

Background: Mitochondrial dysfunction is associated with neuronal disorders, and mitochondrial dynamics are altered in neurodegenerative diseases.
Results: Inhibition of mortalin potentiates amyloid-β-mediated mitochondrial dysfunction and cytotoxicity.
Conclusion: Inhibition of mortalin could lead to mitochondrial dysfunction through mitochondrial fragmentation.
Significance: Activation of mortalin may antagonize the progression of Aβ-mediated neuronal injury in which mitochondrial dysfunction has a key role.
Keywords: Alzheimers Disease, Cell Death, Mitochondria, Mitochondrial Apoptosis, Neurodegenerative Diseases, Drp1, Amyloid-β, Mitochondrial Fission, Mortalin
Abstract
Mitochondrial dynamics greatly influence the biogenesis and morphology of mitochondria. Mitochondria are particularly important in neurons, which have a high demand for energy. Therefore, mitochondrial dysfunction is strongly associated with neurodegenerative diseases. Until now various post-translational modifications for mitochondrial dynamic proteins and several regulatory proteins have explained complex mitochondrial dynamics. However, the precise mechanism that coordinates these complex processes remains unclear. To further understand the regulatory machinery of mitochondrial dynamics, we screened a mitochondrial siRNA library and identified mortalin as a potential regulatory protein. Both genetic and chemical inhibition of mortalin strongly induced mitochondrial fragmentation and synergistically increased Aβ-mediated cytotoxicity as well as mitochondrial dysfunction. Importantly we determined that the expression of mortalin in Alzheimer disease (AD) patients and in the triple transgenic-AD mouse model was considerably decreased. In contrast, overexpression of mortalin significantly suppressed Aβ-mediated mitochondrial fragmentation and cell death. Taken together, our results suggest that down-regulation of mortalin may potentiate Aβ-mediated mitochondrial fragmentation and dysfunction in AD.
Introduction
Mitochondria, essential organelles for both life and death, are highly dynamic. They continuously undergo balanced fission and fusion processes, which are termed mitochondrial dynamics. Mitochondrial dynamics greatly affect the mitochondrial functions such as biogenesis as well as their morphology (1, 2). Imbalanced mitochondrial dynamics are directly linked to many human diseases including cancer, diabetes, and neurodegenerative diseases (3–5). Neurons are particularly dependent on mitochondrial function because of their higher metabolic activity and complex morphology (6). Mitochondria are pivotal for synaptic plasticity and the primary producers of reactive oxygen species (ROS), which contribute to mitochondrial dysfunction. Thus, disruptions of mitochondrial function and mitochondrial dynamics are prominent early events in neurodegenerative diseases such as Alzheimer disease (AD),2 Parkinson disease (PD), Huntington disease, and amyotrophic lateral sclerosis (1, 6). Both the treatment of amyloid-β (Aβ) and the overexpression of either APP (Aβ precursor protein) or APPsw mutant efficiently induce mitochondrial fragmentation and synaptic injury in neuronal cells (7–9).
Mitochondrial fission and fusion processes are regulated by evolutionarily conserved molecular machinery. The large GTPase proteins, MFN1/2 (mitofusin-1/-2) and Opa1 (optic atrophy type 1) assists in the mitochondrial fusion process. Another GTPase protein, Drp1 (dynamin-related protein 1) promotes mitochondrial fission by interacting with mitochondrial outer membrane proteins such as Fis1 and mitochondrial fission factor. Loss-of-function mutations of MFN2 and Opa1 are directly linked to neurodegenerative diseases such as Charcot-Marie-Tooth subtype 2A and autosomal dominant optic atrophy (10, 11). Additionally, a mutation of Drp1 identified in an infant with lethal abnormal brain development also emphasizes the importance of mitochondrial dynamics in neurons (12). As a regulatory mechanism, various post-translational modifications such as phosphorylation, nitrosylation, sumoylation, ubiquitination, or GlcNAcylation of Drp1, proteolytic cleavage of Opa1, and ubiquitination or phosphorylation of MFN1/2 have explained the intricate mechanisms of mitochondrial dynamics in different signals (9, 13–27). Moreover, several other regulatory proteins have been identified. Knockdown of mitochondrial fission factor, GDAP1 (ganglioside-induced differentiation-associated protein-1), MiD49/51 (mitochondrial dynamics protein 49/−51), endophilin B1, or MTP18 (mitochondrial protein 18 kDa) resulted in elongated mitochondria, suggesting that these proteins are involved in the mitochondrial fragmentation processes (28–31). On the other hand, inhibition of prohibitin-2, SLP2 (stomatin-like protein 2), and mitofusion-binding protein promotes mitochondrial fragmentation, indicating that these proteins regulate the mitochondrial fusion process (32–34). Nonetheless, the precise mechanism that coordinates these complex processes of mitochondrial dynamics still remains unclear.
To further understand the regulatory machinery of mitochondrial dynamics, we established a cell-based functional screening system that identified mortalin as a potential regulatory molecule from siRNA library screening. The suppression of mortalin highly induced mitochondrial fragmentation and the suppression synergistically increased Aβ-mediated mitochondrial dysfunctions and cell death. However, up-regulation of mortalin remarkably reduced Aβ-mediated mitochondrial fragmentation and cytotoxicity. Importantly, we found that the expression level of mortalin was reduced in AD patients and in the AD-model mice. Taken together, our results suggest that down-regulation of mortalin exacerbates Aβ-mediated mitochondrial fragmentation and dysfunction in AD.
EXPERIMENTAL PROCEDURES
Cell Culture and Measurement of Mitochondrial Length
SK-N-MC and SH-SY5Y neuroblastoma cells were obtained from the American Type Culture Collection (ATCC). Drp1-deficient mouse embryo fibroblast (MEF) cells were generously provided by Dr. Katsuyoshi Mihara (Kyushu University, Japan) (35). All cells were cultured at 37 °C in a 5% CO2 incubator and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Invitrogen). To generate stable cell lines (SK/mito-YFP, SY5Y/GFP, and SY5Y/GFP-MOT), SK-N-MC were transfected with pmito-YFP and SH-SY5Y were transfected with either pEGFP or pEGFP-mortalin using Lipofectamine 2000 according to the manufacturer's protocol (Invitrogen). Stable transfectants were selected by growth in selection medium containing G418 (1 mg/ml) for 10 days. After single cell dropping, the stable clones were selected under a fluorescence microscope and confirmed by Western blot analysis. Mean mitochondrial length in SK/mito-YFP cells was determined by measuring at least 20 individual mitochondria from images of multiple cells obtained by fluorescence microscopy.
Primary Cortical Neuron Culture
Cortices of 14-embryonic day old ICT mouse were dissected and triturated in Hanks' balanced salt solution containing glucose (5 mg/ml) and sucrose (7 mg/ml). Four hemispheres were plated onto a culture plate coated with poly-l-lysine (100 μg/ml) (Sigma) and laminin (1 μg/ml) (Sigma). Neurons were grown in DMEM (Invitrogen) containing 5% FBS (Hyclone, South Logan, UT), 5% horse serum (HS) (Invitrogen), glutamine (2 mm), and penicillin/streptomycin (Lonza, Basel, Switzerland). To kill proliferating glial cells and make pure neuronal culture, we treated the culture with arabinofuranosyl cytidine (10 μm) at 3 days in vitro (DIV). All experiments were performed at DIV 7.
Reagents
A YFP-fused MitoTracker plasmid (pmito-YFP) was provided by Dr. Yoon, GS (Ajou University, Korea) (36). Mortalin expression plasmid (pEGFP-mortalin) and MKT077 were kindly provided from Dr. Renu Wadhwa (National Institute of Advanced Industrial Science and Technology, Japan) (37, 38). Amyloid-β(1–42) (Aβ) was purchased from American Peptide Co. (Sunnyvale, CA). A MitoTracker® probe and Hoechst 33342 dye were purchased from Invitrogen. The validated siRNA targeting for mortalin (number 1, 5′-GACUAUCGCUCCAUGCCAA-3′ and number 2, 5′-AAACGCAAGUGGAAAUUAA-3′) and negative scrambled siRNA (5′-CCUACGCCACCAAUUUCGU-3′) were purchased from Dharmacon (Thermo Scientific) and previously validated Drp1 siRNA (5′-GAGGUUAUUGAACGACUCA-3′) and Opa1 siRNA (5′-CUGGAAAGACUAGUGUGUU-3′) were synthesized from Bioneer (Daejeon, Korea) (39).
Cell-based Functional Screening with siRNA Library
For the siRNA screening, we listed mitochondrial proteins based on a mitochondrial protein database (MitoProteom database). Among ∼850 genes, we collected around 500 target genes by exclusion of unknown genes. Using the collected genes, we synthesized a custom siRNA library/mitochondrial siRNA library using the Dharmacon siGENOME SMART pool system (Dhamacon, Thermo Scientific). SK/mito-YFP (SK-N-MC stably expressing mito-YFP) cells were seeded in 96-well plates at ∼1500 cells/well. After 24 h, each siRNA was transiently transfected into the cells with a final concentration ∼50 pmol. After 3 and 7 days, mitochondrial morphology was observed under a fluorescence microscope to screen mitochondria dynamics regulator. siRNAs for OPA1 and Drp1 were used as positive controls for each experiment. The screening experiment was repeated two times with consistent results.
Western Blotting
For Western blotting, all lysates were prepared with protein sample buffer (62.5 mm Tris-HCl, pH 6.8, 25% glycerol, 2% SDS, 5% β-mercaptoethanol, 0.01% bromphenol blue) (Bio-Rad). Then the samples separated by SDS-PAGE were transferred to PVDF membrane (Bio-Rad). After blocking with 4% skim milk in TBST (25 mm Tris, 3 mm 140 mm NaCl, 0.05% Tween 20), the membranes were incubated overnight with specific primary antibodies at 4 °C. Anti-Drp1 antibody was from BD Biosciences (San Jose, CA); anti-mortalin antibody was from BD Biosciences; anti-Actin antibody was from Millipore (Temecula, CA). For protein detection, the membranes were incubated with HRP-conjugated secondary antibodies (Pierce).
ROS Measurement
Intracellular ROS levels were assayed using a fluorescent dye, dichloro-dihydro-fluorescein diacetate (DCFH-DA) (Invitrogen), which is converted to highly fluorescent 2′,7′-dichlorofluorescein (DCF) in the presence of oxidant. Briefly, cells plated in 96-well plate were transfected with siRNA. After 3 or 7 days from transfection, the cells were further treated with Aβ for 24 h. Then, the cells were incubated with DCFH-DA (20 μm) in serum-free medium for 30 min and measured the fluorescence (excitation/emission wavelength 358/485) (Victor X3, PerkinElmer Life Sciences). Relative ROS ratio was presented as the change in fluorescence of drug-treated samples compared with that of control samples.
Measurement of Mitochondria Membrane Potential and ATP Level
Mitochondrial membrane potential was examined with a unique fluorescent cationic dye, JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide, BD Biosciences) that detects loss of signal of mitochondrial membrane potential. The fluorescence intensity was measured using plate reader (PerkinElmer Life Sciences) at excitation and emission wavelengths of 485 and 535 nm, respectively, for the monomeric form as well as 535 and 590 nm for J-aggregate forms, respectively. Images were obtained using IX71 (Olympus, Tokyo, Japan) fluorescence microscopes and the cellular total ATP level was detected with an ATP bioluminescence detection kit (Promega, Madison, WI) according to the manufacturer's protocol.
Cell Proliferation Analysis
For the cell proliferation assay, cells seeded in 96-well plates were transfected with mortalin siRNA. After transfection, the cell proliferation rate was measured daily using a Cell Counting Kit-8 (CCK8) solution reagent (10 μm) (Dojindo Laboratories, Kumamoto, Japan) for 2 h. The absorbance was measured with spectrophotometer (Victor-X3, PerkinElmer Life Sciences).
Apoptotic Cell Death Analysis
Apoptotic cell death was determined using an Annexin V-FITC/PI Apoptosis Detection Kit (BD Pharmingen) according to the manufacturer's protocol. Briefly, cells transfected with mortalin siRNA or treated with MKT077 were exposed to Aβ for 24 h. Then, the cells were stained with Annexin V-FITC and propdium iodide (BD Pharmingen). After staining, the cell death ratio was analyzed using a flow cytometer (BD Pharmingen).
Mouse and Human AD Samples
Triple transgenic AD mice (3xTgAD mice; swAPP, PS1-M146V, Tau-P301L) (40) that had been backcrossed to C57BL/6 mice for 8 generations were maintained in our animal facility under pathogen-free conditions. 3-, 6-, and 12-month-old male mice were euthanized, and brains were removed for analysis (n = 5). Inferior parietal lobule and cerebellum specimens from the brains of 5 patients with AD and 5 control subjects that had been enrolled in the University of Kentucky Alzheimer's Disease Center Autopsy Program were used for this study. All patients with AD met both clinical diagnostic criteria and neuropathological diagnostic criteria of AD (41, 42). The control subjects had no history or neuropathological signs of a brain disorder. At autopsy, tissue specimens were rapidly removed and frozen, and were stored at −80 °C.
Statistical Analysis
Data were obtained from least three independent experiments, and presented as mean ± S.E. Statistical evaluation of the results was performed with one-way analysis of variance. Data were considered significant at a value of p < 0.05.
RESULTS
Down-regulation of Mortalin Induces Drp1-dependent Mitochondrial Fragmentation and Dysfunction
It has been previously reported that there are ∼850 proteins in mitochondria (43). For cell-based functional screening, we generated a mitochondrial siRNA library as described under “Experimental Procedures.” In addition, we also established a cell-based functional screening system using SK-N-MC cells that stably expressed the YFP-fused mitochondria tracker, mito-YFP (SK/mito-YFP). Initially to identify novel genetic modulators of mitochondrial dynamics, we screened the siRNA library. Both Drp1 siRNA and Opa1 siRNA were used as positive controls in each experiment. Based on the screening results, we identified mortalin as a potent regulator of mitochondrial dynamics (Table 1).
TABLE 1.
List of mitochondrial dynamics modulator
| Gene name (siRNA) | Mitochondrial fragmentation | Mitochondrial elongation |
|---|---|---|
| Drp1 | +++ | |
| Mortalin | +++ | |
| PHB1 | ++ | |
| PHB2 | +++ | |
| ATP5I | +++ | |
| STOML2 | ++ | |
| TXN | ++ | |
| VDAC1 | ++ | |
| Opa1 | +++ | |
| LETM1 | +++ | |
| VCP | + |
To confirm the screening results, SK/mito-YFP cells were transiently transfected with a scrambled or specific siRNA against mortalin. The results showed that down-regulation of mortalin induced pronounced fragmentation of mitochondria (Fig. 1, A–D). Because Drp1 is a key regulator in mitochondrial fission, we next examined the effects of Drp1 on mortalin-mediated mitochondrial fragmentation. Mortalin siRNA was co-transfected with either scrambled or Drp1 siRNA in SK/mito-YFP cells. The knockdown of Drp1 was confirmed by Western blot analysis. The results showed that reduced Drp1 expression completely suppressed mitochondrial fragmentation induced by mortalin knockdown (Fig. 1E). Mitochondrial dynamics regulate mitochondrial functions as well as their morphology. Excessive mitochondrial fragmentation induces mitochondrial dysfunctions. Therefore, we addressed the effect of mortalin knockdown on mitochondrial function. Because the electrochemical gradient of mitochondrial membrane is essential for ATP synthesis, we detected both mitochondrial membrane potential and total cellular ATP levels following mortalin knockdown. The suppression of mortalin expression markedly induced mitochondrial membrane depolarization but reduced the cellular ATP levels, suggesting that knockdown of mortalin disrupts the mitochondrial membrane potential in neuroblastoma cells (Fig. 2, A and B). In addition, the loss of mitochondrial membrane potential was associated with excessive ROS generation. Thus, we further elucidated mitochondrial dysfunction in mortalin down-regulated cells. The suppression of mortalin expression by their siRNAs resulted in enhanced ROS production in neuroblastoma cells, whereas ROS inhibitors significantly suppressed ROS production, which was induced by down-regulation of mortalin (Fig. 2, C and D). Moreover, we investigated the effect of mortalin knockdown on proliferation. The down-regulation of mortalin suppressed cell proliferation in neuroblastoma cells, respectively (Fig. 2E). Taken together, our data suggest that down-regulation of reduced mortalin induces mitochondrial fragmentation and mitochondrial dysfunction.
FIGURE 1.

Down-regulation of mortalin induces Drp1-dependent mitochondrial fragmentation and dysfunction. A, SK-N-MC cells stably expressing mito-YFP (SK/mito-YFP) were transfected with either a control scrambled siRNA (Sc) or a specific siRNA against mortalin (siMOT), and then mitochondrial morphology was observed under a fluorescence microscope. Both Drp1 siRNA (siDrp1) and Opa1 siRNA (siOpa1) were used as controls. Representative fluorescence pictures of mitochondrial morphology by siRNAs are shown (bar size, 20 μm). B and C, SK/mito-YFP cells were transfected with either control scrambled siRNA (Sc) or specific siRNA against mortalin (si#1 and si#2). After 5 days, mortalin expression was analyzed with Western blotting with anti-mortalin antibody (B). The mitochondrial morphology was observed under a fluorescence microscope (C). D, SK/mito-YFP were transfected with either a control scrambled siRNA (Sc) or a specific siRNA against mortalin (siMOT), and then the mitochondrial length was measured. E, Drp1 siRNA was co-transfected with mortalin siRNA (si#1 and si#2) or Sc in SK/mito-YFP cells. After 7 days, mitochondrial fragmentation was measured under a fluorescence microscopy. Reduced expression of Drp1 by siRNA was confirmed by Western blotting. Data are represented as the mean ± S.E. (n > 3; *, p < 0.01).
FIGURE 2.

Down-regulation of mortalin induces mitochondrial dysfunction. A–C, SK-N-MC cells were transfected with a control scrambled (Sc) or mortalin siRNA (si #1 and si #2). After 7 days, the alteration of mitochondrial membrane potential was monitored by the MitoProbe JC-1 assay (A). The cellular total ATP level was examined by an ATP bioluminescence detection assay (B). The intracellular ROS level was measured by a DCFH-DA fluorescence ROS detection assay (C). D, SK-N-MC cells were transfected with Sc or mortalin siRNA (siMOT) and additionally incubated with ROS inhibitors (NAC and Trolox). After 7 days, mitochondrial fragmentation was measured under a fluorescence microscopy. E, SK-N-MC cells were transfected with a Sc or mortalin siRNA (si #1 and si #2). The cell proliferation rate was determined daily using a cell proliferation assay kit. Data are represented as the mean ± S.E. (n > 3; *, p < 0.01; **, p < 0.05).
MKT077, a Mortalin Chemical Inhibitor Induces Both Mitochondrial Fragmentation and Mitochondrial Dysfunction
With the use of a chemical inhibitor, we investigated the effect of mortalin inhibition on mitochondrial fragmentation and dysfunction. MKT077 is a rhodacyanine dye analog that directly inhibits the function of mortalin (44). To investigate the effect of MKT077 on mitochondrial dynamics, SK/mito-YFP cells were incubated with MKT077 and the mitochondria were monitored (Fig. 3A). Similar to the knockdown experiments, treatment with MKT077 strongly increased mitochondrial fragmentation in a dose-dependent manner (Fig. 3B). Additionally, mitochondrial fragmentation induced by MKT077 was significantly blocked both in Drp1 down-regulated SK-N-MC cells and in Drp1-deficient MEF cells compared with that of the control cells (Fig. 3, C and D). These results suggest that MKT077 also induces Drp1-dependent mitochondrial fragmentation. We further investigated the effect of MKT077 on mitochondrial dysfunction. Wild type (WT) and Drp1-deficient MEF cells were exposed to MKT077, and mitochondrial dysfunction was investigated. Although MKT077 treatment enhanced mitochondrial membrane depolarization and ROS production in WT MEF cells, they were efficiently suppressed in Drp1-deficient MEF cells (Fig. 3, E and F). We further confirmed the effect of mortalin inhibition in primary cultured neurons. Consistently, treatment of MKT077 strongly induced mitochondrial fragmentation in mouse primary cultured neurons (Fig. 4, A and B). Moreover, treatment of MKT significantly increased mitochondrial membrane depolarization as well as ROS production in mouse primary cultured neurons (Fig. 4, C–F). These data suggest that MKT077 promotes mitochondria dysfunction via Drp1 activity.
FIGURE 3.

MKT077, a mortalin chemical inhibitor induces Drp1-mediated mitochondrial fragmentation and mitochondrial dysfunction. A, SK/mito-YFP cells were treated with MKT077 (MKT, 10 μm) for 4 h and then imaged using a fluorescence microscope. B, SK/mito-YFP cells were incubated with increasing concentrations of MKT, and the fragmented mitochondria were observed after 4 h. C, SK/mito-YFP cells transfected with either scrambled siRNA (Sc) or siRNA against Drp1 (siDrp1) were exposed to MKT (10 μm) for 6 h. D, mitochondria in wild type MEF (WT) and Drp1-deficient MEF (Drp1−/−) treated with MKT077 (10 μm) were labeled with a fluorescence MitoTracker (100 nm). Then, cells with fragmented mitochondria were observed by fluorescence microscopy. E, depolarized mitochondrial membrane was analyzed by MitoProbe JC-1 dye in WT and Drp1−/− MEF cells after MKT077 (10 μm) treatment. F, WT and Drp1−/− MEF cells treated with MKT077 (10 μm) were stained with DCF-DA, a ROS labeling dye and the intracellular ROS level was assessed by flow cytometric analysis. Data are represented as the mean ± S.E. (n > 3; *, p < 0.01 or **, p < 0.05).
FIGURE 4.

MKT077 also induces mitochondrial fragmentation and mitochondrial dysfunction in primary cultured neuronal cells. A and B, pure cortical neuronal cultures (DIV 7) were treated with MKT077 (10 μm) for 4 h and stained with a MitoTracker probe. Mitochondrial morphology by MKT077 (A) and mitochondrial length was measured (B) by fluorescence. C and D, pure cortical neuronal cultures (DIV 7) were treated with MKT077 (10 or 20 μm) for 4 h and stained with CM-H2DCFDA (2 μm) for 20 min. The cellular ROS level was observed (C), and measured with a fluorescence microplate reader (D). E and F, MKT077-treated cells were stained with JC-1 staining solution and mitochondria dysfunction was measured by a fluorescence microplate reader (F), and images were observed under fluorescence microscope (E). Data are represented as the mean ± S.E. (n > 3; *, p < 0.02; **, p < 0.05).
Down-regulation of Mortalin Potentiates Aβ-mediated Mitochondria Dysfunction and Cytotoxicity
Aβ promotes mitochondrial dysfunction, which contributes to Alzheimer disease pathology. Moreover, it was recently reported that mitochondrial import of mortalin is influenced by Aβ (45). Thus, we examined the role of mortalin knockdown on Aβ-mediated mitochondria dysfunction. SK-N-MC cells transfected with either scrambled siRNA or mortalin siRNA were incubated with Aβ. Then, both the total cellular ATP level and ROS production were measured. Our results showed that the cellular ATP level decreased appreciably by down-regulation of mortlain compared with Aβ-treated control cells (Fig. 5A). Consistently, mortalin knockdown also significantly increased ROS generation compared with that of control cells following Aβ treatment (Fig. 5B).
FIGURE 5.

Inhibition of mortalin potentiates Aβ-mediated mitochondria dysfunction and cell death. A and B, SK-N-MC cells were transiently transfected with scrambled (Sc) or mortalin siRNA (si #1 and si #2). After 3 days the cells were further treated with Aβ (10 μm) or not. The cellular ATP and ROS levels were determined using an ATP bioluminescence detection assay (A) or a ROS detection assay (B), respectively. C, SH-SY5Y cells transiently transfected with scrambled or mortalin siRNA (si #1 and si #2) were treated with Aβ (10 μm) then, apoptotic cell death was measured by a flow cytometric analysis with Annexin V staining. D-F, SK-N-MC cells were treated with MKT (10 μm) in the presence or absence of Aβ (10 μm) for 20 h. Then the total ATP cellular level, ROS production, and cell death ratio were also determined. Data are represented by the mean ± S.E. (n > 3). Data are considered significant at p values of (*) <0.02 or (**) <0.05.
Because mitochondrial dysfunction is highly associated with cytotoxicity, we next investigated the effect of mortalin knockdown on Aβ-induced cytotoxicity. SH-SY5Y cells transfected with mortalin siRNA were exposed to Aβ, and the cell death rate was determined by flow cytometry. The results suggested that down-regulation of mortalin more enhanced the Aβ-induced cell death than that of control cells (Fig. 5C). We further investigated the effect of mortalin inhibition on Aβ-induced cell death and mitochondrial dysfunction using a chemical inhibitor (MKT077). SK-N-MC cells were exposed to MKT077 in the presence or absence of Aβ, and then the cellular ATP and ROS levels as well as cytotoxicity were examined. Similar to genetic inhibition, MKT077 treatment synergistically affected the Aβ-mediated reduction of ATP levels, induction of ROS generation, and cell death (Fig. 5, D–F). Collectively, these data suggest that mortalin inhibition exacerbates Aβ-induced mitochondrial dysfunction in neuroblastoma cells.
Over-expression of Mortalin Prevents Aβ-mediated Cytotoxicity in Neuroblastoma Cells
Our previous results imply that the inhibition of mortalin is involved in mitochondrial dysfunction, which is associated with cytotoxicity. To demonstrate the effect of mortalin over-expression on mitochondrial morphology and cell death, we established a cell line (SH-SY5Y/mortalin) stably expressing mortain (Fig. 6A). Then we examined the effect of mortalin on Aβ-mediated cell death as well as mitochondrial fragmentation. Consistent with mortalin knockdown experiments, both Aβ-mediated mitochondrial fragmentation and cell death were significantly suppressed in mortalin over-expressing cells compared with that of control cells (Fig. 6, B and C). These results further suggest that over-expression of mortalin prevents Aβ-induced cytotoxicity in neuroblastoma cells.
FIGURE 6.
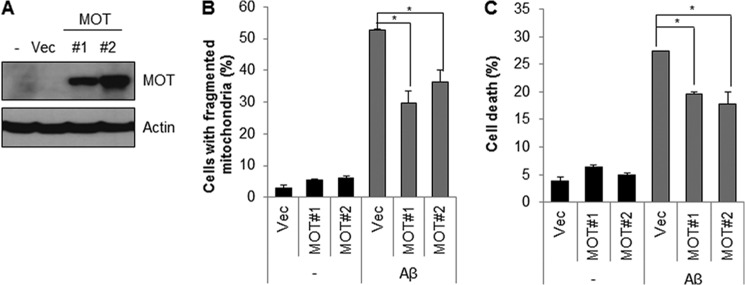
Ectopic expression of mortalin protects Aβ-induced mitochondrial fragmentation and cell death. A, SH-SY5Y cells stably expressing mortalin were generated in SH-SY5Y cells (SY5Y/MOT). Overexpression was confirmed by Western blot analysis with anti-GFP antibody. B, SY5Y/MOT (MOT #1 and MOT #2) cells treated with Aβ (10 μm) for 5 h were stained with MitoTracker dye (100 nm). Then, the cells with fragmented mitochondria were counted under a fluorescence microscope. C, SY5Y/MOT cells were incubated with Aβ (10 μm) for 24 h and stained with propidium iodide. The apoptotic cell death was measured. Data are represented as the mean ± S.E. (n = 5). Data were considered significant at a (*) p < 0.02 value.
Mortalin Is Down-regulated in Brain Samples from 3xTg-AD Mice and Human Alzheimer Patients
In recent reports, mortalin has been implicated in neurodegenerative diseases, such as Parkinson disease (46, 47). However, the implication of mortalin expression with AD has not been elucidated. Therefore, we investigated the expressional regulation of mortalin by employing a triple transgenic mouse model of AD (3xTg-AD) harboring three mutant genes: Aβ precursor protein (APPSwe), presenilin-1 (PS1M146V), and tauP301L (40). The endogenous expression of mortalin gradually increased during normal brain development in mice. In contrast, mortalin expression was not increased in the brain of a 3xTg-AD mouse by 12 months (Fig. 7A). Importantly, the expression of mortalin in brain tissues from human AD patients was considerably decreased compared with that of age-matched normal controls (Fig. 7B). Taken together, these results suggest that mortalin is down-regulated in human AD patients as well as in the AD mice models.
FIGURE 7.
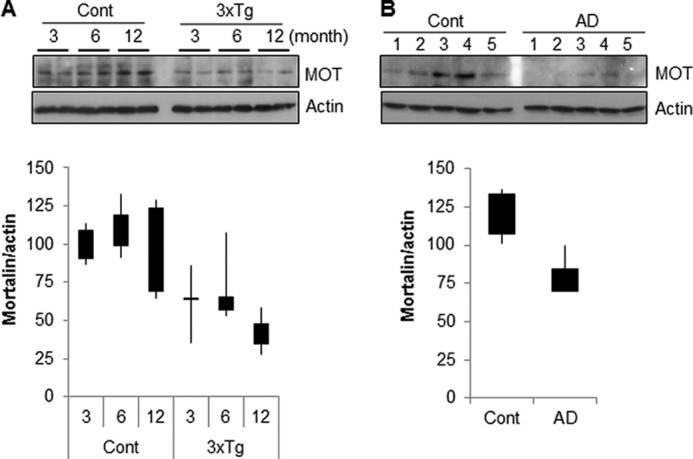
Mortalin expression is down-regulated in Alzheimer disease patient and mice model. A, whole brain extracts from wild type mice (3, 6, and 12 months) and age-matched triple transgenic mice were assessed by Western blotting with mortalin and actin antibodies (upper panel). The relative expression value was calculated using densitometer analysis (lower panel, n = 5). B, the expression level of mortalin was examined in brain tissues from normal and age-matched Alzheimer disease patients (upper panel) and the relative expression value was analyzed using densitometry (lower panel).
DISCUSSION
Mitochondrial dysfunction is associated with neuropathies, and mitochondrial dynamics are altered in neurodegenerative diseases (1, 48). In this study, we synthesized a siRNA library that consisted of mitochondrial proteins and screened the library to identify novel regulators of mitochondrial dynamics. From the screening, we found several already known mitochondrial dynamics modulators such as prohibitin-2, LETM-1 (leucine zipper-EF hand containing transmembrane protein-1), VCP (valosin-containing protein), VDAC, and some mitochondrial ATPase subunits as well as mortalin (Table 1) (33, 49–51). In this article, we demonstrated that the inhibition of mortalin strongly induced mitochondrial fragmentation and dysfunction. In addition, the expression of mortalin was reduced in brain tissues from the AD mice and AD patients. Mortalin is a mitochondrial chaperone protein and a member of the heat shock protein 70 (HSP70) family (52, 53). However, unlike most other HSP70 members, mortalin is not inducible by heat shock but is sensitive to oxidative stress, glucose deprivation, low-level of radiation, and some cytotoxins (54), suggesting that mortalin is a multifunctional protein in various stress conditions. Moreover, mortalin is a key regulatory protein for the import of mitochondrial ATPase components (55). In fact, HSP70 chaperone activity is important to protein quality control and function in mitochondria (56). According to this notion, our results indicated that mortalin has a crucial role in mitochondrial function. The mitochondrial fragmentation and dysfunction caused by mortalin inhibition were dependent on Drp1, suggesting that mitochondrial chaperon activity modulates function of the mitochondrial dynamics (Figs. 1–3). The immune precipitation assay, to examine the direct interaction between Drp1 and mortalin, suggested that mortalin was not directly interacted with Drp1 (data not shown). However, increased ROS by mortalin inhibition may be a key mediator in Drp1 activation in mortalin inhibition-induced mitochondrial fragmentation (Figs. 2D, 3F, and 4C).
Interestingly, recent proteomic analysis with post-mortem PD substantia nigra showed down-regulation of mortalin in PD brain, and mortalin is also deceased in the 6-hydroxydopamine-treated PD rat models (57, 58). Moreover, several PD-associated genetic variants of mortalin have been identified in PD patients (49). The PD-related mutants of mortalin increase both mitochondrial fragmentation and mitochondrial oxidative stress, which eventually impair mitochondrial homeostasis. The mitochondrial dysfunction mediated by down-regulation of mortalin is recovered by over-expression of Parkin, implying that the dysfunction of mortalin is highly associated with PD (59). Moreover, we found that mitochondrial translocation of Parkin was slightly enhanced by treatment of MKT077 (data not shown). However, the roles of mortalin in AD are not well understood. In this study, we examined the expressional regulation of mortalin in AD patients. Our expression analysis revealed that mortalin is down-regulated in brain tissues from 3xTg-AD mouse and AD patients (Fig. 7). Recently, it was shown that ectopic expression of mortalin attenuates Aβ-mediated oxidative stress and neurotoxicity, whereas the suppression of mortalin promotes mitochondrial dysfunction and neuronal injury (60, 61). According to previous results (60, 61), we also observed that the knockdown (by siRNA) and inhibition (by MKT077) of mortalin potentiated Aβ-induced mitochondrial dysfunction and cell death (Fig. 5). Although, the underlying mechanism for mortalin down-regulation in AD and the role of mortalin in Aβ-related neuronal injury are needed to further elucidation in neurodegenerative diseases, our results suggest that down-regulation of mortalin potentiates Aβ-mediated mitochondrial fragmentation and dysfunction in AD. Mortalin expression was differentially regulated in the hippocampus of human APOE4-targeted replacement mice, and the oxidation of mortalin was increased in APOE knock-out mice (62, 63). Oxidized mortalin induced mitochondrial aggregation and resulted in cell death in yeast (64). Therefore, future investigation of the transcriptional and translational regulation of mortalin in AD is needed.
Unlike PD or AD, mortalin is abundantly expressed in many human cancers (65). The up-regulation of mortalin in primary cells reduces cellular senescence and apoptosis but increases the life span of worms (66, 67). Indeed, we observed that over-expression of mortalin suppressed Aβ-induced cell death in neuroblastoma cells (Fig. 5C). As a chaperon, mortalin negatively regulates p53, a tumor suppressor protein (65, 68). MKT077 inhibits the chaperone function of mortalin and disrupts the binding of mortalin and p53, resulting in p53 accumulation (44, 69). The p53 protein is up-regulated in the superior temporal gyrus of AD patients and that increases Tau phosphorylation (70, 71). Although the relationship between mortalin and p53 in AD remains unclear, previous results suggest that up-regulation of mortalin reduces neuronal damage in neurodegenerative diseases. Indeed, Wang et al. (72) recently concluded that activation of HSP70 reduces neurotoxicity mediated by aggregation of the polyglutamine (poly-Q) protein, which is associated with Huntington disease.
In conclusion, we have suggested that mortalin is down-regulated in AD that could lead to mitochondrial dysfunction through mitochondrial fragmentation. Thus, up-regulation or activation of mortalin through drug targeting may antagonize the progression of AD in which mitochondrial dysfunction plays a key role.
Acknowledgments
We deeply thank Dr. Gyesoon Yoon (Ajou University, Korea) and Dr. Renu Wadhwa (NIAIST, Japan) for kindly providing mito-YFP plasmid and mortalin cDNA. We also appreciate to Dr. M. Katsuyoshi (Kyushu University, Japan) for providing Drp1-deficient MEF cells.
This work was supported by Korean Health 21 R&D Project, Ministry of Health & Welfare, Republic of Korea Grant A092042 and the Korea-UK Collaborative Alzheimer's Disease Research Project, Ministry of Health & Welfare, Republic of Korea Grant A120196.
- AD
- Alzheimer disease
- PD
- Parkinson disease
- MEF
- mouse embryo fibroblast
- DIV
- days in vitro
- ROS
- reactive oxygen species
- DCFA-DA
- 2′,7′-dichlorofluorescein diacetate
- EGFP
- enhanced green fluorescent protein
- DCF
- dichlorofluorescein
- Drp1
- dynamin-related protein 1.
REFERENCES
- 1. Cho D. H., Nakamura T., Lipton S. A. (2010) Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol. Life Sci. 67, 3435–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Westermann B. (2010) Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 11, 872–884 [DOI] [PubMed] [Google Scholar]
- 3. Chan D. C. (2006) Mitochondria. Dynamic organelles in disease, aging, and development. Cell 125, 1241–1252 [DOI] [PubMed] [Google Scholar]
- 4. Chen H., Chan D. C. (2009) Mitochondrial dynamics–fusion, fission, movement, and mitophagy in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liesa M., Palacín M., Zorzano A. (2009) Mitochondrial dynamics in mammalian health and disease. Physiol. Rev. 89, 799–845 [DOI] [PubMed] [Google Scholar]
- 6. Knott A. B., Perkins G., Schwarzenbacher R., Bossy-Wetzel E. (2008) Mitochondrial fragmentation in neurodegeneration. Nat. Rev. Neurosci. 9, 505–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barsoum M. J., Yuan H., Gerencser A. A., Liot G., Kushnareva Y., Gräber S., Kovacs I., Lee W. D., Waggoner J., Cui J., White A. D., Bossy B., Martinou J. C., Youle R. J., Lipton S. A., Ellisman M. H., Perkins G. A., Bossy-Wetzel E. (2006) Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 25, 3900–3911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang X., Zuo X., Kucejova B., Chen X. J. (2008) Reduced cytosolic protein synthesis suppresses mitochondrial degeneration. Nat. Cell Biol. 10, 1090–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cho D. H., Nakamura T., Fang J., Cieplak P., Godzik A., Gu Z., Lipton S. A. (2009) S-Nitrosylation of Drp1 mediates β-amyloid-related mitochondrial fission and neuronal injury. Science 324, 102–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Alexander C., Votruba M., Pesch U. E., Thiselton D. L., Mayer S., Moore A., Rodriguez M., Kellner U., Leo-Kottler B., Auburger G., Bhattacharya S. S., Wissinger B. (2000) OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215 [DOI] [PubMed] [Google Scholar]
- 11. Züchner S., Mersiyanova I. V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E. L., Zappia M., Nelis E., Patitucci A., Senderek J., Parman Y., Evgrafov O., Jonghe P. D., Takahashi Y., Tsuji S., Pericak-Vance M. A., Quattrone A., Battaloglu E., Polyakov A. V., Timmerman V., Schröder J. M., Vance J. M. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451 [DOI] [PubMed] [Google Scholar]
- 12. Chang C. R., Manlandro C. M., Arnoult D., Stadler J., Posey A. E., Hill R. B., Blackstone C. (2010) A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J. Biol. Chem. 285, 32494–32503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gawlowski T., Suarez J., Scott B., Torres-Gonzalez M., Wang H., Schwappacher R., Han X., Yates J. R., 3rd, Hoshijima M., Dillmann W. (2012) Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J. Biol. Chem. 287, 30024–30034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Palmer C. S., Osellame L. D., Laine D., Koutsopoulos O. S., Frazier A. E., Ryan M. T. (2011) MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen Y., Dorn G. W. (2013) PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Taguchi N., Ishihara N., Jofuku A., Oka T., Mihara K. (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529 [DOI] [PubMed] [Google Scholar]
- 17. Chang C. R., Blackstone C. (2007) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J. Biol. Chem. 282, 21583–21587 [DOI] [PubMed] [Google Scholar]
- 18. Cribbs J. T., Strack S. (2007) Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Han X. J., Lu Y. F., Li S. A., Kaitsuka T., Sato Y., Tomizawa K., Nairn A. C., Takei K., Matsui H., Matsushita M. (2008) CaM kinase I α-induced phosphorylation of Drp1 regulates mitochondrial morphology. J. Cell Biol. 182, 573–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meuer K., Suppanz I. E., Lingor P., Planchamp V., Göricke B., Fichtner L., Braus G. H., Dietz G. P., Jakobs S., Bähr M., Weishaupt J. H. (2007) Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 14, 651–661 [DOI] [PubMed] [Google Scholar]
- 21. Cereghetti G. M., Stangherlin A., Martins de Brito O., Chang C. R., Blackstone C., Bernardi P., Scorrano L. (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harder Z., Zunino R., McBride H. (2004) Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr. Biol. 14, 340–345 [DOI] [PubMed] [Google Scholar]
- 23. Figueroa-Romero C., Iñiguez-Lluhí J. A., Stadler J., Chang C. R., Arnoult D., Keller P. J., Hong Y., Blackstone C., Feldman E. L. (2009) SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J. 23, 3917–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zunino R., Schauss A., Rippstein P., Andrade-Navarro M., McBride H. M. (2007) The SUMO protease SENP5 is required to maintain mitochondrial morphology and function. J. Cell Sci. 120, 1178–1188 [DOI] [PubMed] [Google Scholar]
- 25. Braschi E., Zunino R., McBride H. M. (2009) MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 10, 748–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yonashiro R., Ishido S., Kyo S., Fukuda T., Goto E., Matsuki Y., Ohmura-Hoshino M., Sada K., Hotta H., Yamamura H., Inatome R., Yanagi S. (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 25, 3618–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karbowski M., Neutzner A., Youle R. J. (2007) The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 178, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Niemann A., Ruegg M., La Padula V., Schenone A., Suter U. (2005) Ganglioside-induced differentiation associated protein 1 is a regulator of the mitochondrial network. New implications for Charcot-Marie-Tooth disease. J. Cell Biol. 170, 1067–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tondera D., Czauderna F., Paulick K., Schwarzer R., Kaufmann J., Santel A. (2005) The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J. Cell Sci. 118, 3049–3059 [DOI] [PubMed] [Google Scholar]
- 30. Otera H., Wang C., Cleland M. M., Setoguchi K., Yokota S., Youle R. J., Mihara K. (2010) Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palmer C. S., Osellame L. D., Stojanovski D., Ryan M. T. (2011) The regulation of mitochondrial morphology. Intricate mechanisms and dynamic machinery. Cell Signal. 23, 1534–1545 [DOI] [PubMed] [Google Scholar]
- 32. Eura Y., Ishihara N., Oka T., Mihara K. (2006) Identification of a novel protein that regulates mitochondrial fusion by modulating mitofusin (Mfn) protein function. J. Cell Sci. 119, 4913–4925 [DOI] [PubMed] [Google Scholar]
- 33. Merkwirth C., Dargazanli S., Tatsuta T., Geimer S., Löwer B., Wunderlich F. T., von Kleist-Retzow J. C., Waisman A., Westermann B., Langer T. (2008) Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev. 22, 476–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tondera D., Grandemange S., Jourdain A., Karbowski M., Mattenberger Y., Herzig S., Da Cruz S., Clerc P., Raschke I., Merkwirth C., Ehses S., Krause F., Chan D. C., Alexander C., Bauer C., Youle R., Langer T., Martinou J. C. (2009) SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 28, 1589–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S. O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y., Taguchi N., Morinaga H., Maeda M., Takayanagi R., Yokota S., Mihara K. (2009) Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 11, 958–966 [DOI] [PubMed] [Google Scholar]
- 36. Yoon Y. S., Yoon D. S., Lim I. K., Yoon S. H., Chung H. Y., Rojo M., Malka F., Jou M. J., Martinou J. C., Yoon G. (2006) Formation of elongated giant mitochondria in DFO-induced cellular senescence. Involvement of enhanced fusion process through modulation of Fis1. J. Cell Physiol. 209, 468–480 [DOI] [PubMed] [Google Scholar]
- 37. Wadhwa R., Takano S., Robert M., Yoshida A., Nomura H., Reddel R. R., Mitsui Y., Kaul S. C. (1998) Inactivation of tumor suppressor p53 by mot-2, an hsp70 family member. J. Biol. Chem. 273, 29586–29591 [DOI] [PubMed] [Google Scholar]
- 38. Wadhwa R., Sugihara T., Yoshida A., Nomura H., Reddel R. R., Simpson R., Maruta H., Kaul S. C. (2000) Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. 60, 6818–6821 [PubMed] [Google Scholar]
- 39. Park S. J., Shin J. H., Kim E. S., Jo Y. K., Kim J. H., Hwang J. J., Kim J. C., Cho D. H. (2012) Mitochondrial fragmentation caused by phenanthroline promotes mitophagy. FEBS Lett. 586, 4303–4310 [DOI] [PubMed] [Google Scholar]
- 40. Oddo S., Caccamo A., Kitazawa M., Tseng B. P., LaFerla F. M. (2003) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol. Aging 24, 1063–1070 [DOI] [PubMed] [Google Scholar]
- 41. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease, NIARI (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol. Aging 18, S1-S2 [PubMed] [Google Scholar]
- 42. Jo D. G., Arumugam T. V., Woo H. N., Park J. S., Tang S. C., Mughal M., Hyun D. H., Park J. H., Choi Y. H., Gwon A. R., Camandola S., Cheng A., Cai H., Song W., Markesbery W. R., Mattson M. P. (2010) Evidence that γ-secretase mediates oxidative stress-induced β-secretase expression in Alzheimer's disease. Neurobiol. Aging 31, 917–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taylor S. W., Fahy E., Zhang B., Glenn G. M., Warnock D. E., Wiley S., Murphy A. N., Gaucher S. P., Capaldi R. A., Gibson B. W., Ghosh S. S. (2003) Characterization of the human heart mitochondrial proteome. Nat. Biotechnol. 21, 281–286 [DOI] [PubMed] [Google Scholar]
- 44. Walker C., Böttger S., Low B. (2006) Mortalin-based cytoplasmic sequestration of p53 in a nonmammalian cancer model. Am. J. Pathol. 168, 1526–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Devi L., Prabhu B. M., Galati D. F., Avadhani N. G., Anandatheerthavarada H. K. (2006) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shi M., Jin J., Wang Y., Beyer R. P., Kitsou E., Albin R. L., Gearing M., Pan C., Zhang J. (2008) Mortalin. A protein associated with progression of Parkinson disease? J. Neuropathol. Exp. Neurol. 67, 117–124 [DOI] [PubMed] [Google Scholar]
- 47. Burbulla L. F., Schelling C., Kato H., Rapaport D., Woitalla D., Schiesling C., Schulte C., Sharma M., Illig T., Bauer P., Jung S., Nordheim A., Schöls L., Riess O., Krüger R. (2010) Dissecting the role of the mitochondrial chaperone mortalin in Parkinson's disease. Functional impact of disease-related variants on mitochondrial homeostasis. Hum. Mol. Genet. 19, 4437–4452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Su B., Wang X., Zheng L., Perry G., Smith M. A., Zhu X. (2010) Abnormal mitochondrial dynamics and neurodegenerative diseases. Biochim. Biophys. Acta 1802, 135–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tamai S., Iida H., Yokota S., Sayano T., Kiguchiya S., Ishihara N., Hayashi J., Mihara K., Oka T. (2008) Characterization of the mitochondrial protein LETM1, which maintains the mitochondrial tubular shapes and interacts with the AAA-ATPase BCS1L. J. Cell Sci. 121, 2588–2600 [DOI] [PubMed] [Google Scholar]
- 50. Park J., Kim Y., Choi S., Koh H., Lee S. H., Kim J. M., Chung J. (2010) Drosophila Porin/VDAC affects mitochondrial morphology. PLoS One 5, e13151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kim N. C., Tresse E., Kolaitis R. M., Molliex A., Thomas R. E., Alami N. H., Wang B., Joshi A., Smith R. B., Ritson G. P., Winborn B. J., Moore J., Lee J. Y., Yao T. P., Pallanck L., Kundu M., Taylor J. P. (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78, 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wadhwa R., Kaul S. C., Ikawa Y., Sugimoto Y. (1993) Identification of a novel member of mouse hsp70 family. Its association with cellular mortal phenotype. J. Biol. Chem. 268, 6615–6621 [PubMed] [Google Scholar]
- 53. Bhattacharyya T., Karnezis A. N., Murphy S. P., Hoang T., Freeman B. C., Phillips B., Morimoto R. I. (1995) Cloning and subcellular localization of human mitochondrial hsp70. J. Biol. Chem. 270, 1705–1710 [DOI] [PubMed] [Google Scholar]
- 54. Kaul S. C., Deocaris C. C., Wadhwa R. (2007) Three faces of mortalin. A housekeeper, guardian and killer. Exp. Gerontol. 42, 263–274 [DOI] [PubMed] [Google Scholar]
- 55. Schneider H. C., Berthold J., Bauer M. F., Dietmeier K., Guiard B., Brunner M., Neupert W. (1994) Mitochondrial Hsp70/MIM44 complex facilitates protein import. Nature 371, 768–774 [DOI] [PubMed] [Google Scholar]
- 56. Kampinga H. H., Craig E. A. (2010) The HSP70 chaperone machinery. J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jin J., Hulette C., Wang Y., Zhang T., Pan C., Wadhwa R., Zhang J. (2006) Proteomic identification of a stress protein, mortalin/mthsp70/GRP75. Relevance to Parkinson disease. Mol. Cell Proteomics 5, 1193–1204 [DOI] [PubMed] [Google Scholar]
- 58. Chiasserini D., Tozzi A., de Iure A., Tantucci M., Susta F., Orvietani P. L., Koya K., Binaglia L., Calabresi P. (2011) Mortalin inhibition in experimental Parkinson's disease. Mov. Disord. 26, 1639–1647 [DOI] [PubMed] [Google Scholar]
- 59. Yang H., Zhou X., Liu X., Yang L., Chen Q., Zhao D., Zuo J., Liu W. (2011) Mitochondrial dysfunction induced by knockdown of mortalin is rescued by Parkin. Biochem. Biophys. Res. Commun. 410, 114–120 [DOI] [PubMed] [Google Scholar]
- 60. Qu M., Zhou Z., Xu S., Chen C., Yu Z., Wang D. (2011) Mortalin overexpression attenuates β-amyloid-induced neurotoxicity in SH-SY5Y cells. Brain Res. 1368, 336–345 [DOI] [PubMed] [Google Scholar]
- 61. Qu M., Zhou Z., Chen C., Li M., Pei L., Yang J., Wang Y., Li L., Liu C., Zhang G., Yu Z., Wang D. (2012) Inhibition of mitochondrial permeability transition pore opening is involved in the protective effects of mortalin overexpression against β-amyloid-induced apoptosis in SH-SY5Y cells. Neurosci. Res. 72, 94–102 [DOI] [PubMed] [Google Scholar]
- 62. Choi J., Forster M. J., McDonald S. R., Weintraub S. T., Carroll C. A., Gracy R. W. (2004) Proteomic identification of specific oxidized proteins in ApoE-knockout mice. Relevance to Alzheimer's disease. Free Radic. Biol. Med. 36, 1155–1162 [DOI] [PubMed] [Google Scholar]
- 63. Osorio C., Sullivan P. M., He D. N., Mace B. E., Ervin J. F., Strittmatter W. J., Alzate O. (2007) Mortalin is regulated by APOE in hippocampus of AD patients and by human APOE in TR mice. Neurobiol. Aging 28, 1853–1862 [DOI] [PubMed] [Google Scholar]
- 64. Kawai A., Nishikawa S., Hirata A., Endo T. (2001) Loss of the mitochondrial Hsp70 functions causes aggregation of mitochondria in yeast cells. J. Cell Sci. 114, 3565–3574 [DOI] [PubMed] [Google Scholar]
- 65. Takano S., Wadhwa R., Yoshii Y., Nose T., Kaul S. C., Mitsui Y. (1997) Elevated levels of mortalin expression in human brain tumors. Exp. Cell Res. 237, 38–45 [DOI] [PubMed] [Google Scholar]
- 66. Yokoyama K., Fukumoto K., Murakami T., Harada S., Hosono R., Wadhwa R., Mitsui Y., Ohkuma S. (2002) Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 516, 53–57 [DOI] [PubMed] [Google Scholar]
- 67. Kaul S. C., Yaguchi T., Taira K., Reddel R. R., Wadhwa R. (2003) Overexpressed mortalin (mot-2)/mthsp70/GRP75 and hTERT cooperate to extend the in vitro lifespan of human fibroblasts. Exp. Cell Res. 286, 96–101 [DOI] [PubMed] [Google Scholar]
- 68. Dundas S. R., Lawrie L. C., Rooney P. H., Murray G. I. (2005) Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival. J. Pathol. 205, 74–81 [DOI] [PubMed] [Google Scholar]
- 69. Deocaris C. C., Widodo N., Shrestha B. G., Kaur K., Ohtaka M., Yamasaki K., Kaul S. C., Wadhwa R. (2007) Mortalin sensitizes human cancer cells to MKT-077-induced senescence. Cancer Lett. 252, 259–269 [DOI] [PubMed] [Google Scholar]
- 70. Hooper C., Chapple J. P., Lovestone S., Killick R. (2007) The Notch-1 intracellular domain is found in sub-nuclear bodies in SH-SY5Y neuroblastomas and in primary cortical neurons. Neurosci. Lett. 415, 135–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Buizza L., Cenini G., Lanni C., Ferrari-Toninelli G., Prandelli C., Govoni S., Buoso E., Racchi M., Barcikowska M., Styczynska M., Szybinska A., Butterfield D. A., Memo M., Uberti D. (2012) Conformational altered p53 as an early marker of oxidative stress in Alzheimer's disease. PLoS One 7, e29789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang A. M., Miyata Y., Klinedinst S., Peng H. M., Chua J. P., Komiyama T., Li X., Morishima Y., Merry D. E., Pratt W. B., Osawa Y., Collins C. A., Gestwicki J. E., Lieberman A. P. (2013) Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat. Chem. Biol. 9, 112–118 [DOI] [PMC free article] [PubMed] [Google Scholar]