil-Insig− mice using a fecal dual isotope ratio method (46). Interestingly, fractional cholesterol absorption was not significantly different between control (0.29 ± 0.03) and Vil-Insig− (0.40 ± 0.04) mice. These data suggest that the molecular pathways controlling intestinal sterol synthesis and absorption are not coordinately regulated.
The finding of lipid accumulation in the crypt bases of the Vil-Insig− mice has two major implications: first, the excess lipids are exiting the upper villus enterocytes, presumably via chylomicrons; second, a population of cells in the crypt base, which is composed largely of intestinal stem cells and paneth cells, appears to be accumulating lipids to a high degree. This lipidosis suggests that Insigs, and by extension SREBPs, may play a role in the biology of these cells. The finding of normal villus height and structure in the Vil-Insig− mice suggests that the stem cell compartment is functioning normally at a gross level and that the lipidosis appears to be benign in terms of stem cell renewal and differentiation. This supports the idea that the intestinal function of Insigs is to suppress the cholesterologenic capacity of differentiated enterocytes. Further studies will be required to determine the identity of these lipid-laden cells and whether their function has been impacted.
In summary, these studies demonstrate that the SREBP pathway is an active player in the control of enterocyte lipid metabolism in vivo. Open questions include whether this regulation in intestine contributes to the pathogenesis of diseases of lipid metabolism, such as dyslipidemia, fatty liver, and obesity. The potential role of the SREBP pathway in other aspects of intestinal biology, such as cell growth or immune function, as has been proposed for other tissues (47), is also an area warranting further study.
Acknowledgments
We thank Michael Brown and Joseph Goldstein for mentorship and support; Jay Horton for critical review of the manuscript; Stephen Turley for helpful advice and technical assistance; Norma Anderson, Christina Li, Isis Soto, Jeff Cormier, Wenzhu Fan, and Min Ding for technical assistance; and the University of Texas Southwestern-Richardson Molecular Pathology Core Facility for tissue sectioning and staining.
This work was supported, in whole or in part, by National Institutes of Health Grant HL-20948.
- ER
- endoplasmic reticulum
- ABCA1
- ABCG5, and ABCG8, ATP binding cassette transporter A1, G5, and G8, respectively
- FAS
- fatty acid synthase
- HMGR
- HMG-CoA reductase
- IDOL
- inducible degrader of the LDL receptor
- LXR
- liver X receptor
- nSREBP
- nuclear SREBP
- SREBP
- sterol regulatory element-binding protein.
REFERENCES
- 1. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs. Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang T., Espenshade P. J., Wright M. E., Yabe D., Gong Y., Aebersold R., Goldstein J. L., Brown M. S. (2002) Crucial step in cholesterol homeostasis. Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110, 489–500 [DOI] [PubMed] [Google Scholar]
- 3. Ye J., DeBose-Boyd R. A. (2011) Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb. Perspect. Biol. 3, a004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sever N., Yang T., Brown M. S., Goldstein J. L., DeBose-Boyd R. A. (2003) Accelerated degradation of HMG CoA reductase mediated by binding of insig-1 to its sterol-sensing domain. Mol. Cell 11, 25–33 [DOI] [PubMed] [Google Scholar]
- 5. Jo Y., Debose-Boyd R. A. (2010) Control of cholesterol synthesis through regulated ER-associated degradation of HMG CoA reductase. Crit. Rev. Biochem. Mol. Biol. 45, 185–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yabe D., Brown M. S., Goldstein J. L. (2002) Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 99, 12753–12758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sever N., Song B. L., Yabe D., Goldstein J. L., Brown M. S., DeBose-Boyd R. A. (2003) Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 278, 52479–52490 [DOI] [PubMed] [Google Scholar]
- 8. Engelking L. J., Liang G., Hammer R. E., Takaishi K., Kuriyama H., Evers B. M., Li W. P., Horton J. D., Goldstein J. L., Brown M. S. (2005) Schoenheimer effect explained. Feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J. Clin. Invest. 115, 2489–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Engelking L. J., Evers B. M., Richardson J. A., Goldstein J. L., Brown M. S., Liang G. (2006) Severe facial clefting in Insig-deficient mouse embryos caused by sterol accumulation and reversed by lovastatin. J. Clin. Invest. 116, 2356–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xie C., Turley S. D., Dietschy J. M. (2009) ABCA1 plays no role in the centripetal movement of cholesterol from peripheral tissues to the liver and intestine in the mouse. J. Lipid Res. 50, 1316–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spady D. K., Dietschy J. M. (1983) Sterol synthesis in vivo in 18 tissues of the squirrel monkey, guinea pig, rabbit, hamster, and rat. J. Lipid Res. 24, 303–315 [PubMed] [Google Scholar]
- 12. Dietschy J. M., Turley S. D., Spady D. K. (1993) Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J. Lipid Res. 34, 1637–1659 [PubMed] [Google Scholar]
- 13. Dietschy J. M., Gamel W. G. (1971) Cholesterol synthesis in the intestine of man: regional differences and control mechanisms. J. Clin. Invest. 50, 872–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Engelking L. J., McFarlane M. R., Li C. K., Liang G. (2012) Blockade of cholesterol absorption by ezetimibe reveals a complex homeostatic network in enterocytes. J. Lipid Res. 53, 1359–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang D. Q. (2007) Regulation of intestinal cholesterol absorption. Annu. Rev. Physiol. 69, 221–248 [DOI] [PubMed] [Google Scholar]
- 16. Radhakrishnan A., Goldstein J. L., McDonald J. G., Brown M. S. (2008) Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol. A delicate balance. Cell Metab. 8, 512–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Madison B. B., Dunbar L., Qiao X. T., Braunstein K., Braunstein E., Gumucio D. L. (2002) Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283 [DOI] [PubMed] [Google Scholar]
- 18. Engelking L. J., Kuriyama H., Hammer R. E., Horton J. D., Brown M. S., Goldstein J. L., Liang G. (2004) Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J. Clin. Invest. 113, 1168–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Horton J. D., Shimano H., Hamilton R. L., Brown M. S., Goldstein J. L. (1999) Disruption of LDL receptor gene in transgenic SREBP-1a mice unmasks hyperlipidemia resulting from production of lipid-rich VLDL. J. Clin. Invest. 103, 1067–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liang G., Yang J., Horton J. D., Hammer R. E., Goldstein J. L., Brown M. S. (2002) Diminished hepatic response to fasting/refeeding and LXR agonists in mice with selective deficiency of SREBP-1c. J. Biol. Chem. 277, 9520–9528 [DOI] [PubMed] [Google Scholar]
- 21. Yang J., Goldstein J. L., Hammer R. E., Moon Y. A., Brown M. S., Horton J. D. (2001) Decreased lipid synthesis in livers of mice with disrupted Site-1 protease gene. Proc. Natl. Acad. Sci. U.S.A. 98, 13607–13612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shimano H., Shimomura I., Hammer R. E., Herz J., Goldstein J. L., Brown M. S., Horton J. D. (1997) Elevated levels of SREBP-2 and cholesterol synthesis in livers of mice homozygous for a targeted disruption of the SREBP-1 gene. J. Clin. Invest. 100, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsuda M., Korn B. S., Hammer R. E., Moon Y. A., Komuro R., Horton J. D., Goldstein J. L., Brown M. S., Shimomura I. (2001) SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 15, 1206–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rashid S., Curtis D. E., Garuti R., Anderson N. N., Bashmakov Y., Ho Y. K., Hammer R. E., Moon Y. A., Horton J. D. (2005) Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc. Natl. Acad. Sci. U.S.A. 102, 5374–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shimano H., Horton J. D., Hammer R. E., Shimomura I., Brown M. S., Goldstein J. L. (1996) Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J. Clin. Invest. 98, 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Churukian C. J. (1999) Lillie's oil red O method for neutral lipids. J. Histotechnol. 22, 309–311 [Google Scholar]
- 27. Braunstein E. M., Qiao X. T., Madison B., Pinson K., Dunbar L., Gumucio D. L. (2002) Villin. A marker for development of the epithelial pyloric border. Dev. Dyn. 224, 90–102 [DOI] [PubMed] [Google Scholar]
- 28. Yabe D., Komuro R., Liang G., Goldstein J. L., Brown M. S. (2003) Liver-specific mRNA for Insig-2 down-regulated by insulin. Implications for fatty acid synthesis. Proc. Natl. Acad. Sci. U.S.A. 100, 3155–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sato R., Inoue J., Kawabe Y., Kodama T., Takano T., Maeda M. (1996) Sterol-dependent transcriptional regulation of sterol regulatory element-binding protein-2. J. Biol. Chem. 271, 26461–26464 [DOI] [PubMed] [Google Scholar]
- 30. Chen G., Liang G., Ou J., Goldstein J. L., Brown M. S. (2004) Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. U.S.A. 101, 11245–11250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Repa J. J., Liang G., Ou J., Bashmakov Y., Lobaccaro J. M., Shimomura I., Shan B., Brown M. S., Goldstein J. L., Mangelsdorf D. J. (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev. 14, 2819–2830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schultz J. R., Tu H., Luk A., Repa J. J., Medina J. C., Li L., Schwendner S., Wang S., Thoolen M., Mangelsdorf D. J., Lustig K. D., Shan B. (2000) Role of LXRs in control of lipogenesis. Genes Dev. 14, 2831–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Horton J. D., Shah N. A., Warrington J. A., Anderson N. N., Park S. W., Brown M. S., Goldstein J. L. (2003) Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc. Natl. Acad. Sci. U.S.A. 100, 12027–12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Venkateswaran A., Laffitte B. A., Joseph S. B., Mak P. A., Wilpitz D. C., Edwards P. A., Tontonoz P. (2000) Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR α. Proc. Natl. Acad. Sci. U.S.A. 97, 12097–12102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Turley S. D., Valasek M. A., Repa J. J., Dietschy J. M. (2010) Multiple mechanisms limit the accumulation of unesterified cholesterol in the small intestine of mice deficient in both ACAT2 and ABCA1. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G1012–G1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Repa J. J., Turley S. D., Quan G., Dietschy J. M. (2005) Delineation of molecular changes in intrahepatic cholesterol metabolism resulting from diminished cholesterol absorption. J. Lipid Res. 46, 779–789 [DOI] [PubMed] [Google Scholar]
- 37. Temel R. E., Brown J. M. (2012) Biliary and nonbiliary contributions to reverse cholesterol transport. Curr. Opin. Lipidol. 23, 85–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van der Velde A. E., Brufau G., Groen A. K. (2010) Transintestinal cholesterol efflux. Curr. Opin. Lipidol. 21, 167–171 [DOI] [PubMed] [Google Scholar]
- 39. van der Veen J. N., van Dijk T. H., Vrins C. L., van Meer H., Havinga R., Bijsterveld K., Tietge U. J., Groen A. K., Kuipers F. (2009) Activation of the liver X receptor stimulates trans-intestinal excretion of plasma cholesterol. J. Biol. Chem. 284, 19211–19219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Feingold K. R. (1989) Importance of small intestine in diabetic hypercholesterolemia. Diabetes 38, 141–145 [DOI] [PubMed] [Google Scholar]
- 41. Feingold K. R., Lear S. R., Moser A. H. (1984) De novo cholesterol synthesis in three different animal models of diabetes. Diabetologia 26, 234–239 [DOI] [PubMed] [Google Scholar]
- 42. Feingold K. R., Zsigmond G., Hughes-Fulford M., Lear S. R., Moser A. H. (1985) The effect of diabetes mellitus on the lymphatic transport of intestinal sterols. Metabolism 34, 1105–1109 [DOI] [PubMed] [Google Scholar]
- 43. Shimomura I., Bashmakov Y., Shimano H., Horton J. D., Goldstein J. L., Brown M. S. (1997) Cholesterol feeding reduces nuclear forms of sterol regulatory element binding proteins in hamster liver. Proc. Natl. Acad. Sci. U.S.A. 94, 12354–12359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stange E. F., Dietschy J. M. (1983) Absolute rates of cholesterol synthesis in rat intestine in vitro and in vivo. A comparison of different substrates in slices and isolated cells. J. Lipid Res. 24, 72–82 [PubMed] [Google Scholar]
- 45. Stange E. F., Dietschy J. M. (1983) Cholesterol synthesis and low density lipoprotein uptake are regulated independently in rat small intestinal epithelium. Proc. Natl. Acad. Sci. U.S.A. 80, 5739–5743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schwarz M., Russell D. W., Dietschy J. M., Turley S. D. (1998) Marked reduction in bile acid synthesis in cholesterol 7 α-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J. Lipid Res. 39, 1833–1843 [PubMed] [Google Scholar]
- 47. Shao W., Espenshade P. J. (2012) Expanding roles for SREBP in metabolism. Cell Metab. 16, 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]