

Background: Complex phosphorylation mechanisms negatively regulate stathmin microtubule-destabilizing activity.
Results: Stathmin is co-regulated by JNK and PKA to maintain the integrity of interphase microtubules during hyperosmotic stress.
Conclusion: Stress-activated JNK and PKA pathways are integrated in the regulation of the microtubule array during cell stress.
Significance: Stress signaling regulation of microtubule destabilizing stathmin is critical in determining cytoskeleton organization in response to cell stress.
Keywords: Cytoskeleton, Jun N-terminal Kinase (JNK), Protein Kinase A (PKA), Protein Phosphorylation, Signal Transduction, Osmotic Stress, Stathmin
Abstract
Dynamic microtubule changes after a cell stress challenge are required for cell survival and adaptation. Stathmin (STMN), a cytoplasmic microtubule-destabilizing phosphoprotein, regulates interphase microtubules during cell stress, but the signaling mechanisms involved are poorly defined. In this study ectopic expression of single alanine-substituted phospho-resistant mutants demonstrated that STMN Ser-38 and Ser-63 phosphorylation were specifically required to maintain interphase microtubules during hyperosmotic stress. STMN was phosphorylated on Ser-38 and Ser-63 in response to hyperosmolarity, heat shock, and arsenite treatment but rapidly dephosphorylated after oxidative stress treatment. Two-dimensional PAGE and Phos-tag gel analysis of stress-stimulated STMN phospho-isoforms revealed rapid STMN Ser-38 phosphorylation followed by subsequent Ser-25 and Ser-63 phosphorylation. Previously, we delineated stress-stimulated JNK targeting of STMN. Here, we identified cAMP-dependent protein kinase (PKA) signaling as responsible for stress-induced STMN Ser-63 phosphorylation. Increased cAMP levels induced by cholera toxin triggered potent STMN Ser-63 phosphorylation. Osmotic stress stimulated an increase in PKA activity and elevated STMN Ser-63 and CREB (cAMP-response element-binding protein) Ser-133 phosphorylation that was substantially attenuated by pretreatment with H-89, a PKA inhibitor. Interestingly, PKA activity and subsequent phosphorylation of STMN were augmented in the absence of JNK activation, indicating JNK and PKA pathway cross-talk during stress regulation of STMN. Taken together our study indicates that JNK- and PKA-mediated STMN Ser-38 and Ser-63 phosphorylation are required to preserve interphase microtubules in response to hyperosmotic stress.
Introduction
Stathmin (STMN)2 is the archetypal member of the STMN-like protein family of tubulin-associating proteins that also includes SCG10, SCLIP, RB3′, and RB3″ (1). These proteins, via their formation of a ternary complex with two α/β-tubulin heterodimers (T2S), sequester tubulin and so promote microtubule destabilization (2, 3). At the cellular level, this enhanced microtubule destabilization is critical for the control of microtubule organization and dynamics and contributes to processes required for cell division, migration, differentiation, and proliferation (1). Interestingly, STMN deletion in mice results in behavioral defects, and early onset axon degeneration and dysregulated STMN is associated with motor neuron loss, indicating important STMN functions in brain development and neuronal maintenance (4–6). Furthermore, altered expression and post-translational modification of STMN have been functionally linked to enhanced proliferation and invasiveness in a broad range of cancer types in addition to emerging roles in host-pathogen interactions (7–9). These studies highlight the significance of STMN function and regulation in development and disease progression.
STMN was initially identified as a cytosolic protein phosphorylated in response to extracellular stimuli (10). STMN is phosphorylated on four conserved serine residues (Ser-16, Ser-25, Ser-38, and Ser-63), and mutagenesis studies coupled with in vitro tubulin polymerization assays have revealed the contribution of site-specific serine phosphorylation to enhance microtubule stabilization by preventing the formation of the STMN-tubulin T2S complex (11). For example, STMN Ser-16 or Ser-63 phosphorylation was sufficient to reduce STMN inhibition of microtubule assembly, whereas the effects of STMN Ser-25 and Ser-38 phosphorylation were more marginal. Importantly, the phosphorylation of all four serine residues was required to inhibit STMN activity completely in vitro (11).
STMN is phosphorylated in response to cell stress stimuli such as heat shock, hyperosmolarity (osmotic stress (OS)), chemical stress, inflammatory cytokines, proteasome inhibition, and hypoxia (12–16). The multisite phosphorylation of STMN differs depending on the cellular and signaling context, and a number of different protein kinases are known to target specific STMN phosphorylation sites in cells. STMN Ser-16 can be phosphorylated by PAK1, Ca2+/calmodulin-dependent kinase II/IV, or cAMP-dependent protein kinase (PKA) (17–20), whereas proline-flanked Ser-25 and Ser-38 residues are targeted by mitogen-activated protein kinases and cyclin-dependent kinases (21–23). The multisite phosphorylation of STMN generates complex combinations of STMN phospho-isomers that contribute to overall STMN regulation of microtubule stability and organization.
STMN Ser-16 and Ser-25 phosphorylation have been linked to cancer cell metastasis, migration, and neurite outgrowth (20, 24, 25), whereas STMN Ser-25 and Ser-38 phosphorylation are associated with cell stress signaling (12, 14, 26). In contrast, the kinases that target STMN Ser-63 are less well characterized, although active PKA in vitro or the ectopic overexpression of PKA in cells can promote STMN Ser-63 phosphorylation (17, 27). The biological context and consequence of PKA signaling to STMN are unclear and are also further complicated by interdependent relationships of the STMN phosphorylation sites. For example, STMN Ser-16 and Ser-63 targeting by mitotic kinases requires prior Ser-25 and Ser-38 phosphorylation (28). Similarly, our recent studies highlighted that the efficient phosphorylation of STMN Ser-25 in response to OS required prior phosphorylation of STMN Ser-38 (14). Therefore, although we have previously characterized JNK-dependent STMN Ser-25 and Ser-38 phosphorylation in response to cell stress, the signaling pathway(s) that regulates STMN Ser-63 and its contributions to microtubule regulation during cell stress remains enigmatic.
In this study we investigated the relative importance of STMN-specific serine phosphorylation toward its activity. Our combined use of mobility shift detection and site-specific phospho-STMN antibodies allowed our characterization of STMN phosphorylation in response to cell stress, revealing the complexities of the STMN phospho-isomers stimulated under these conditions. We have also defined a role for PKA in the phosphorylation and regulation of STMN function during hyperosmotic stress and uncovered signaling cross-talk between JNK and PKA regulation of STMN. Our studies highlight the complex interplay of phosphorylation to regulate STMN activity in the maintenance of interphase microtubules in the context of cellular stress.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
All antibodies were obtained from commercial sources: phospho-STMN Ser-16, Ser-25, Ser-38, and Ser-63 antibodies (Abcam), α-tubulin antibody (Santa Cruz Biotechnology), STMN antibodies (Cell Signaling Technology (CST) #3352 or Sigma #O0138), and phospho-c-Jun S73 antibody (CST). Protein kinase inhibitors JNK inhibitor VIII, aurora kinase inhibitor III, staurosporine (broad spectrum serine/threonine inhibitor), bisindolylmaleimide I (protein kinase C (PKC) inhibitor), and H-89 (PKA inhibitor) were purchased from Calbiochem and dissolved in DMSO. Phos-tagTM was purchased from Wako Chemicals. Cell culture serum, medium, and supplements were obtained from Invitrogen. Nerve growth factor (NGF), sorbitol, and all routine laboratory chemicals were from Sigma.
Cell Culture and Treatment
PC12 rat pheochromocytoma cells were maintained in Dulbecco's modified Eagles medium (DMEM) supplemented with 10% (v/v) horse serum, 5% (v/v) fetal calf serum (FCS), and 100 units/ml penicillin/streptomycin and cultured in a humidified 5% CO2 environment. COS-1 monkey kidney fibroblasts were grown in DMEM supplemented with 10% (v/v) FCS and 100 units/ml penicillin/streptomycin. PC12 cells were plated onto 60-mm collagen I-coated dishes (Iwaki), whereas COS-1 cells were plated on uncoated dishes. Liposomal-mediated transfection was performed with LipofectamineTM 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. Cells were serum-starved overnight and pretreated with kinase inhibitors (30 min) before cell treatments/stress stimulation.
Analysis of Microtubule Arrays
COS-1 cell treatments were performed on uncoated glass coverslips. STMN phosphorylation site mutant constructs (STMN-S16A, STMN-S25A, STMN-S38A, STMN-S63A, and STMN-AAAA) were made by site-directed mutagenesis as previously described (14). Mutant constructs were then cloned into pXJ40-myc and subjected to restriction digestion and sequencing analysis before further use.
Cells were then washed in phosphate-buffered saline (PBS) before fixation with 4% (w/v) paraformaldehyde (20 min, room temperature). Sample preparation and confocal microscopy were as previously described (29). Cells were permeabilized (0.2% (v/v) Triton X-100 in PBS) and pre-blocked (10% (v/v) FCS in PBS) before incubation with primary antibodies and Cy2/Cy3-conjugated secondary antibodies diluted in 1% (w/v) bovine serum albumin (BSA) in PBS. The cells were then mounted (GelMount, Biomeda Corp.), and images were captured on a Leica TCS SP2 confocal microscope using a 100 × 1.35 NA objective.
Cell Lysate Preparation, Electrophoresis, and Immunoblotting
PC12 cells were washed with ice-cold PBS before lysis in radioimmune precipitation assay buffer (50 mm Tris-HCl (pH 7.3), 150 mm NaCl, 0.1 mm EDTA, 1% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100, 0.2% (w/v) NaF, and 100 μm Na3VO4) supplemented with Complete protease inhibitors (Roche Applied Science). Cell lysates were incubated (10 min, on ice) before removal of cell debris by centrifugation (14,000 × g, 15 min, 4 °C). Protein concentrations were then determined by Bradford assay. Protein lysates in Laemmli buffer were resolved by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes, blocked (5% (w/v) nonfat milk powder in Tris-buffered saline with 0.01% (v/v) Tween 20), and immunoblotted with the appropriate primary and horseradish peroxidase-conjugated secondary antibodies (Millipore). Proteins were visualized with enhanced chemiluminescence (Pierce), and bands were quantified by densitometric analysis (Bio-Rad ChemidocTM MP Imager). Phos-tagTM gels were prepared as per SDS-PAGE gels with the addition of 50 μm Phos-tagTM reagent and 5 μm MnCl2. After electrophoresis, Phos-tagTM gels were soaked in transfer buffer containing 1 mm EDTA (10 min with gentle agitation) followed by another 10 min in buffer without EDTA to remove manganese ions from the gel before transfer to PVDF membranes.
Phosphatase Treatment
Cell lysates were prepared in radioimmune precipitation assay buffer lacking EDTA, NaF, and Na3VO4. Lysates (100 μg of total protein as estimated by Bradford assay) were treated with 400 units of λ-phosphatase (New England Biolabs) for 25 min at 30 °C. Laemmli buffer was then added, and proteins were resolved for immunoblotting as described above.
Two-dimensional PAGE
Protein lysates from PC12 cells were prepared in lysis buffer (40 mm Tris-HCl (pH 8.0), 8 m urea, 4% (w/v) CHAPS) and clarified by centrifugation (14,000 × g, 15 min, 4 °C), and protein concentrations were determined by the Bradford assay. First-dimension isoelectric focusing was performed using 11-cm linear immobilized pH 4–7 gradient drystrips, 1% pH 4–7 carrier ampholytes, and 20 mm DTT on an EttanTM IPGphorTM (Amersham Biosciences). Run conditions were as specified by the manufacturer. Second-dimension separation was performed on 8–16% gradient Tris-HCl polyacrylamide gels (Bio-Rad). After electrophoresis, proteins were transferred onto PVDF membranes and immunoblotted with total STMN antibody.
PKA Activity and cAMP Assays
PKA activity in cell lysates was assayed using a PKA assay kit (Millipore) according to the manufacturer's description. Briefly, cell lysates (20 μg) were incubated with 80 μm Kemptide, 10 μCi of [γ-32P]ATP in kinase reaction buffer (20 mm MOPS (pH 7.2), 25 mm β-glycerophosphate, 5 mm EGTA, 1 mm sodium orthovanadate, 1 mm dithiothreitol) with or without PKA peptide inhibitor for 10 min at 30 °C. Samples were spotted onto P81 phosphocellulose paper, and phosphorylated peptide was separated from [γ-32P]ATP by washing in 0.75% phosphoric acid. Radioactivity was quantified using liquid scintillation counting. Residual activity in the presence of PKA peptide inhibitor was subtracted to calculate PKA activity.
cAMP was measured in whole-cell lysates using a competition enzyme-linked immunoassay (cAMP XPTM Assay kit, CST) according to the manufacturer's instructions. Absorbance measurements from OS-treated cell lysates were expressed as a percentage of forskolin (20 μm, 20 min) treatment using the equation (Astress − Abasal(0 min))/(Aforskolin − Abasal(0 min)).
Tubulin Polymer Assay
Fractionation and immunoblot analysis of soluble and polymerized tubulin from stress-stimulated cell lysates were performed as described previously (30) with slight modifications. Cells were lysed in a microtubule stabilization buffer (100 mm PIPES (pH 6.9), 5 mm MgCl2, 1 mm EGTA, 30% (v/v) glycerol, 0.1% (v/v) Nonidet P40, 0.1% (v/v) Triton X-100, 0.1% (v/v) Tween 20, 0.1% (v/v) β-mercaptoethanol, 100 μm GTP, 1 mm ATP, and 2 μm taxol) supplemented with protease inhibitors. Lysates were precleared (1000 × g, 5 min) before polymerized tubulin pelleted by ultracentrifugation (100,000 × g, 30 min, 37 °C). Soluble fractions were removed, and pellets were washed and depolymerized with 2 mm CaCl2 (4 °C). Soluble and polymerized tubulin were resolved on SDS-PAGE, detected, and quantified by immunoblotting. Tubulin polymer levels in pellet fractions were expressed as a % of total tubulin ((tubulinPellet)/(tubulinSoluble + tubulinPellet)).
In Vitro STMN Phosphorylation and Tubulin Turbidity Assay
Purified recombinant STMN (GST fusion or Precision Protease cleaved to remove GST tag) was incubated with active kinase (JNK1 and/or PKA) in a kinase reaction buffer (20 mm HEPES (pH 7.6), 20 mm MgCl2·6H2O, 20 μm ATP, 20 mm β-glycerophosphate, and supplemented with 25 μm Na3VO4 and 100 μm DTT) for 120 min at 30 °C. STMN serine phosphorylation was determined by immunoblot analysis with phospho antibodies. In vitro tubulin polymerization in the presence or absence of STMN was performed as described previously (30). Briefly, purified porcine tubulin (300 μg, Cytoskeleton Inc.) was incubated with STMN to a final volume of 100 μl in tubulin assembly buffer (80 mm PIPES (pH 6.8), 0.5 mm EGTA, 2 mm MgCl2, 1 mm GTP, and 10% (v/v) glycerol) on clear 96-well plates. Tubulin polymerization was initiated with incubation at 37 °C, and absorbance (340 nm) was measured every minute on a temperature-controlled plate reader (POLARstar Optima, BMG Labtech).
Statistical Analysis
All values are presented as the mean ± S.E. Values were compared using two-way analysis of variance or unpaired Student's t tests as appropriate. p values <0.05 were considered significant.
RESULTS
STMN Ser-38 and Ser-63 Are Essential for the Maintenance of Interphase Microtubules during Hyperosmolarity
STMN phosphorylation has been linked to global microtubule destabilization (31). We thus examined the impact of a STMN-AAAA mutant in which all four regulatory serine phosphorylation sites (Ser-16, Ser-25, Ser-38, and Ser-63) had been alanine-substituted. The effects of STMN-WT (wild type) and STMN-AAAA highlighted the importance of phosphorylation as a key negative regulatory modification of STMN activity in maintaining the organization of the cellular microtubule array (Fig. 1A). To assess the contributions of each serine, both under control and stress conditions, we expressed single-alanine-substituted STMN mutants. In contrast to STMN-AAAA, the expression of single-alanine-substituted STMN mutants (S16A, S25A, S38A, or S63A) did not substantially perturb the microtubule array under basal conditions as indicated by the density and radial arrangement of microtubules that did not differ markedly from cells expressing wild-type STMN or adjacent non-transfected cells (Fig. 1A).
FIGURE 1.

Site-specific STMN serine phosphorylation contributions toward microtubule maintenance during hyperosmolarity. A, Myc-tagged STMN (STMN WT), phospho-resistant STMN (STMN AAAA), or single serine-to-alanine-substituted mutants (STMN S16A, S25A, S38A, or S63A) were transiently expressed in COS-1 cells. At 48 h post-transfection, cells were fixed and stained with anti-Myc for ectopically expressed STMN (green), anti-α-tubulin for microtubules (red), and DAPI for DNA (blue). B, COS-1 cells were transfected as in A and stimulated with sorbitol (0.5 m, 60 min) before fixation and immunofluorescence analysis. C, the proportion of control or sorbitol-treated (0.5 m, 60 min) COS-1 cells, expressing Myc-tagged STMN or STMN mutants, with normal or destabilized microtubule (MT) arrays. The total cell numbers were quantified from three independent experiments. D, the expression levels of Myc-tagged WT or mutant STMN in sorbitol-treated (OS, 0.5 m, 60 min) COS-1 cells were determined by immunoblot analysis. E, soluble and polymerized tubulin fractions were prepared from sorbitol-treated (0.5 m, 60 min) COS-1 cells expressing Myc-tagged STMN or STMN mutants and immunoblotted to assess levels of tubulin and STMN expression. The levels of polymerized tubulin as a proportion of total tubulin was calculated from two independent experiments with mean values plotted. Con, control.
We then interrogated how these phosphorylation sites may contribute to the response to hyperosmolar stress conditions (OS) initiated by the addition of 0.5 m sorbitol to the culture media. We found that cells expressing STMN-WT maintained their microtubule arrays, as did cells expressing STMN S16A or STMN S25A (Fig. 1B). In contrast, the microtubules in cells expressing STMN S38A or S63A were completely destabilized and resembled cells expressing the STMN-AAAA mutant (Fig. 1B, arrows). Our quantitation indicated that the proportion of cells expressing STMN S38A or S63A with perturbed microtubule arrays increased after osmotic stress (Fig. 1C). In contrast, sorbitol treatment did not substantially alter the proportion of STMN-WT-, STMN S16A-, or STMN S25A-expressing cells with destabilized microtubule arrays. Similarly, biochemical interrogation of tubulin polymer levels indicated reduced polymerized tubulin in pelleted fractions prepared from sorbitol-stimulated cells expressing STMN S38A or S63A specifically (Fig. 1E). In comparison to STMN WT, tubulin polymer levels were marginally reduced in cells expressing STMN S16A or S25A, but this did not change with sorbitol treatment (Fig. 1E). Cells expressing phospho-deficient STMN-AAAA mutants were essentially devoid of polymerized tubulin regardless of cell treatment. Fluorescence intensity (Fig. 1, A and B) or immunoblot detection (Fig. 1, D and E) using an anti-myc tag antibody to detect the exogenous STMN expression, indicated comparable levels of STMN constructs in both microscopic and immunoblot experiments. The importance of STMN Ser-38 and Ser-63 in maintaining the cytoplasmic microtubule array during OS thus prompted our further investigation of their phosphorylation during stress.
Stresses Differentially Impact STMN Site-specific and Global Phosphorylation Profiles
Evaluation of the phosphorylation of STMN has been facilitated by the availability of phospho-specific antibodies (14, 25). Using these antibodies, we showed that OS induced rapid and robust STMN Ser-38 and Ser-63 phosphorylation in PC12 cells, whereas STMN Ser-25 phosphorylation was more modest, and STMN Ser-16 was only detected at 60 min OS (Fig. 2A). As a control, we included the differentiation stimulus, NGF (50 ng/ml), which stimulated potent STMN Ser-16 and Ser-25 phosphorylation but with only minimal elevation of STMN Ser-38 and Ser-63 phosphorylation at 60 and 120 min (Fig. 2A). These results emphasized the clear differences in STMN phosphorylation profiles under stress and growth conditions, and so we broadened our evaluation to consider other environmental stress stimuli. This revealed that chemotoxicity induced with arsenite exposure or heat shock also stimulated robust STMN Ser-38 phosphorylation in PC12 cells (Fig. 2B). In addition, STMN Ser-25 and Ser-63 phosphorylation were increased in response to heat shock or arsenite albeit at more modest levels compared with OS but comparable to the levels observed upon NGF stimulation (Fig. 2B). Under the conditions tested, the growth stimulus epidermal growth factor (EGF) did not enhance STMN phosphorylation at any of the sites examined (Fig. 2B). Furthermore, oxidative stress induced with free radical generators, glucose oxidase, or hydrogen peroxide (H2O2), reduced basal STMN phosphorylation at all four regulatory serine residues (Fig. 2C).
FIGURE 2.
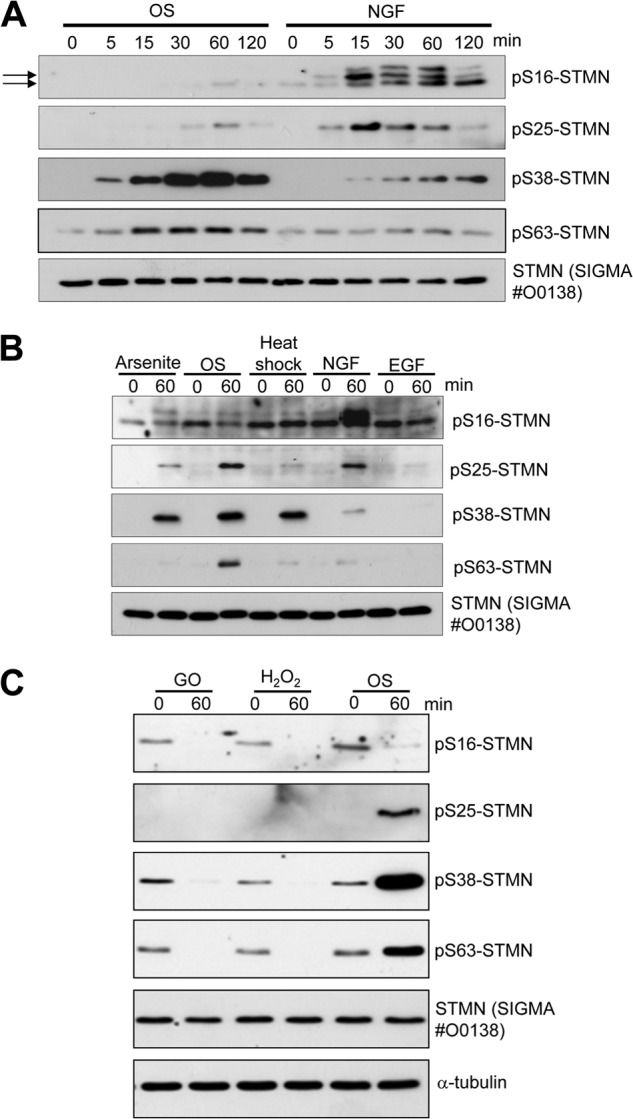
STMN phosphorylation in response to cellular stress. A, PC12 cells were treated with OS (sorbitol, 0.5 m) or NGF (50 ng/ml) for the indicated times. Protein lysates were then prepared and blotted with site-specific phosphoserine STMN antibodies or a pan-STMN antibody, #O0138 (Sigma) as indicated. Arrows indicate bands corresponding to STMN. B, PC12 cells were treated with arsenite (300 μm), heat shock (42 °C), sorbitol (OS, 0.5 m), EGF (10 ng/ml), or NGF (50 ng/ml) for 60 min before cell lysis and blotting with phosphoserine STMN antibodies. C, PC12 cells were treated (60 min) with glucose oxidase (GO, 25 milliunits/ml), hydrogen peroxide (H2O2, 1 mm), or OS (sorbitol, 0.5 m), and STMN phosphorylation was determined by immunoblot analysis.
These findings highlight the potent stimulation of multisite STMN phosphorylation by environmental stress stimuli such as hyperosmolarity, chemotoxins, and heat shock but a marked STMN dephosphorylation by oxidative stress. However, a significant disadvantage of this phospho-antibody immunoblotting approach remains in its inability to report on the proportion of the STMN phosphorylated at each site and whether populations of STMN are multiply phosphorylated. To probe these issues more closely, we adopted two approaches to phosphorylation site assessment by separation of different phosphorylated STMN species, namely the traditional approach of iso-electric focusing/two-dimensional gel separation (27) and the more recently developed mobility shift assessments accentuated by the inclusion of the Phos-tag reagent that binds phosphorylated proteins specifically to retard their mobility on SDS-PAGE (32). This latter method also has the capacity to reveal detailed protein phosphorylation profiles by resolving distinct phospho-isoforms arising from both the specific residues and number of residues phosphorylated (33). For each method we used total STMN antibodies to detect the separated STMN population. With the iso-electric focusing/two-dimensional gel separation approach, the phosphorylated STMN species differed markedly for control cells, cells subjected to OS, and cells stimulated with NGF (Fig. 3). According to published separation profiles for STMN phospho-isoforms (27), NGF stimulated an increase in dual-phosphorylated STMN (phospho-Ser-16 and phospho-Ser-25) phospho-isoform, but this STMN phospho-isoform was not present in detectable levels after OS (Fig. 3). NGF also induced a prominent triphosphorylated STMN (phospho-Ser-16 and phospho-Ser-25 and phospho-Ser-38), whereas the levels of this phospho-isoform were lower for OS, but different tri-phosphorylated STMN species (phospho-Ser-16/Ser-25 and phospho-Ser-38 and phospho-Ser-63) were more prominent (Fig. 3).
FIGURE 3.
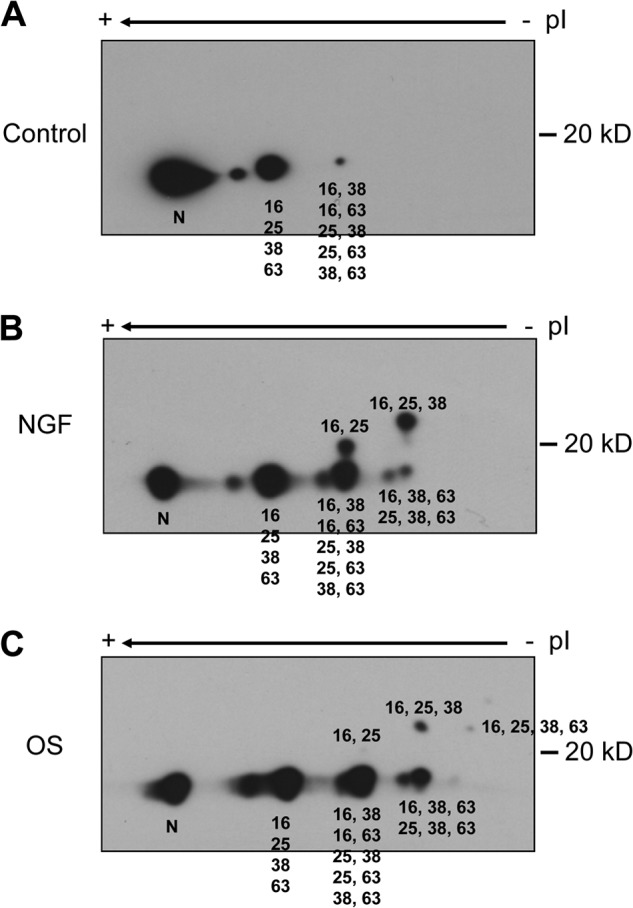
Two-dimensional PAGE analysis of STMN phospho-isoforms. PC12 cells were treated with vehicle (0.1% (w/v) BSA in PBS; A), NGF (50 ng/ml; B), or OS (sorbitol, 0.5 m; C) for 60 min. Protein lysates were then prepared and resolved on two-dimensional PAGE. STMN phospho-isoforms were assigned according to size and the isoelectric point as previously described (27). N, nonphosphorylated STMN.
Using the mobility-shift technique on Phos-tag gels, we evaluated the response to OS or NGF treatment for 30 and 60 min. Immunoblotting for STMN from OS-treated cells revealed multiple slower migrating bands that were distinct STMN phospho-isoforms sensitive to λ-phosphatase treatment (Fig. 4). NGF treatment also reduced STMN mobility on Phos-tag gels, but the different band profile indicated distinct STMN phospho-isoforms under these conditions (Fig. 4). The substantial decrease in unphosphorylated STMN on Phos-tag gels, when compared with control or NGF-treated samples, indicated a high stoichiometry of STMN phosphorylation during OS (Fig. 4).
FIGURE 4.
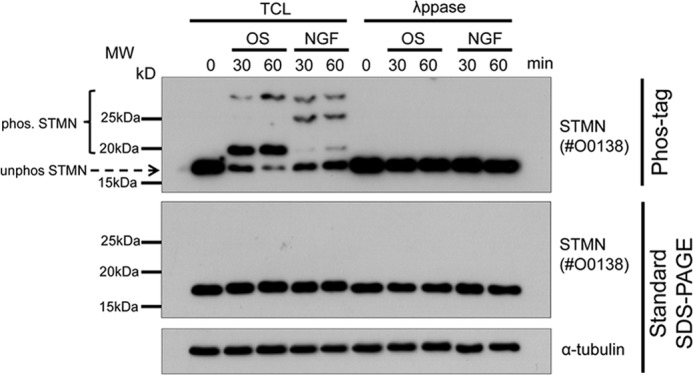
Mobility shift of phosphorylated STMN on Phos-tag SDS-PAGE. PC12 cells were stimulated with OS (sorbitol, 0.5 m) or NGF (50 ng/ml) for 30 or 60 min. Unstimulated cells (0 min) were included as a control. Protein lysates were treated with λ-phosphatase (λ-ppase) or left untreated (TCL) before resolving using Phos-tag or standard SDS-PAGE and immunoblotted for STMN or α-tubulin for protein loading. The mobility shift of phosphorylated STMN bands (bracket) and the position of unphosphorylated STMN (arrow) are also indicated.
An additional advantage of the Phos-tag system lies in the ability to profile multiple samples in parallel on the same gel for more detailed evaluations and cross-comparisons. To investigate the kinetics of STMN phospho-isoform formation, we analyzed samples taken over a time course of OS exposure (5–120 min). We noted a non-phosphorylated STMN band, which we designated as NP and confirmed by λ-phosphatase treatment, and three clearly resolved STMN phospho-isoform bands that we designated as SP1, SP2, and SP3 (Fig. 5A). In addition, two slower migrating phospho-STMN bands appeared under longer exposure and were designated SP4 and SP5, respectively (Fig. 5B). In response to OS, the SP1 band was observed at the earliest time point tested (5 min), increased in intensity with time, and was the most prominent STMN phospho-isoform by 120 min OS (Fig. 5A). Interestingly, we observed the formation of the SP2 band after 15 min OS, but the decrease in its intensity at 30 min of OS likely reflects subsequent phosphorylation on additional sites to form multiphosphorylated STMN species (Fig. 5A). Consistent with this interpretation, the appearance of SP3 coincided with the decrease of SP2 (Fig. 5A). OS-stimulated increases in SP4 and SP5 were also observed between 30 and 120 min (Fig. 5B). In response to OS, SP1 and SP3 represented the main phospho-isoforms present in terms of abundance (Fig. 5A). Thus, our mobility shift analyses provided insights into the progression of STMN multisite phosphorylation over the OS time course.
FIGURE 5.
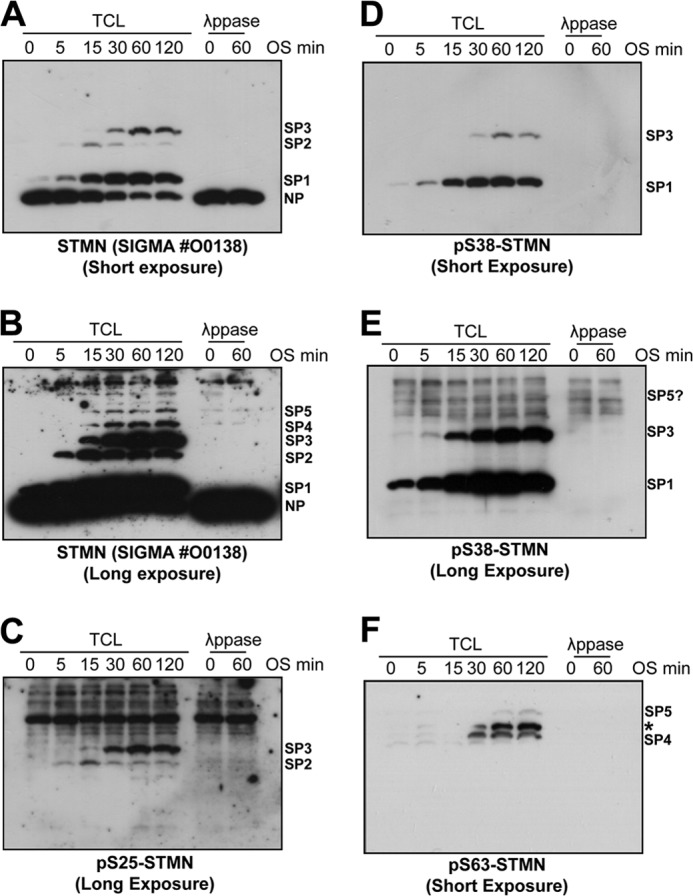
Phos-tag mobility shift analysis of STMN phospho-isoforms induced by hyperosmotic stress. A, PC12 cells were stimulated with sorbitol (0.5 m) for 0–120 min. Total cell protein lysates (TCL) were prepared and resolved on Phos-tag gels before immunoblotting for STMN (#O0138, Sigma). λ-Phosphatase (λppase) treatment of lysates confirmed the phosphorylation status of bands with reduced mobility in response to OS treatment. B, the chemiluminescence signal from STMN blot in A was imaged for extended duration (long exposure) to a reveal lower abundance phospho-STMN bands. Stress-induced, λppase-sensitive bands represent distinct STMN phospho-isoforms, which are annotated as (SP1–SP5). Non-phosphorylated STMN was annotated as NP. C, protein lysates in A were blotted for STMN Ser-25 phosphorylation. D, lysates were blotted for STMN Ser-38 phosphorylation. E, a long exposure of the blot depicted in D. F, PC12 lysates were blotted for STMN Ser-63 phosphorylation.
We then extended these analyses by additionally immunoblotting with site-specific phospho-STMN antibodies. Our blots with Ser(P)-16 STMN antibody failed to detect bands in OS-treated cell lysates on Phos-tag (data not shown), consistent with marginal phosphorylation of Ser-16 in response to stress. The SP1 phospho-isoform was specifically detected by the Ser(P)-38-STMN antibody and not by phospho-STMN antibodies directed at the other serine residues (Fig. 5, C–F). This indicated that the OS-stimulated SP1 band was singly phosphorylated STMN, specifically phospho-Ser-38 STMN. Similarly, the SP2 band appeared to be singly phosphorylated STMN but in this case was phospho-Ser-25 STMN (Fig. 5C). These interpretations were consistent with specific STMN serine phosphorylation contributing differently to gel mobility shifts, with STMN Ser-25 phosphorylation causing a greater reduction in STMN mobility (27). The SP3 phospho-isoform represented dual-phosphorylated STMN (2P) at Ser-25 and Ser-38, specifically (Fig. 5, C–E), supporting the notion that the reduction in SP2 at 30 min of OS reflected sequential phosphorylation of singly phosphorylated STMN proteins. An extended exposure of the Ser(P)-38-STMN blots revealed a number of nonspecific higher molecular weight bands that complicated detection of slower migrating STMN phospho-isomers, but we did identify a λ-phosphatase-sensitive, slower migrating, low abundance band in the region corresponding to migration rate of the SP5 phospho-isoform (Fig. 5E). Blotting for STMN Ser-63 phosphorylation revealed the slowest migrating SP4 and SP5 bands were phosphorylated at Ser-63, supporting the interpretation that this band represents the multisite-phosphorylated STMN (Fig. 5F). A third nonspecific band (annotated with * in Fig. 5F) was also detected on the Ser(P)-63-STMN blot, but this band was also detected as a nonspecific 37-kDa protein when resolved on standard SDS-PAGE gels (data not shown). Thus, confirmation of SP4 and SP5 bands on the Ser(P)63-STMN blot as STMN phospho-isoforms was additionally determined with STMN siRNA knockdown (data not shown). Our analysis thus revealed the profile and progression of STMN multisite serine phosphorylation during OS, with rapid Ser-38 phosphorylation followed by subsequent Ser-25 and Ser-63 phosphorylation.
Evaluation of STMN Ser-38 and Ser-63 Phosphorylation Reveals Contributions by JNK and PKA and Their Cross-talk
With our previous observation of OS-enhanced STMN phospho-Ser-38 mediated by the stress-activated kinase JNK (14), we next evaluated the phosphorylation of STMN Ser-38 with more directed evaluation of the contributions of this phospho-isoform to the total STMN population during OS. Because the polyclonal STMN antibody available from CST (#3352) was directed against a sequence encompassing the phospho-Ser-38 STMN site, we evaluated the performance of this antibody in detection of the phospho-Ser-38- and non-phospho-STMN forms. In an in vitro kinase assay, phosphorylation of the recombinant purified GST-STMN fusion protein by active JNK1 reduced CST antibody #3352 detection of recombinant STMN by Western blot compared with the unphosphorylated protein (Fig. 6A). In contrast, immunoblotting with STMN antibody #O0138 (Sigma) revealed equivalent loading, and immunoblotting with phospho-STMN Ser-38 confirmed robust phosphorylation at this site (Fig. 6A). Similarly, when we subjected PC12 cells to OS, we observed the reduction in CST #3352 antibody detection of total STMN, increased STMN Ser-38 phosphorylation, but unchanged detection by the #O0138 Sigma antibody (Fig. 6B). Furthermore, λ-phosphatase treatment of protein lysates from OS-stimulated PC12 cells before immunoblot analysis returned STMN detection by the CST #3352 antibody, confirming that phosphorylation modification impacted on detection by this antibody (Fig. 6B). In a time course analysis after OS stimulation (0–60 min), we also observed that the loss of STMN detection by the CST #3352 antibody was inversely related to STMN Ser-38 phosphorylation (Fig. 6, C and D), indicating the phosphorylation of STMN Ser-38 specifically as underlying the reduced detection of STMN by the CST #3352 antibody. Thus, our results indicate that the CST #3352 antibody only recognizes STMN that is unphosphorylated on Ser-38. As the detection of STMN with the CST #3352 antibody was rapidly reduced to near undetectable levels by 60 min of osmotic stress (Fig. 6, C and D), a substantial proportion of endogenous STMN was phosphorylated at Ser-38 and highlights substantial stress signaling to the total cellular STMN pool.
FIGURE 6.
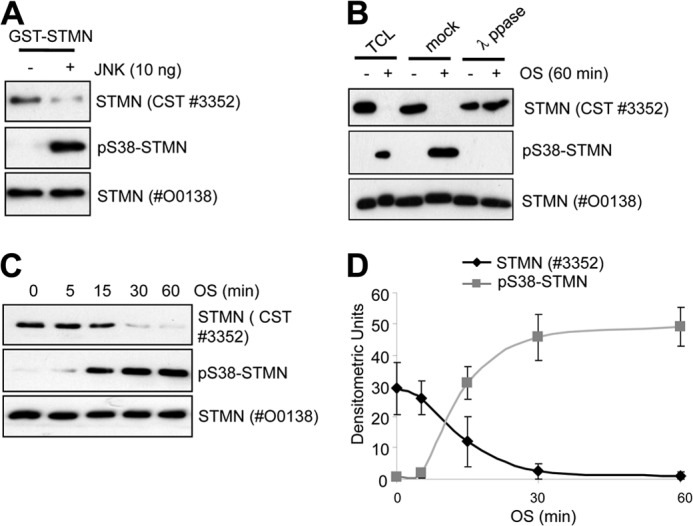
Antibody detection of unphosphorylated STMN revealed a high stoichiometry of STMN Ser-38 phosphorylation in response to hyperosmolarity. A, GST-STMN was phosphorylated by active JNK1 in an in vitro kinase assay. As a control, GST-STMN was incubated without kinase. Samples were then immunoblotted with the indicated antibodies. B, PC12 cells were stimulated with OS (sorbitol, 0.5 m, 60 min) or left untreated. Protein lysates were then treated with λ-phosphatase, mock-treated in the absence of phosphatase, or left untreated before immunoblot analysis. C, PC12 cells were stimulated with OS (sorbitol, 0.5 m) for the indicated times (0–60 min) before immunoblotting as indicated. D, phospho-Ser-38 STMN and STMN detected with the antibody from CST (#3352) were quantitated by densitometric analysis and plotted as a time course of sorbitol treatment. Values are mean ± S.E. from three independent experiments.
We next considered the regulation of STMN Ser-63 phosphorylation under OS conditions. STMN Ser-63 is not a proline-flanked residue and, therefore, not expected to be targeted by proline-directed serine/threonine kinases such as those of the MAPK family. However, a prior report of priming phosphorylation on the proline-flanked Ser-25 and Ser-38 residues for subsequent STMN targeting (28) prompted us to investigate the relationship between STMN Ser-38 and Ser-63 phosphorylation in response to OS. STMN Ser-38 is targeted predominantly by JNK in response to OS (14); therefore, we determined the effect of JNK inhibition on OS-stimulated STMN Ser-63 phosphorylation. For JNK inhibition, we utilized JNK inhibitor VIII, a potent small molecule inhibitor of JNK1/2 isoforms with superior kinase selectivity compared with other JNK inhibitory compounds (e.g. SP600125, JNK inhibitor V) as determined previously (34). Pretreatment of PC12 cells with JNK Inhibitor VIII (50 μm) robustly inhibited OS-stimulated c-Jun phosphorylation and attenuated OS-stimulated STMN Ser-38 phosphorylation (Fig. 7, A and B). Strikingly, stress-stimulated STMN Ser-63 phosphorylation was phosphorylated more rapidly and to a much higher degree with JNK inhibition (Fig. 7, A and B). This highlighted that STMN Ser-63 targeting did not require STMN Ser-38 phosphorylation. Rather, our results indicated a distinct signaling mechanism targeting STMN Ser-63 that was up-regulated after JNK inhibition.
FIGURE 7.
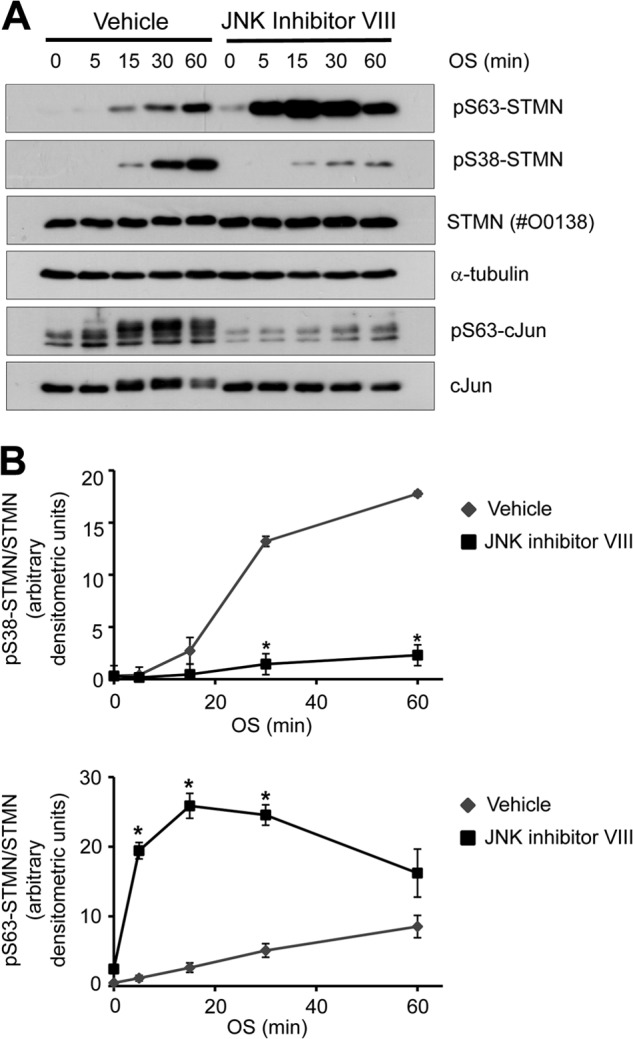
OS-stimulated STMN Ser-63 phosphorylation is augmented in the absence of JNK activity. A, PC12 cells were pretreated with JNK inhibitor VIII (50 μm, 30 min) or a vehicle control (DMSO, 0.1% v/v) before OS treatment (sorbitol 0.5 m, 5–120 min). Protein lysates were then blotted for Ser(P)-38-STMN (pS38-STMN), Ser(P)63-STMN ((pS36-STMN)), pan-STMN expression, and α-tubulin. B, phosphorylated STMN bands were quantitated, normalized for protein loading, and expressed as -fold activation over DMSO-treated unstimulated controls. Values are the mean ± S.E. from three independent experiments. *, p < 0.05 compared with DMSO vehicle.
To identify the upstream signaling events responsible for stress-induced targeting of STMN Ser-63, we initially tested a serine/threonine kinase inhibitor with broad-spectrum activity, staurosporine. Pretreatment of PC12 cells with staurosporine before OS resulted in the marked reduction of STMN Ser-63 phosphorylation (Fig. 8A). The inhibition of basal and stress-stimulated STMN Ser-63 phosphorylation was evident at 20 nm staurosporine, and significantly reduced phosphorylation was observed at 50 or 100 nm staurosporine (Fig. 8B).
FIGURE 8.
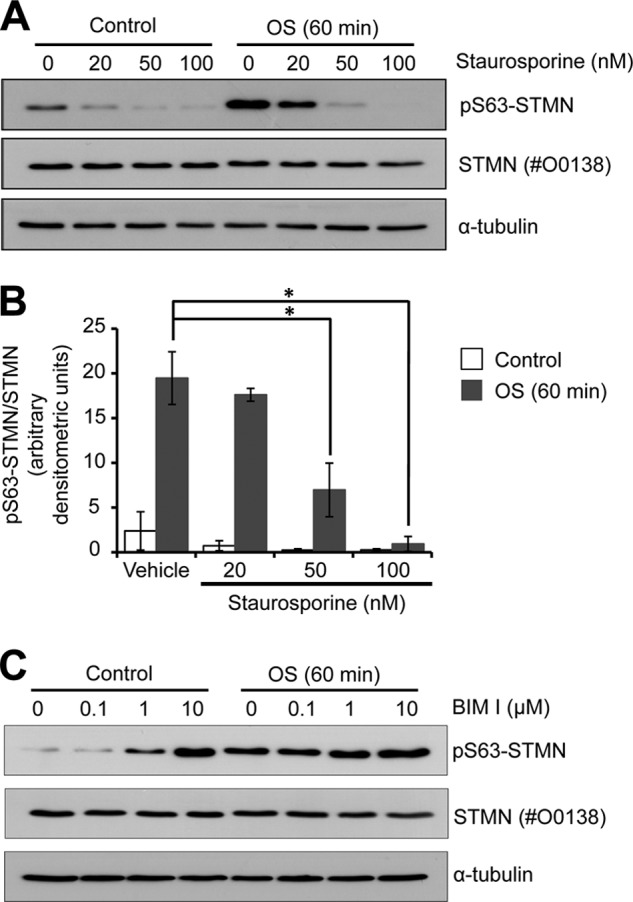
OS-stimulated STMN Ser-63 phosphorylation is attenuated by broad spectrum serine/threonine kinase inhibition. A, PC12 cells were pretreated for 30 min with staurosporine (0–100 nm) before OS (sorbitol, 0.5 m, 60 min) or left unstimulated (Control). Protein lysates were prepared and immunoblotted for STMN Ser-63 phosphorylation, STMN expression, and α-tubulin levels as a loading control. B, phospho-Ser-63 STMN bands were quantitated by densitometric analysis, normalized for STMN expression, and plotted against increasing concentrations of staurosporine. Values are the mean ± S.E. from three independent experiments. *, p < 0.05. C, PC12 cells were pretreated (30 min) with bisindolylmaleimide I (BIM I, 0–10 μm) before OS (sorbitol, 0.5 m, 60 min) or left untreated (control) and subjected to immunoblot analysis.
As protein kinases targeted by staurosporine include PKC and PKA, we next tested the PKC inhibitor bisindolylmaleimide I, but this did not reduce OS-stimulated STMN phosphorylation (Fig. 8C), suggesting that PKC was not involved in stress-stimulated STMN Ser-63 targeting. In contrast, pretreatment with the PKA inhibitor, H-89 (10 μm), significantly attenuated STMN Ser-63 phosphorylation in response to OS (Fig. 9A). OS-stimulated STMN Ser-63 phosphorylation was significantly reduced in the presence of H-89 to 28% that of vehicle control levels (DMSO) (Fig. 9, B and C). In addition, the modest increase in NGF-stimulated STMN Ser-63 phosphorylation was significantly inhibited in the presence of H-89 (45% of levels in DMSO treated cells; Fig. 9, B and C). Our study highlights PKA signaling to STMN particularly at the Ser-63 target site in both growth factor and stress-stimulated conditions. We had additionally tested the effect of myristoylated PKI (5–24), a cell permeable peptide inhibitor of PKA, but this compound failed to inhibit the phosphorylation of established PKA target proteins such as cAMP-response element-binding protein (CREB) in our cellular studies (data not shown). Therefore, we were unable to utilize the PKI inhibitor to test PKA activity toward STMN. In contrast, we demonstrated that H-89 pretreatment attenuated OS-stimulated CREB phosphorylation (Ser-133) (Fig. 9D), a direct downstream target of PKA (35). This indicates that PKA is activated in response to OS and validates the specificity of the H-89 as a potent PKA inhibitor. In support of this, our direct measurements of PKA activity in cell lysates revealed significant and sustained increases in activity after sorbitol treatment (15–60 min) (Fig. 9E). Furthermore, increasing cAMP levels with cholera toxin (1 ng/ml) treatment resulted in substantially elevated CREB Ser-133 and STMN Ser-63 phosphorylation that was sensitive to H-89 treatment (Fig. 9D). Interestingly, sorbitol treatment did not induce a detectable change in cAMP levels (Fig. 9F), consistent with a previous report (36). This suggests that OS stimulated PKA activation independently of raised global cAMP concentrations. Thus, our results indicate that STMN is targeted predominantly by JNK and PKA under conditions of cellular stress.
FIGURE 9.

STMN Ser-63 is phosphorylated by PKA in response to hyperosmolarity. A, PC12 cells were pretreated with the H-89 inhibitor (0–10 μm, 30 min) or DMSO (0.1% v/v) before OS (sorbitol, 0.5 m, 60 min). Protein lysates were immunoblotted for Ser(P)63-STMN and STMN expression. α-Tubulin levels were also determined to assess protein loading. B, PC12 cells were pretreated with H-89 (10 μm, 30 min) or DMSO (0.1% v/v) followed by sorbitol (OS, 0.5 m) or NGF treatment (50 ng/ml) for 60 min and blotted as in A. Con, control. C, densitometric analysis of phospho-Ser-63 STMN bands in B was performed and plotted in a bar graph. Values are the mean ± S.E. from three independent experiments. *, p < 0.05 compared with DMSO vehicle. D, PC12 cells were pretreated with H-89 (10 μm, 30 min) or DMSO (0.1% v/v) followed by OS (sorbitol, 0.5 m) or cholera toxin (CTX, 1 ng/ml) treatment for 60 min. Control cells were left unstimulated. Protein lysates were prepared and blotted for Ser(P)63-STMN, phosphorylated CREB (p-CREB) and total protein expression. E, PC12 cells were treated with sorbitol (OS, 0.5 m, 5–60 min) or forskolin (FSK, 20 μm, 20 min). PKA activity was then measured in cell lysates using a radiolabeled in vitro kinase activity assay, and values are expressed as a percentage of forskolin-stimulated activity (100%). Values are the mean ± S.E. from three independent experiments. *, p < 0.05 compared with DMSO vehicle. F, sorbitol-stimulated PC12 cells (OS, 0.5 m; 5 or 15 min) were lysed, and cAMP levels were determined using a competition HRP-linked immunoassay. Basal cAMP levels were determined in untreated cells (0 min), and forskolin treatment (20 μm, 20 min) was included as a positive control. Absorbance values (mean = 4) were expressed relative to forskolin (100%). G, bacterially expressed recombinant GST-STMN was phosphorylated in vitro in the presence of active JNK or PKA before immunoblot analysis of STMN serine phosphorylation with site-specific phospho-STMN antibodies. H, tubulin (300 μg) was polymerized at 37 °C in the absence (Buffer) or presence of recombinant STMN (35 μg) or STMN phosphorylated with JNK, PKA, or JNK and PKA in combination. Tubulin polymerization status was monitored by measuring absorbance (340 nm) at 1-min intervals for 90 min. This study was performed twice with identical findings.
To determine if JNK- and PKA-mediated phosphorylation was sufficient to inactivate STMN, we performed an in vitro tubulin polymerization assay with recombinant STMN. First, we verified in vitro STMN serine residues targeted by JNK and PKA and observed STMN Ser-25 and Ser-38 phosphorylation in the presence of JNK, whereas STMN Ser-16 and Ser-63 were specifically targeted by PKA as expected (Fig. 9G). In the presence of both kinases, STMN is hyperphosphorylated on all four serine residues (Fig. 9G). In turbidity assays, tubulin polymerization in the presence of JNK-phosphorylated STMN was not substantially different from unphosphorylated STMN, whereas PKA-phosphorylation alone partially reversed STMN inhibition of tubulin assembly (Fig. 9H). In contrast, in the presence STMN phosphorylated by both JNK and PKA, the initiation of tubulin assembly was increased and tubulin polymerized to the same extent as a buffer-only control (Fig. 9H). Thus, although STMN phosphorylation by JNK was not sufficient to substantially alter STMN activity, JNK targeting in addition to PKA was required to fully attenuate the inhibitory effects of STMN on tubulin assembly in vitro.
In response to cell stress, we noted that the increase in STMN Ser-63 phosphorylation was more rapidly stimulated with JNK inhibition (Fig. 7). Similarly, basal and short term (5 and 15 min) OS-stimulated CREB phosphorylation were augmented in the presence of a JNK inhibitor (Fig. 10, A and B). This correlated with more rapid STMN Ser-63 phosphorylation (Fig. 7), indicating that PKA regulation of STMN is altered in the absence of JNK activity. In support of this, osmotic stress-stimulated PKA activity is augmented by JNK inhibition (Fig. 10C). We observed an approximately 2-fold increase with JNK inhibitor VIII co-treatment with sorbitol compared with stress stimulation with a vehicle control (Fig. 10C). The increased PKA activation with JNK inhibition was sufficient to markedly elevated STMN Ser-63 phosphorylation observed in Fig. 7, as H-89 pretreatment attenuated augmented STMN Ser-63 phosphorylation from JNK inhibition to levels comparable with basal (Fig. 10D). Taken together, our studies have revealed phosphorylation of STMN on Ser-63 as a significant downstream target of activated endogenous PKA in response to OS. Interestingly, PKA signaling to STMN was augmented with JNK inhibition indicating cross-talk between these signaling pathways in the regulation of STMN during hyperosmotic stress.
FIGURE 10.
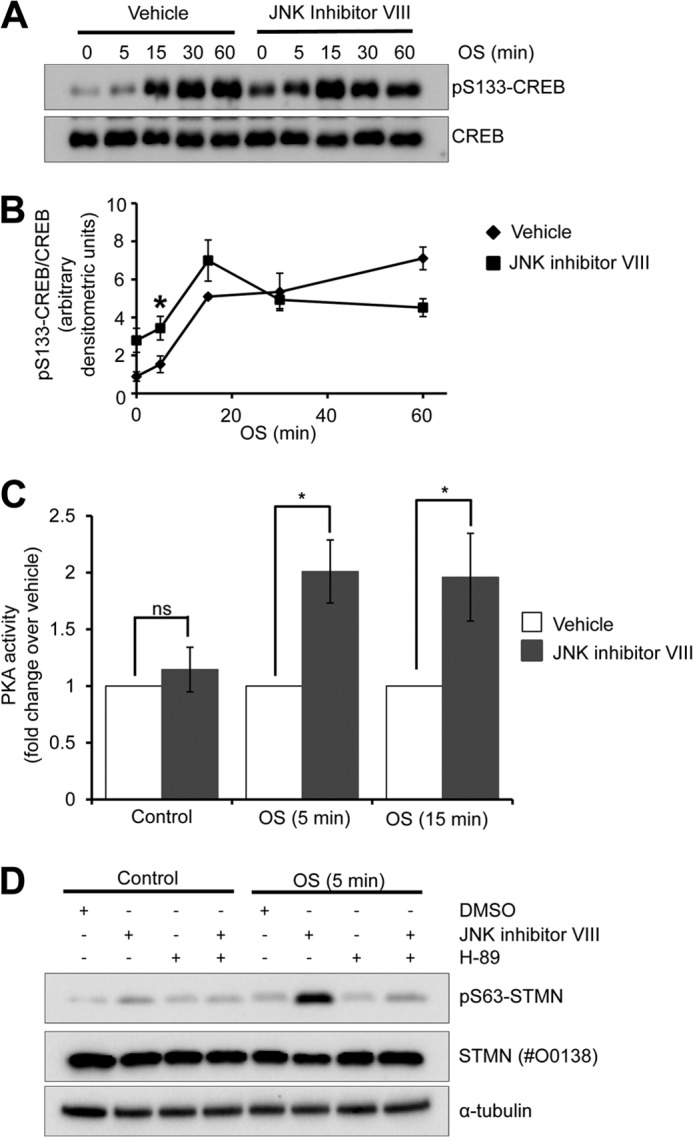
Enhanced STMN Ser-63 phosphorylation during JNK inhibition is mediated by PKA. A, PC12 cells were pretreated with JNK Inhibitor VIII (50 μm, 30 min) or a vehicle control (DMSO, 0.1% v/v) before OS treatment (sorbitol 0.5 m, 5–60 min). Protein lysates were then blotted for CREB and phosphorylated (Ser-133)-CREB. B, phosphorylated CREB bands were quantified by densitometric analysis, normalized for total protein expression, and plotted against duration of OS treatment. Values are the mean ± S.E. from three independent experiments (*, p < 0.05 compared with DMSO vehicle). C, PKA activity was measured in cell lysates prepared from PC12 cells pretreated with DMSO (vehicle, 0.1% v/v) or JNK Inhibitor VIII (50 μm, 30 min) followed by 0, 5, or 15 min sorbitol stimulation (OS, 0.5 m). Values are the mean ± S.E. from three independent experiments (*, p < 0.05 compared with DMSO vehicle). D, PC12 cells were pretreated DMSO (0.1% v/v), JNK inhibitor VIII (50 μm, 30 min), H-89 (10 μm, 30 min), or JNK inhibitor VIII and H-89 in combination before OS treatment (sorbitol 0.5 m, 5 min). Protein lysates were then immunoblotted for Ser(P)63-STMN, total STMN, or α-tubulin expression.
DISCUSSION
Microtubule reorganization under the control of complex regulatory networks is fundamental for cellular responses to stress stimuli including altered osmolarity, temperature, and pH (37, 38). In this study the contribution of distinct STMN serine residue phosphorylation toward maintaining microtubule integrity was revealed through alanine substitution experiments. We also characterized stress-induced STMN multisite phosphorylation and profiled the generation of distinct STMN phospho-isoforms using mobility shift detection in combination with immunoblot analysis with site-specific phosphoserine STMN antibodies. In addition, we have also identified PKA activation as a stress-induced signaling pathway targeting STMN and novel cross-talk with JNK in the control of microtubule architecture under conditions of cellular stress.
Consistent with our previous studies (14), we found that OS, chemical, and heat stress but not oxidative stress stimuli induced rapid STMN phosphorylation. Interestingly, STMN Ser-38 was phosphorylated at high stoichiometry with >90% of the total cellular pool post-translationally modified after OS treatment. This is likely mediated predominantly by stress-activated JNK signaling as JNK inhibition substantially reduces OS-induced phosphorylation at that site (14). The STMN Ser-38 residue is located in a disordered linker region that does not interact with tubulin (39), and prior in vitro studies have suggested that Ser-38 phosphorylation had marginal effects in on STMN-tubulin binding and microtubule-destabilizing activity (11, 28), which were confirmed by our current investigations (Fig. 9H). However, that cellular STMN is rapidly and extensively phosphorylated at Ser-38 suggests that the targeting of this specific serine residue has significant functions in cellular responses to osmotic stress.
We found distinct differences in stress-stimulated STMN phospho-isoforms when compared with nerve growth factor treatment with a predominance of single site-phosphorylated (1P) STMN at Ser-38 highlighting the significance of this residue in the context of cell stress. In support of this, the ectopic expression of STMN mutants resistant to Ser-38 phosphorylation specifically led to microtubule loss only under stress conditions, highlighting a key contribution of STMN Ser-38 phosphorylation in maintaining the microtubule array during cell stress. This is also consistent with the notion that JNK-mediated inactivation of STMN constitutes part of cellular responses to stress.
We also demonstrated prominent phosphorylation of STMN Ser-63 in response to OS. Previous studies have indicated that in vitro phosphorylation of STMN on Ser-63 is sufficient to attenuate microtubule destabilizing activity with greater reduction observed with Ser-16 and Ser-63 dual phosphorylation (28, 31). In our hands in vitro phosphorylation of STMN Ser-16 and Ser-63 by PKA partially attenuated STMN activity, and a combination of JNK and PKA-mediated hyperphosphorylation of STMN was required to completely restore tubulin polymerization in vitro consistent with prior reports (11). In ectopic expression cell studies, pseudophosphorylation of Ser-63 phosphorylation alone is not sufficient to inactivate STMN activity leading to a block in mitotic progression (28, 40). In cells we found that the expression of STMN S63A resulted in microtubule depolymerization during osmotic stress, highlighting the requirement for Ser-63 phosphorylation to inactivate STMN during cell stress. In addition, ectopic expression of STMN S38A was sufficient to destabilize microtubules after osmotic stress. This is in contrast to the marginal contribution of STMN Ser-38 phosphorylation toward control of microtubule plasticity in vitro (11, 41), highlighting a complex cellular role for STMN phospho-isoforms. We conclude that both STMN Ser-38 and Ser-63 phosphorylation are required to effectively inactivate STMN and preserve interphase microtubules during cell stress. Similarly, dual phosphorylation at Ser-38 and Ser-63 is sufficient to inactivate STMN for the proper formation of the mitotic spindle (40). Thus, our finding supports STMN Ser-38 and Ser-63 dual phosphorylation as major contributors toward the inactivation of cellular STMN to facilitate microtubule formation in mitosis and interphase.
In exploring the signaling events involved, we identified a role for the PKA signaling pathway. We found that elevated cAMP levels, induced by cholera toxin, was sufficient to trigger STMN Ser-63 phosphorylation and that stress induced increased PKA activity and STMN Ser-63 phosphorylation that was ablated with the H-89 compound, a reasonably specific inhibitor of PKA (42). STMN is a well established PKA substrate, with previous studies demonstrating PKA phosphorylation of STMN Ser-16 and Ser-63 in in vitro kinase and ectopic expression studies (17, 27, 43). In contrast, the function of endogenous PKA-regulated STMN activity is less well defined. STMN is phosphorylated in primary mouse neurons in response to increase intracellular cAMP levels induced by forskolin treatment and subsequent activation of adenyl cyclase (44). In addition, PKA targeting of membrane-associated SCG10 and phosphorylation of Ser-97 on the conserved STMN-like domain have been shown to regulate microtubule dynamics associated with growth cone guidance (45). These studies highlight important neurological functions of endogenous PKA/STMN signaling. Our studies also indicate that STMN is a target of endogenous PKA during OS.
PKA is activated by broad cellular processes to target multiple substrates with specificity conferred through the assembly of various combinations of regulatory and catalytic subunits and the subcompartmentalization of the signaling module by kinase-anchoring proteins (46, 47). PKA signaling is also regulated by cellular tonicity (36). Hypertonic stress induced cAMP-independent PKA activation for the phosphorylation and regulation of transcription factors to alter gene expression (36). Consistent with this previous study, we demonstrated that OS stimulated PKA activity but did not increase intracellular cAMP levels. This was involved in the direct regulation of microtubule organization through STMN phosphorylation as PKA inhibition attenuated stress-induced phosphorylation of STMN Ser-63, which we showed was required to preserved interphase microtubules. Thus, our study demonstrated PKA targeting of STMN in the context of cell stress for the regulation of the microtubule cytoskeleton.
In investigating the relationship between JNK and PKA targeting of cellular STMN, unlike in the context of mitotically induced STMN hyperphosphorylation (28), we ruled out a possible role for JNK-mediated STMN Ser-38 phosphorylation as a priming phosphorylation event for subsequent Ser-63 phosphorylation. In contrast, JNK inhibition augmented PKA activity, which resulted in more rapid and augmented PKA-mediated STMN Ser-63 phosphorylation. This indicated that JNK activity moderated PKA signaling to STMN during OS. Cross-talk between cAMP/PKA and ERK MAPKs has been described with pathway intersection likely at the level of Ras-Raf proteins and facilitated by protein kinase A-anchoring proteins (48, 49). JNK can be regulated by PKA signaling, although the outcome (JNK activation or inhibition) is dependent on cellular context (50–54). The reciprocal effect of JNK activity on PKA signaling is less well explored, whereas our study indicates that stress-activated JNK negatively regulates PKA activity and signaling to downstream substrates. The precise mechanism involved in JNK/PKA cross-talk is currently unknown.
Taken together, our study implicates JNK- and PKA-mediated phosphorylation of STMN as significant signaling events to constrain STMN activity to maintain microtubule array organization and function under conditions of hyperosmolarity. Future studies will determine upstream events involved in JNK/PKA cross-talk and if these mechanisms co-operate to regulate microtubules in a broader cellular context.
This work was funded by National Health and Medical Research Council of Australia Grants 628335 (to D. C. H. N.) and 566804 (to M. A. B.).
- STMN
- stathmin
- OS
- osmotic stress
- PKA
- protein kinase A/cAMP-dependent protein kinase
- SP
- STMN phospho-isomer
- CST
- Cell Signaling Technology
- CREB
- cAMP-response element-binding protein.
REFERENCES
- 1. Cassimeris L. (2002) The oncoprotein 18/stathmin family of microtubule destabilizers. Curr. Opin. Cell Biol. 14, 18–24 [DOI] [PubMed] [Google Scholar]
- 2. Belmont L. D., Mitchison T. J. (1996) Identification of a protein that interacts with tubulin dimers and increases the catastrophe rate of microtubules. Cell 84, 623–631 [DOI] [PubMed] [Google Scholar]
- 3. Jourdain L., Curmi P., Sobel A., Pantaloni D., Carlier M. F. (1997) Stathmin. A tubulin-sequestering protein which forms a ternary T2S complex with two tubulin molecules. Biochemistry 36, 10817–10821 [DOI] [PubMed] [Google Scholar]
- 4. Liedtke W., Leman E. E., Fyffe R. E., Raine C. S., Schubart U. K. (2002) Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am. J. Pathol. 160, 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shumyatsky G. P., Malleret G., Shin R. M., Takizawa S., Tully K., Tsvetkov E., Zakharenko S. S., Joseph J., Vronskaya S., Yin D., Schubart U. K., Kandel E. R., Bolshakov V. Y. (2005) stathmin, a gene enriched in the amygdala, controls both learned and innate fear. Cell 123, 697–709 [DOI] [PubMed] [Google Scholar]
- 6. Wen H. L., Lin Y. T., Ting C. H., Lin-Chao S., Li H., Hsieh-Li H. M. (2010) Stathmin, a microtubule-destabilizing protein, is dysregulated in spinal muscular atrophy. Hum. Mol. Genet 19, 1766–1778 [DOI] [PubMed] [Google Scholar]
- 7. Belletti B., Baldassarre G. (2011) Stathmin. A protein with many tasks. New biomarker and potential target in cancer. Expert Opin. Ther. Targets 15, 1249–1266 [DOI] [PubMed] [Google Scholar]
- 8. Byrne F. L., Yang L., Phillips P. A., Hansford L. M., Fletcher J. I., Ormandy C. J., McCarroll J. A., Kavallaris M. (2013) RNAi-mediated stathmin suppression reduces lung metastasis in an orthotopic neuroblastoma mouse model. Oncogene 10.1038/onc.2013.11 [DOI] [PubMed] [Google Scholar]
- 9. Chen P. W., Lin S. J., Tsai S. C., Lin J. H., Chen M. R., Wang J. T., Lee C. P., Tsai C. H. (2010) Regulation of microtubule dynamics through phosphorylation on stathmin by Epstein-Barr virus kinase BGLF4. J. Biol. Chem. 285, 10053–10063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sobel A., Boutterin M. C., Beretta L., Chneiweiss H., Doye V., Peyro-Saint-Paul H. (1989) Intracellular substrates for extracellular signaling. Characterization of a ubiquitous, neuron-enriched phosphoprotein (stathmin). J. Biol. Chem. 264, 3765–3772 [PubMed] [Google Scholar]
- 11. Honnappa S., Jahnke W., Seelig J., Steinmetz M. O. (2006) Control of intrinsically disordered stathmin by multisite phosphorylation. J. Biol. Chem. 281, 16078–16083 [DOI] [PubMed] [Google Scholar]
- 12. Beretta L., Dubois M. F., Sobel A., Bensaude O. (1995) Stathmin is a major substrate for mitogen-activated protein kinase during heat shock and chemical stress in HeLa cells. Eur. J. Biochem. 227, 388–395 [DOI] [PubMed] [Google Scholar]
- 13. Hu J. Y., Chu Z. G., Han J., Dang Y. M., Yan H., Zhang Q., Liang G. P., Huang Y. S. (2010) The p38/MAPK pathway regulates microtubule polymerization through phosphorylation of MAP4 and Op18 in hypoxic cells. Cell. Mol. Life Sci. 67, 321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ng D. C., Zhao T. T., Yeap Y. Y., Ngoei K. R., Bogoyevitch M. A. (2010) c-Jun N-terminal kinase phosphorylation of stathmin confers protection against cellular stress. J. Biol. Chem. 285, 29001–29013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santamaría E., Mora M. I., Muñoz J., Sánchez-Quiles V., Fernández-Irigoyen J., Prieto J., Corrales F. J. (2009) Regulation of stathmin phosphorylation in mouse liver progenitor-29 cells during proteasome inhibition. Proteomics 9, 4495–4506 [DOI] [PubMed] [Google Scholar]
- 16. Vancompernolle K., Boonefaes T., Mann M., Fiers W., Grooten J. (2000) Tumor necrosis factor-induced microtubule stabilization mediated by hyperphosphorylated oncoprotein 18 promotes cell death. J. Biol. Chem. 275, 33876–33882 [DOI] [PubMed] [Google Scholar]
- 17. Gradin H. M., Larsson N., Marklund U., Gullberg M. (1998) Regulation of microtubule dynamics by extracellular signals. cAMP-dependent protein kinase switches off the activity of oncoprotein 18 in intact cells. J. Cell Biol. 140, 131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. le Gouvello S., Manceau V., Sobel A. (1998) Serine 16 of stathmin as a cytosolic target for Ca2+/calmodulin-dependent kinase II after CD2 triggering of human T lymphocytes. J. Immunol. 161, 1113–1122 [PubMed] [Google Scholar]
- 19. Melander Gradin H., Marklund U., Larsson N., Chatila T. A., Gullberg M. (1997) Regulation of microtubule dynamics by Ca2+/calmodulin-dependent kinase IV/Gr-dependent phosphorylation of oncoprotein 18. Mol. Cell. Biol. 17, 3459–3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wittmann T., Bokoch G. M., Waterman-Storer C. M. (2004) Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J. Biol. Chem. 279, 6196–6203 [DOI] [PubMed] [Google Scholar]
- 21. Brattsand G., Marklund U., Nylander K., Roos G., Gullberg M. (1994) Cell-cycle-regulated phosphorylation of oncoprotein 18 on Ser-16, Ser-25, and Ser-38. Eur. J. Biochem. 220, 359–368 [DOI] [PubMed] [Google Scholar]
- 22. Hayashi K., Pan Y., Shu H., Ohshima T., Kansy J. W., White C. L., 3rd, Tamminga C. A., Sobel A., Curmi P. A., Mikoshiba K., Bibb J. A. (2006) Phosphorylation of the tubulin-binding protein, stathmin, by Cdk5 and MAP kinases in the brain. J. Neurochem. 99, 237–250 [DOI] [PubMed] [Google Scholar]
- 23. Marklund U., Brattsand G., Shingler V., Gullberg M. (1993) Serine 25 of oncoprotein 18 is a major cytosolic target for the mitogen-activated protein kinase. J. Biol. Chem. 268, 15039–15047 [PubMed] [Google Scholar]
- 24. Di Paolo G., Pellier V., Catsicas M., Antonsson B., Catsicas S., Grenningloh G. (1996) The phosphoprotein stathmin is essential for nerve growth factor-stimulated differentiation. J. Cell Biol. 133, 1383–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li N., Jiang P., Du W., Wu Z., Li C., Qiao M., Yang X., Wu M. (2011) Siva1 suppresses epithelial-mesenchymal transition and metastasis of tumor cells by inhibiting stathmin and stabilizing microtubules. Proc. Natl. Acad. Sci. U.S.A. 108, 12851–12856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mizumura K., Takeda K., Hashimoto S., Horie T., Ichijo H. (2006) Identification of Op18/stathmin as a potential target of ASK1-p38 MAP kinase cascade. J. Cell Physiol. 206, 363–370 [DOI] [PubMed] [Google Scholar]
- 27. Beretta L., Dobránsky T., Sobel A. (1993) Multiple phosphorylation of stathmin. Identification of four sites phosphorylated in intact cells and in vitro by cyclic AMP-dependent protein kinase and p34cdc2. J. Biol. Chem. 268, 20076–20084 [PubMed] [Google Scholar]
- 28. Larsson N., Marklund U., Gradin H. M., Brattsand G., Gullberg M. (1997) Control of microtubule dynamics by oncoprotein 18. Dissection of the regulatory role of multisite phosphorylation during mitosis. Mol. Cell. Biol. 17, 5530–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ng D. C., Ng I. H., Yeap Y. Y., Badrian B., Tsoutsman T., McMullen J. R., Semsarian C., Bogoyevitch M. A. (2011) Opposing actions of extracellular signal-regulated kinase (ERK) and signal transducer and activator of transcription 3 (STAT3) in regulating microtubule stabilization during cardiac hypertrophy. J. Biol. Chem. 286, 1576–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ng D. C., Lin B. H., Lim C. P., Huang G., Zhang T., Poli V., Cao X. (2006) Stat3 regulates microtubules by antagonizing the depolymerization activity of stathmin. J. Cell Biol. 172, 245–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Horwitz S. B., Shen H. J., He L., Dittmar P., Neef R., Chen J., Schubart U. K. (1997) The microtubule-destabilizing activity of metablastin (p19) is controlled by phosphorylation. J. Biol. Chem. 272, 8129–8132 [DOI] [PubMed] [Google Scholar]
- 32. Kinoshita E., Kinoshita-Kikuta E., Takiyama K., Koike T. (2006) Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol. Cell Proteomics 5, 749–757 [DOI] [PubMed] [Google Scholar]
- 33. Kinoshita-Kikuta E., Kinoshita E., Koike T. (2012) Separation and identification of four distinct serine-phosphorylation states of ovalbumin by Phos-tag affinity electrophoresis. Electrophoresis 33, 849–855 [DOI] [PubMed] [Google Scholar]
- 34. Anastassiadis T., Deacon S. W., Devarajan K., Ma H., Peterson J. R. (2011) Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1039–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gonzalez G. A., Montminy M. R. (1989) Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59, 675–680 [DOI] [PubMed] [Google Scholar]
- 36. Ferraris J. D., Persaud P., Williams C. K., Chen Y., Burg M. B. (2002) cAMP-independent role of PKA in tonicity-induced transactivation of tonicity-responsive enhancer/ osmotic response element-binding protein. Proc. Natl. Acad. Sci. U.S.A. 99, 16800–16805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang C., Zhang L., Chen W. (2011) Plant cortical microtubules are putative sensors under abiotic stresses. Biochemistry 76, 320–326 [DOI] [PubMed] [Google Scholar]
- 38. White E. (2011) Mechanical modulation of cardiac microtubules. Pflugers Arch. 462, 177–184 [DOI] [PubMed] [Google Scholar]
- 39. Gigant B., Curmi P. A., Martin-Barbey C., Charbaut E., Lachkar S., Lebeau L., Siavoshian S., Sobel A., Knossow M. (2000) The 4 Å X-ray structure of a tubulin:stathmin-like domain complex. Cell 102, 809–816 [DOI] [PubMed] [Google Scholar]
- 40. Gavet O., Ozon S., Manceau V., Lawler S., Curmi P., Sobel A. (1998) The stathmin phosphoprotein family. Intracellular localization and effects on the microtubule network. J. Cell Sci. 111, 3333–3346 [DOI] [PubMed] [Google Scholar]
- 41. Manna T., Thrower D. A., Honnappa S., Steinmetz M. O., Wilson L. (2009) Regulation of microtubule dynamic instability in vitro by differentially phosphorylated stathmin. J. Biol. Chem. 284, 15640–15649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., Zarrinkar P. P. (2011) Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 [DOI] [PubMed] [Google Scholar]
- 43. Holmfeldt P., Stenmark S., Gullberg M. (2007) Interphase-specific phosphorylation-mediated regulation of tubulin dimer partitioning in human cells. Mol. Biol. Cell 18, 1909–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chneiweiss H., Beretta L., Cordier J., Boutterin M. C., Glowinski J., Sobel A. (1989) Stathmin is a major phosphoprotein and cyclic AMP-dependent protein kinase substrate in mouse brain neurons but not in astrocytes in culture. Regulation during ontogenesis. J. Neurochem. 53, 856–863 [DOI] [PubMed] [Google Scholar]
- 45. Togano T., Kurachi M., Watanabe M., Grenningloh G., Igarashi M. (2005) Role of Ser-50 phosphorylation in SCG10 regulation of microtubule depolymerization. J. Neurosci. Res. 80, 475–480 [DOI] [PubMed] [Google Scholar]
- 46. Scott J. D., Pawson T. (2009) Cell signaling in space and time. Where proteins come together and when they're apart. Science 326, 1220–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Torgersen K. M., Vang T., Abrahamsen H., Yaqub S., Taskén K. (2002) Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell. Signal. 14, 1–9 [DOI] [PubMed] [Google Scholar]
- 48. Gerits N., Kostenko S., Shiryaev A., Johannessen M., Moens U. (2008) Relations between the mitogen-activated protein kinase and the cAMP-dependent protein kinase pathways. Comradeship and hostility. Cell. Signal. 20, 1592–1607 [DOI] [PubMed] [Google Scholar]
- 49. Smith F. D., Langeberg L. K., Cellurale C., Pawson T., Morrison D. K., Davis R. J., Scott J. D. (2010) AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 12, 1242–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chae H. J., Chae S. W., Kim H. R. (2004) Cyclic adenosine monophosphate inhibits nitric oxide-induced apoptosis of cardiac muscle cells in a c-Jun N-terminal kinase-dependent manner. Immunopharmacol. Immunotoxicol. 26, 249–263 [DOI] [PubMed] [Google Scholar]
- 51. Hewer R. C., Sala-Newby G. B., Wu Y. J., Newby A. C., Bond M. (2011) PKA and Epac synergistically inhibit smooth muscle cell proliferation. J. Mol. Cell. Cardiol. 50, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hsueh Y. P., Lai M. Z. (1995) c-Jun N-terminal kinase but not mitogen-activated protein kinase is sensitive to cAMP inhibition in T lymphocytes. J. Biol. Chem. 270, 18094–18098 [DOI] [PubMed] [Google Scholar]
- 53. Xu H., Washington S., Verderame M. F., Manni A. (2008) Activation of protein kinase A (PKA) signaling mitigates the antiproliferative and antiinvasive effects of α-difluoromethylornithine in breast cancer cells. Breast Cancer Res. Treat. 107, 63–70 [DOI] [PubMed] [Google Scholar]
- 54. Zhang J., Wang Q., Zhu N., Yu M., Shen B., Xiang J., Lin A. (2008) Cyclic AMP inhibits JNK activation by CREB-mediated induction of c-FLIP(L) and MKP-1, thereby antagonizing UV-induced apoptosis. Cell Death Differ. 15, 1654–1662 [DOI] [PubMed] [Google Scholar]