

Background: Peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1) α and β are transcriptional regulators of mitochondrial metabolism.
Results: Loss of PGC-1 coactivators in mouse heart causes a defect in phospholipid biosynthesis resulting in mitochondrial ultrastructural abnormalities.
Conclusion: PGC-1 coactivators coordinately regulate mitochondrial metabolism and structure.
Significance: The mitochondrial phenotype of PGC-1-deficient mice resembles that of humans with genetic defects in mitochondrial phospholipid biosynthesis (Barth syndrome).
Keywords: Cardiac Metabolism, Cardiolipin, Gene Regulation, Heart, Mitochondria, Polyunsaturated Fatty Acids, Lipidomics, Phospholipids, Transcriptional Coregulators
Abstract
The energy demands of the adult mammalian heart are met largely by ATP generated via oxidation of fatty acids in a high capacity mitochondrial system. Peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1)-α and -β serve as inducible transcriptional coregulators of genes involved in mitochondrial biogenesis and metabolism. Whether PGC-1 plays a role in the regulation of mitochondrial structure is unknown. In this study, mice with combined deficiency of PGC-1α and PGC-1β (PGC-1αβ−/−) in adult heart were analyzed. PGC-1αβ−/− hearts exhibited a distinctive mitochondrial cristae-stacking abnormality suggestive of a phospholipid abnormality as has been described in humans with genetic defects in cardiolipin (CL) synthesis (Barth syndrome). A subset of molecular species, containing n-3 polyunsaturated species in the CL, phosphatidylcholine, and phosphatidylethanolamine profiles, was reduced in PGC-1αβ-deficient hearts. Gene expression profiling of PGC-1αβ−/− hearts revealed reduced expression of the gene encoding CDP-diacylglycerol synthase 1 (Cds1), an enzyme that catalyzes the proximal step in CL biosynthesis. Cds1 gene promoter-reporter cotransfection experiments and chromatin immunoprecipitation studies demonstrated that PGC-1α coregulates estrogen-related receptors to activate the transcription of the Cds1 gene. We conclude that the PGC-1/estrogen-related receptor axis coordinately regulates metabolic and membrane structural programs relevant to the maintenance of high capacity mitochondrial function in heart.
Introduction
The integrity and function of cellular organelles depend, in part, on their membrane structure, including the phospholipid (PL)2 component. This is particularly true for the mitochondrion, which is supported by a complex membranous structure that comprises the cristae, a labyrinth system that increases surface area allowing for the electron transport complex that supports capacity ATP production. In tissues with high mitochondrial density, such as heart, a significant proportion of cellular PL resides in the mitochondria. In addition, the mitochondrial membrane includes a class of specialized phospholipid, cardiolipin (CL) (1). The importance of PL for mitochondrial integrity and function is underscored by the phenotype of Barth syndrome, which can be caused by a genetic defect in CL remodeling resulting in alterations in mitochondrial ultrastructure and respiratory function causing cardiac and skeletal myopathies (2).
Little is known about how levels of mitochondrial phospholipids are regulated in accordance with oxidative capacity and organellar turnover. The mitochondrial component of highly oxidative tissues such as heart is remarkably dynamic. Mitochondrial biogenesis and fusion/fission rates vary in response to developmental cues and diverse physiological conditions (3–5). The production and remodeling of CL and other PL species must be regulated in accordance with changes in mitochondrial number and turnover. It is also likely that mitochondrial PL synthesis and turnover rates are regulated in accordance with energy demands.
Over the past decade, the transcriptional coregulators, peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1) α and β, have been shown to serve as “master regulators” of mitochondrial biogenesis and function. Extensive studies in cells and in mice have revealed the importance of the PGC-1 coactivators as “boosters” of transcription factors involved in the regulation of nuclear and mitochondrial genes encoding enzymes and proteins involved in mitochondrial energy transduction and ATP synthesis (6, 7). Loss- and gain-of-function studies in mice have demonstrated the importance of PGC-1α and -β in mitochondrial biogenesis in the developing heart and other mitochondrion-rich tissues (8–13). More recently, the PGC-1 coactivators have been shown to regulate the expression of mitochondrial fusion and fission (14). The PGC-1 coactivators have also been implicated in human disease. For example, the expression and activity of PGC-1α has been shown to be reduced in heart failure and diabetes (15, 16).
In this study, we identified a unique mitochondrial cristae abnormality in the hearts of adult mice with combined PGC-1α and -β deficiency. Lipidomic analysis of the hearts of these mice revealed a distinct pattern of reduced levels of CL together with a subset of PL species. Gene regulatory studies demonstrated that this abnormal CL phenotype is related to a defect in the PGC-1-mediated transcriptional control of CDP-diacylglycerol synthase (Cds1), an enzyme that catalyzes a critical upstream step in the biosynthesis of CL. Taken together, these results identify a new role for the PGC-1 coactivators in the regulation of PL synthesis relevant to cardiac mitochondrial structure and function.
EXPERIMENTAL PROCEDURES
Animal Studies
All animal experiments and euthanasia protocols were conducted in strict accordance with the National Institutes of Health guidelines for humane treatment of animals. Generation of PGC-1βf/f mice was described previously (9). To generate adult cardiac-specific inducible PGC-1α- and PGC-1β-deficient mice, PGC-1βf/f mice were bred with mice expressing a mutated estrogen receptor-Cre recombinase fusion protein under control of the MHC promoter to specifically delete PGC-1β in the heart. These mice in turn were crossed with PGC-1α−/− (10) to obtain mice PGC-1α−/−βf/f/Mercre. Cardiac-specific PGC-1β deletion (PGC-1β−/−) or PGC-1αβ double deficiency (PGC-1αβ−/−) was induced by intraperitoneal injection of 50 mg/kg/day of tamoxifen (TAM) for 2 days, at the age of 2 months, and was compared directly with their sex-matched littermates injected with vehicle.
Electron Microscopy
Female PGC-1αβ−/− (n = 3) and PGC-1α−/− (n = 3) mice were euthanized 2 months after TAM injection (performed at 2 months of age). Cardiac papillary muscles were dissected and fixed first in Karnovsky's fixative (2% glutaraldehyde, 1% paraformaldehyde, and 0.08% sodium cacodylate), postfixed in 1% osmium tetroxide, dehydrated in graded ethanol, embedded in Poly Bed plastic resin, and sectioned for electron microscopy. Sections were visualized using an FEI Morgagni transmission electron microscope.
Lipidomics
Male PGC-1αβ−/− (n = 4) and PGC-1α−/− (n = 4), along with PGC-1αβ+/+ control mice (TAM injected, n = 4), were euthanized 1 month after the intraperitoneal injection of TAM or vehicle (injection performed at 2 months of age). Each individual lipid extract from bi-ventricle was reconstituted with a volume of 500 μl/mg of protein of original tissue sample in 1:1 CHCl3/MeOH. The lipid extracts were finally flushed with nitrogen, capped, and stored at −20 °C for electrospray ionization mass spectrometry as described previously (17). A TSQ Vantage triple-quadrupole mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an automated nanospray apparatus (i.e. Nanomate HD, Advion Bioscience Ltd., Ithaca, NY) and Xcalibur system software were utilized in the study (18, 19). The nanomate was controlled by ChipSoft 7.2.0 software. Each lipid extract solution prepared above was diluted to less than 50 pmol of total lipids/μl with CHCl3/MeOH/isopropyl alcohol (1:2:4 by volume) prior to infusion to the mass spectrometer with the nanomate. A mass resolution of 0.7 Thomson was employed for acquisition of the majority of mass spectra except that mass spectra for CL analysis were acquired with a mass resolution of 0.3 Thomson as described previously (20). Identification and quantification of individual lipid molecular species were performed by using a multidimensional mass spectrometry-based shotgun lipidomics as described previously (19, 21). Analysis of 4-hydroxynonenal (HNE) was performed as described previously (22). Data from heart samples were normalized to protein content, and all data are presented as the mean ± S.D. of multiple samples from different animals.
Adenoviral Infection
The adenoviral expression vectors Ad-PGC-1α, Ad-PGC-1β, and Ad-GFP have been described previously (9). The primary neonatal rat cardiac myocytes (NRCMs) were infected with adenovirus 24 h after differentiation and harvested 72 h post-infection. The adenoviral shRNA vectors Ad-shPGC-1α and Ad-shPGC-1β have been described previously (23, 24). The NRCMs were harvested 96 h following the infection of Ad-shPGC adenovirus.
RNA Analyses and qRT-PCR
Total RNA was isolated from mouse bi-ventricle using the RNAzol method (Tel-Test). Total RNA from cultured NRCMs was isolated with RNAqueous kit (Ambion) as per manufacturer's instruction. qRT-PCR was performed as described (25). In brief, total RNA isolated was reverse-transcribed with AffinityScript qPCR cDNA synthesis kit (Agilent Technologies). PCRs were performed in triplicate in a 96-well format using an MX3005P Real Time PCR system (Stratagene). The mouse- or rat-specific primer sets (SYBR Green) used to detect specific gene expression can be found in Table 1. The 36B4 primer set was included in a separate well (in triplicate) and used to normalize the gene expression data.
TABLE 1.
Primer sequences for qRT-PCR analysis and ChIP
| Primers | Forward | Reverse |
|---|---|---|
| Mouse qRT-PCR | ||
| Cds1 | GTCCTGTCTGAGGACGGTG | GAAACCGAGAGCACCAGC |
| Cds2 | TCAGACGCTGTCTCTCCATC | ATGACCGAACTACGGCAGAG |
| Pgs1 | GAAGTTTCCTTCCGACCTCAAG | AGCAGCATAGTGCGAGAGTTC |
| Ptpmt1 | CTATGAACGAGGAGTACGAGACC | AACTGGACTCCTTTGTGGAGATT |
| Crls1 | ACTTCAATCCTTGCTATGCCAC | GGACTGCTGTATTTACCTTGCT |
| Taz | TTCCCTTCTGGGAAGATGTG | ATTTCTTCAGCTTGGGCAAA |
| Lpin1 | CCTAATGATGTACCACCATTCCA | GGGAGTCCTCTGGCAATCTAC |
| Ppap2c | CAGGTCAACTGTTCTGGCTATGT | AATACATGCCAAAGGAGGAGTG |
| Chpt1 | CCTGGGACTCTTTATCTACCAGTC | CAACCAGTCAGGATGTGTTCCTA |
| Cept1 | CTTCTACTGCCCTACAGCTACAGA | TCCCAGAGGAGAGCTACTGTTAGT |
| Pcyt1 | GGCATGACCAGAGTGAAACA | AGCCCTATGTCAGGGTGACT |
| Pcyt2 | AGGAGAGGTACAAGATGGTACAGG | GCTTCACTTCCTCATAGGTGTCTC |
| 36B4 | TGGAAGTCCAACTACTTCCTCAA | ATCTGCTGCATCTGCTTGGAG |
| PGC-1β | TCCAGAAGTCAGCGGCCTTGTGTCA | CTCTGGGACAGGGCAGCACCGA |
| Rat qRT-PCR | ||
| Taz | GCAGACATCTGCTTCACCAA | CCATCCCTTTCTGATAGACACC |
| Cds1 | CCACCATCCTCAGACAGGAC | GGAGATCATGGTCAGGGTGA |
| 36B4 | CACCTTCCCACTGGCTGAA | TCCTCCGACTCTTCCTTTGC |
| Mouse ChIP | ||
| Cds1 (ERR −738) | TGCTGGTTAGTTATGAGGCTTTAAGG | GACACACATCCGTAATTCACACAAAC |
| Cds1 (ERR +1294) | GCTCAAACAGACACAAAAGACAGACA | ACAGAAAACAGAGAAGTTAGTTCCACTACC |
| Cds1 (negative control) | TGTAGGGCTGAGGCCTTGTG | ACATGACTGTCCAAATGTGAGCTG |
Cell Culture, Transfection, and Luciferase Reporter Assays
pcDNA3.1-myc/his.PGC-1α (26), pcDNA3.1-ERRα (27), and pCATCH-PGC-1β (28) vectors have been described previously. pSG5-ERRβ and pSG5-ERRγ were generous gifts from Jean-Marc Vanacker (Institut de Genomique Fonctionnelle de Lyon) and M. Stallcup (University of Southern California), respectively. Mouse mcds1.Luc.1.15 and mcds1.Luc.2.39 promoter reporters were constructed by subcloning the PCR product amplified by forward primer 5′-GGTGTACGCGTGGAGATCAGAGG-3′ with either 5′-CCAGAACGGCATCAACGAGC-3′ or 5′-GTGTAAGCTTTGGGAGGAACTTCC-3′ reverse primer into the MluI/XhoI or MluI/HindIII sites in pGL3-basic reporter plasmid, respectively. Mouse mcds1.Luc.0.73 promoter reporter was constructed by first subcloning a 3-kb PCR-generated fragment into pGL3-Basic using the following primers: forward primer 5′-CCAGTCTGGGTACCTTGGGAGG-3′ and reverse primer 5′-CCAGAACGGCATCAACGAGC-3′. Subsequent digestion with KpnI to remove a 2.5-kb fragment, and ligation of the remaining 5.5-kb backbone, generated the mcds1.Luc.0.73 promoter reporter. Mouse mcds1.Luc.1.97 was constructed by digestion of mcds1.Luc.2.39k with KpnI to remove the 0.42-kb region and re-ligation of the 6.77-kb backbone. C2C12 myoblasts were cultured at 37 °C and 5% CO2 in DMEM supplemented with 10% FBS. Transient transfections in C2C12 myoblasts were performed using Attractene (Qiagen) as per the manufacturer's protocol. Briefly, 350 ng of reporter was cotransfected with 140 ng of PGC-1α, 35 ng of ERR expression vectors, as well as 10 ng of CMV promoter-driven Renilla luciferase (Promega) to control for transfection efficiency. 48 h after cotransfection, luciferase assay was performed using Dual-Glo (Promega) as per manufacturer's recommendations. For C2C12 myotube studies, cotransfection was performed in myoblasts. 24 h following the transfection, cells were refed with 2% horse serum/DMEM to promote differentiation. Luciferase assay was performed 96 h later. All transfection data are presented as the means ± S.E. for at least three separate transfection experiments done in triplicate.
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed as described previously (29, 30). Briefly, C2C12 myoblasts were differentiated for 4 days in 2% horse serum/DMEM, followed by adenoviral infection. 48 h after infection, myotubes were fixed for 30 min with 0.5 mm ethylene glycol bis(succinimidylsuccinate) (Pierce) and then cross-linked with 1% formaldehyde (10 min). Chromatin fragmentation was performed by sonication using a Bioruptor (Diagenode). Sheared protein-DNA complexes were immunoprecipitated by using anti-ERRα (29), anti-PGC-1α (8, 29), or rabbit IgG control (Sigma). Following reversal of cross-linking, isolated and purified DNA fragments were amplified by PCR (Stratagene MX3005P detection system) to detect the enrichment of amplicons corresponding to a 100-bp region encompassing the conserved ERR-response element and a 137-bp region encompassing the upstream ERR-response element of the mouse Cds1 promoter or a 91-bp distal region of the mouse Cds1 gene (negative control). Quantitative analysis was performed by the standard curve method and normalized to IgG control. Specific primers for target regions are listed in Table 1.
Statistical Analysis
Data were analyzed using t tests or analysis of variance where appropriate. The level of significance was set at p < 0.05 in all cases. Data are reported as means ± S.E., except for the lipidomics studies where S.D. was used.
RESULTS
Mitochondrial Ultrastructural Abnormalities in Adult PGC-1αβ-deficient (PGC-1αβ−/−) Mouse Heart
Mice with cardiac-specific targeting of the PGC-1β gene in a generalized PGC-1α-deficient background were generated so that PGC-1αβ deficiency could be induced in adult mice. Three-month-old PGC-1αβ−/− mice were viable 1 month following conditional cardiac-specific deletion of PGC-1β (2.98% of PGC-1β levels remaining compared with PGC-1α−/−). In addition, cardiac function was grossly normal based on echocardiographic analyses (data not shown). Electron microscopic analysis did not reveal significant changes in mitochondrial size or volume density in PGC-1αβ−/− hearts compared with PGC-1α−/− or wild-type controls (Fig. 1A and data not shown). However, a significant subset of mitochondria in cardiac myocytes of the PGC-1αβ−/− mice exhibited a distinctive alteration in the structure and arrangement of the cristae. Specifically, low power field surveys identified electron dense regions in many mitochondria (Fig. 1A). High magnification images of these regions demonstrated tightly stacked or “collapsed” cristae with diminished intercristal space. The abnormal cristae appeared to be disconnected from the inner mitochondrial membrane (Fig. 1B).
FIGURE 1.

Mitochondrial ultrastructural abnormalities in hearts of adult PGC-1αβ−/− mice. Representative electron micrographs of sections taken from the left ventricular papillary muscle from 16-week-old PGC-1α−/− (α−/−) and PGC-1αβ−/− (αβ−/−) mice at two different magnifications. Arrows indicate the structurally abnormal mitochondria with “stacked” cristae (white arrows). Scale bar for each magnification is shown at the bottom right corner: A, 1 μm; B, 5 μm.
CL Deficiency in Adult PGC-1αβ−/− Hearts
Mitochondrial cristae “stacking” abnormalities similar to those observed in the adult PGC-1αβ−/− hearts have been described previously in the heart and skeletal muscle of Barth syndrome patients (2). Barth syndrome is a genetic disorder caused by mutations in the gene encoding tafazzin, a mitochondrial transacylase. Tafazzin is responsible for the remodeling and maturation of CL, a class of phospholipids found exclusively in mitochondrial membranes (1). Therefore, CL levels were assessed in the hearts of 3-month-old PGC-1αβ−/− hearts by lipidomics. Total cardiac CL levels were decreased in PGC-1α−/− mice (data not shown) and to a greater extent in the PGC-1αβ-deficient line (Fig. 2A).
FIGURE 2.
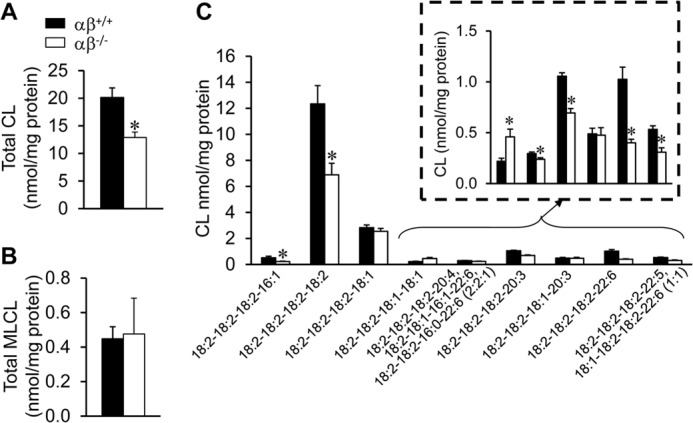
Cardiolipin deficiency in adult PGC-1αβ−/− hearts. Total CL (A) and MLCL (B) levels in 12-week-old wild-type (αβ+/+) and PGC-1αβ−/− (αβ−/−) hearts. C, quantitative analysis of representative CL molecular species determined in hearts of two genotypes as denoted. Bars represent mean ± S.D. *, p < 0.05 compared with αβ+/+.
CL is a unique dimeric phospholipid containing four acyl chains; the composition of which defines multiple molecular species (31). Lipidomic analysis of the adult PGC-1αβ−/− hearts demonstrated that the cardiac CL molecular profile was perturbed. Compared with wild-type mice, the levels of the most abundant CL molecular species were reduced in adult PGC-1αβ−/− heart, including tetra-linoleoyl cardiolipin (18:2–18:2–18:2–18:2 CL), as well as molecular species containing three linoleic acid (18:2) chains (18:2–18:2–18:2–16:1, 18:2–18:2–18:2–20:3, and 18:2–18:2–18:2–22:6 CL) (Fig. 2C).
The CL profile in Barth syndrome associated with tafazzin gene mutations is characterized by an increase in monolysocardiolipin (MLCL) content, in addition to the reduction of CL (32, 33). However, MLCL levels were normal in the PGC-1αβ-deficient heart (Fig. 2B) strongly suggesting that the perturbations in CL level and composition reflect a biochemical abnormality that was not exclusive to tafazzin function.
Alterations in the Phosphatidylcholine and Phosphatidylethanolamine Profiles in PGC-1αβ−/− Hearts
To broaden the analysis of cardiac phospholipids in the PGC-1αβ-deficient heart, global (“shotgun”) lipidomic analysis (19, 34) was conducted. The results of this analysis revealed a significant decrease in total phosphatidylcholine (PC) and phosphatidylethanolamine (PE) levels in PGC-1αβ−/− compared with wild-type hearts (Fig. 3A, data not shown).
FIGURE 3.
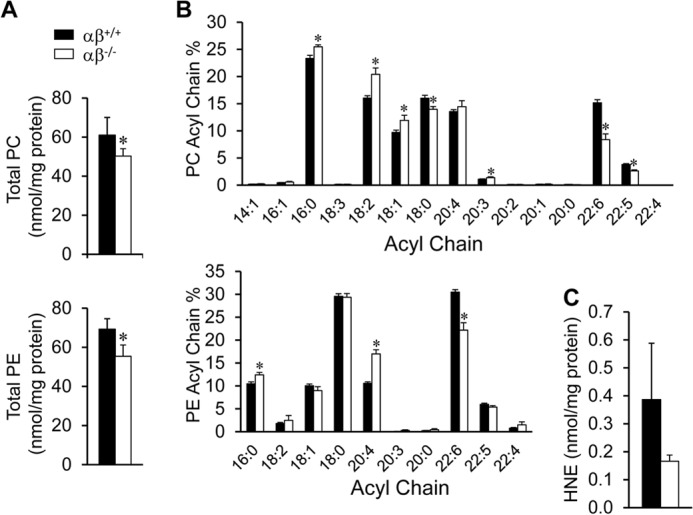
Abnormal PC and PE profiles in PGC-1αβ−/− hearts. A, total PC and PE analyzed from 12-week-old wild-type (αβ+/+) and PGC-1αβ−/− (αβ−/−) hearts. B, percentage of the fatty acyl chain groups in PC and PE. C, HNE content. Bars represent mean ± S.D. *, p < 0.05 compared with αβ+/+.
Further analysis of the PC and PE species in the PGC-1αβ−/− hearts revealed a selective reduction in n-3 polyunsaturated fatty acyl (PUFA)-containing species, including those containing 22:6n-3 and 22:5n-3 acyl chains (Fig. 3B). Analysis of the individual molecular species of PC and PE revealed an interesting pattern (Table 2). Specifically, the overall abundance of 22:6n-3 and 22:5n-3 fatty acids (FA) was decreased (Fig. 3B, Table 2). In contrast to the n-3 PUFA-containing species, levels of n-6 FA-containing species, such as 20:3, 20:4, and 18:2 FA in PC and PE, were increased in the PGC-1αβ−/− hearts (Table 2). Loss of PUFA-containing species was also observed in the CL profile (Table 2).
TABLE 2.
Percentage of fatty acyl chain species within phospholipid CL, PC, and PE profiles in PGC-1 αβ−/− hearts
Means ± S.D. are shown.
| Phospholipid fatty acid (%) | Cardiolipin |
Phosphatidylcholine |
Phosphatidylethanolamine |
|||
|---|---|---|---|---|---|---|
| αβ+/+ | αβ−/− | αβ+/+ | αβ−/− | αβ+/+ | αβ−/− | |
| 16:1n-7 | 0.469 ± 0.166 | 0.125 ± 0.144a | 0.449 ± 0.042 | 0.575 ± 0.116 | ||
| 16:0 | 0.027 ± 0.015 | 0.019 ± 0.024 | 23.337 ± 0.571 | 25.484 ± 0.346a | 10.478 ± 0.408 | 12.424 ± 0.530a |
| 18:3n-3 | 0.159 ± 0.015 | 0.154 ± 0.018 | ||||
| 18:2n-6 | 89.882 ± 1.132 | 87.693 ± 1.298a | 16.041 ± 0.425 | 20.414 ± 1.166a | 1.798 ± 0.237 | 2.477 ± 1.060 |
| 18:1n-9 | 5.830 ± 0.557 | 9.272 ± 1.188a | 9.719 ± 0.417 | 11.928 ± 0.946a | 10.052 ± 0.369 | 8.975 ± 0.844 |
| 18:0 | 16.021 ± 0.527 | 13.977 ± 0.506a | 29.565 ± 0.575 | 29.358 ± 0.820 | ||
| 20:4n-6 | 0.049 ± 0.024 | 0.035 ± 0.043 | 13.568 ± 0.333 | 14.455 ± 1.101 | 10.573 ± 0.324 | 16.999 ± 0.903a |
| 20:3n-6 | 1.752 ± 0.301 | 1.980 ± 0.201 | 1.096 ± 0.051 | 1.382 ± 0.161a | 0.044 ± 0.051 | 0.275 ± 0.138a |
| 20:2n-6 | 0.039 ± 0.044 | 0.049 ± 0.075 | 0.126 ± 0.019 | 0.116 ± 0.032 | ||
| 20:1n-9 | 0.162 ± 0.030 | 0.174 ± 0.064 | ||||
| 20:0 | 0.139 ± 0.009 | 0.083 ± 0.012a | 0.230 ± 0.054 | 0.428 ± 0.218 | ||
| 22:6n-3 | 1.626 ± 0.211 | 0.651 ± 0.201a | 15.171 ± 0.567 | 8.387 ± 1.054a | 30.490 ± 0.531 | 22.185 ± 1.607a |
| 22:5n-3 | 0.324 ± 0.062 | 0.178 ± 0.043a | 3.805 ± 0.159 | 2.625 ± 0.187a | 5.987 ± 0.246 | 5.378 ± 0.315a |
| 22:4n-6 | 0.030 ± 0.002 | 0.020 ± 0.003a | 0.783 ± 0.142 | 1.500 ± 0.653 | ||
a p < 0.05 compared to αβ+/+.
The selective loss of n-3 PUFA-containing phospholipids could indicate increased lipid peroxidation (35), which previously has been reported in PGC-1α-deficient hearts in response to pressure overload (36). To explore this possibility, levels of the lipid peroxidation product HNE were determined. No significant increase of HNE content was detected in the PGC-1αβ−/− hearts (Fig. 3C).
Evidence for Altered Expression of Phospholipid Biosynthesis Pathway Genes in PGC-1αβ−/− Hearts
The observed abnormalities across multiple phospholipid species suggested a defect in phospholipid biosynthesis pathways in the PGC-1αβ-deficient hearts. Therefore, the expression of genes involved in multiple phospholipid biosynthesis pathways was assessed in the PGC-1αβ−/− heart (Fig. 4A). Interestingly, expression of the gene encoding CDS-1, which catalyzes the synthesis of CDP-DAG from phosphatidic acid (Fig. 4A), was reduced in PGC-1α−/− and PGC-1β−/− hearts and to a greater extent in the PGC-1αβ−/− heart (Fig. 4B). No significant change of tafazzin expression was found in the PGC-1αβ−/− heart, although a downward trend was noted. In contrast, there was an increase in the expression of a number of other genes in the phospholipid biosynthesis pathway, including the enzymes in the CL biosynthesis pathway, such as CDP-diacylglycerol synthase 2 (Cds2), phosphatidylglycerophosphate synthase 1 (Pgs1), protein-tyrosine phosphatase, mitochondrial 1 (Ptpmt1), and cardiolipin synthase (Crls1), as well as lipin 1 (Lpin1), phosphatidic acid phosphatase type 2C (Ppap2c), and choline phosphotransferase 1 (Chpt1) in the Kennedy pathway (Fig. 4B).
FIGURE 4.

Altered expression of phospholipid biosynthesis pathway genes in adult PGC-1αβ−/− mice. A, schematic representation of de novo PL biosynthesis and remodeling pathways in mammalian cells. B, results of qRT-PCR analysis of RNA extracted from hearts of 12-week-old wild-type (αβ+/+), PGC-1α−/− (α−/−), PGC-1β−/− (β−/−), and PGC-1αβ−/− (αβ−/−) mice for the following: CDP-DAG synthase (Cds); phosphatidylglycerophosphate synthase 1 (Pgs1); protein-tyrosine phosphatase, mitochondrial 1 (Ptpmt1); cardiolipin synthase (Crls); tafazzin (Taz); lipin 1 (Lpin1); phosphatidic acid phosphatase type 2C (Chptc); choline phosphotransferase 1 (Chpt1); choline/ethanolaminephosphotransferase 1 (Cept1); phosphate cytidylyltransferase 1; choline (Pcyt1); and phosphate cytidylyltransferase 2, ethanolamine (Pcyt2). Bars represent mean ± S.E. *, p < 0.05 compared with αβ+/+; †, p < 0.05 compared with α−/−; and #, p < 0.05 compared with β−/−. C, quantitative analysis of RNA extracted from cultured NRCMs infected with Ad-PGC-1α (Ad-1α) and Ad-PGC-1β (Ad-1β) adenovirus. Bars represent mean ± S.E. *, p < 0.05 compared with NRCMs infected with Ad-GFP control. D, qRT-PCR analysis of RNA extracted from cultured NRCMs infected with Ad-shPGC-1α and Ad-shPGC-1β (PGC-1α/β knockdown (KD)) adenovirus. Bars represent mean ± S.E. *, p < 0.05 compared with NRCMs infected with Ad-control (−). Additional abbreviations used are as follows: MAM, mitochondrion-associated endoplasmic reticulum membrane; Mito, mitochondria; G3P, glycerol 3-phosphate; PA, phosphatidic acid, CDP-DAG, CDP-diacylglycerol; PGP, phosphatidylglycerolphosphate; PG, phosphatidylglycerol; CL, cardiolipin; MLCL, monolysocardiolipin; LPC, lysophosphatidylcholine; PAP, phosphatidic acid phosphatase; DAG, diacylglycerol; Chk, choline kinase; p-choline, phosphorylcholine; Etnk, ethanolamine kinase; p-ethanolamine, phosphorylethanolamine; PS, phosphatidylserine; PC, phosphatidylcholine; PE, phosphatidylethanolamine; Ptdss, phosphatidylserine synthase; Pemt, phosphatidylethanolamine N-methyltransferase; Pisd, phosphatidylserine decarboxylase; AU, arbitrary units.
To further assess the role of PGC-1 coactivators on the regulation of genes involved in phospholipid biosynthesis, we analyzed a transcriptomic dataset generated previously in cultured NRCMs in which PGC-1α or PGC-1β was overexpressed (Ref. 9 and data not shown). Interestingly, expression of Cds1 was dramatically induced by either PGC-1α or PGC-1β overexpression (Fig. 4C). In contrast, tafazzin expression was not significantly induced by the PGC-1 coactivators (Fig. 4C). Consistent with the induction of Cds1 by PGC-1 overexpression, shRNA-mediated knockdown of PGC-1 in NRCMs resulted in a significant decrease in Cds1 expression (Fig. 4D). Taken together, these results suggest that the PGC-1 coactivators regulate the transcription of Cds1, which encodes an enzyme that catalyzes the synthesis of CDP-diacylglycerol (CDP-DAG), a key intermediate supplying the pathway leading to the synthesis of CL and other PL species (Fig. 4A).
PGC-1α Activates Transcription of the Cds1 Gene
We next sought to determine whether PGC-1α activated transcription of the Cds1 gene. For these studies, a mouse Cds1 promoter reporter construct containing a promoter fragment and a portion of intron 1 (mcds1.Luc.2.39) was transfected into C2C12 myocytes in the presence or absence of an expression vector for PGC-1α. PGC-1α markedly induced the activity of mcds1.Luc.2.39 (Fig. 5A). The corresponding Cds1 promoter region was analyzed for consensus DNA-binding sites of transcription factors that could be mediating the effects of PGC-1α. Notably, two conserved putative ERR-binding site sequences were identified (Fig. 5B). ERR is a well characterized PGC-1 coactivator target (27). One of the putative ERR sites located downstream of the transcription start site in the first intron of the Cds1 gene was an excellent match for an ERR-binding site (Fig. 5B) (37). Removal of the upstream ERR-response element in the −738 site (mcds1.Luc.1.97) had little effect on the PGC-1 responsiveness of the Cds1 promoter (Fig. 5C). In contrast, PGC-1α-mediated activation of a Cds1 promoter reporter construct lacking the downstream ERRE site (mcds1.Luc.1.15) was significantly impaired. Deletion of both putative ERR-binding sites (mcds1.Luc.0.73) abolished the activating effects of PGC-1α (Fig. 5C).
FIGURE 5.

Transcriptional activation of the Cds1 gene by PGC-1/ERR. A, transient transfections were performed in C2C12 myoblasts (differentiated into myotubes) using a mouse Cds1 promoter reporter construct, mcds1.luc.2.39, containing a putative ERR-binding site at −738 and +1294, as denoted in B, cotransfected with pcDNA3.1-PGC-1α construct (+) compared with vector backbone alone (−). The bars represent mean ± S.E. promoter-reporter activity shown as relative light units (RLU) normalized to the condition transfected with vector backbone alone. C, results of transient transfection performed with mouse Cds1 reporter mcds1.luc.2.39 and truncation mutants of mcds1.luc.1.97, 1.15, or 0.73 with the pcDNA3.1-PGC-1α construct in C2C12 myoblasts (differentiated into myotubes). D, C2C12 myoblasts were transfected with mouse Cds1 reporter mcds1.luc.2.39 in the presence of pcDNA3.1-PGC-1α or pCATCH-PGC-1β and expression vector of ERRα, ERRβ, or ERRγ. *, p < 0.05 compared with pcDNA3.1 control; †, p < 0.05 compared with PGC-1α or PGC-1β alone. E, quantification of ChIP assays performed with chromatin isolated from C2C12 myoblasts (differentiated into myotubes) infected with Ad-PGC-1α or control virus using anti-PGC-1α, anti-ERRα or IgG (negative control). Schematics above the graphs indicate the relative positions of primers used for amplification (black arrows), and the position of the downstream conserved ERRE. Upstream region, −4799/−4709 primer set that does not contain an ERR-responsive region was used as an intergenic negative control (NC). Bars represent the % of input of ChIP ± S.E. *, p < 0.05 compared with IgG control.
To investigate the interaction of PGC-1α and ERR on the Cds1 gene promoter, cotransfection studies were conducted with mcds1.Luc.2.39 in C2C12 myoblasts, which express low levels of both factors (data not shown). Compared with the level of activation observed upon transfection of expression vectors for either PGC-1α or ERRα alone, cotransfection of both resulted in synergistic coactivation (up to 80-fold, Fig. 5D). Similar results were obtained with ERRβ or ERRγ (Fig. 5D). Similarly, the Cds1 promoter activity was synergistically activated by cotransfection of PGC-1β and ERRs (Fig. 5D). We next sought to determine the occupation of PGC-1α and ERRα on the Cds1 promoter region using ChIP in C2C12 myocytes infected with an adenoviral PGC-1α expression vector. A region containing the downstream ERRE site, spanning +1247 to +1347 bp downstream of the transcription start site, was specifically enriched by the PGC-1α or ERRα antibodies (Fig. 5E). In contrast, an intergenic negative control region was not enriched by either antibody (Fig. 5E). In addition, the region spanning −738 to −730 bp, containing the upstream ERRE site, was not enriched by either PGC-1α or ERRα antibody (data not shown).
DISCUSSION
Herein, we describe a new role for the PGC-1 coactivators in the regulation of cardiac PL biosynthesis. Adult mice with combined PGC-1α and -1β deficiency in heart exhibited mitochondrial cristae stacking abnormalities reminiscent of human Barth syndrome. This mitochondrial phenotype was shown to be associated with reduced levels of sub-species of CL, PC, and PE. Gene regulatory studies demonstrated that the expression of the gene encoding the CL biosynthesis enzyme CDP-diacylglycerol synthase 1, which catalyzes a proximal step in CL synthesis, was downregulated. Finally, the Cds1 gene was shown to be a direct target for PGC-1α through its coactivating function on the transcription factor ERRα.
The PGC-1 coactivators are key regulators of mitochondrial function and biogenesis (6, 7). Studies over the past decade have shown that this family of inducible transcriptional coregulators serve to coordinately regulate the expression of nuclear and mitochondrial genes encoding enzymes and proteins involved in virtually all aspects of mitochondrial energy transduction and ATP synthesis in mitochondrion-rich tissues such as heart, brown adipose tissue, and skeletal muscle (38, 39). In addition, the PGC-1 coactivators are necessary for perinatal mitochondrial biogenesis in heart (9). In this study, we found that the PGC-1 coactivators also serve a key function in cardiac myocyte CL biosynthesis. This role is consistent with the role of the PGC-1 coactivators as master regulators of mitochondrial energy metabolism, given that mitochondrial structural integrity and function are dependent on its CL composition.
We found that the PGC-1 coactivators are necessary for normal expression of Cds1 in the adult heart. CDS-1 catalyzes a proximal step in the PL biosynthetic pathway (Fig. 4A). Cell-based cotransfection studies demonstrated that PGC-1α activates Cds1 gene transcription via a site in the first intron by coregulating the nuclear receptor ERRα. ERRα is a well known target of PGC-1 coregulation (25, 37). Chromatin immunoprecipitation studies confirmed that PGC-1α and ERRα co-occupy the ERR-binding site in the first intron of the Cds1 gene, providing further evidence that this mechanism involves direct transcriptional activation. Somewhat surprisingly, we did not find evidence that PGC-1 regulates other steps in the CL biosynthesis pathway. Indeed, the level of expression of the genes encoding other enzymes in this pathway was normal or up-regulated, suggesting a possible compensatory response. In yeast, CDS-1 has been localized exclusively to the endoplasmic reticulum (40). However, the precise location of CDS-1 in mammalian cells has not been fully determined. Our data strongly suggest that decreased Cds1 gene expression contributes to reduced CL levels. These results suggest that in mammalian cells, CDS-1 may play an important function in mitochondrial PL biosynthesis. Moreover, recognition of Cds1 as a direct PGC-1 target also strongly suggests it plays a role in mitochondrial metabolism and membrane structure. It is important to note, however, that our results do not exclude potential contribution by mitochondrial CDP-diacylglycerol synthase (Cds2) and mitochondrial translocator assembly and maintenance protein, homolog (Saccharomyces cerevisiae) (Tamm41) to CL biosynthesis.
Shotgun lipidomics of the PGC-1αβ−/− heart revealed an interesting pattern of altered PL levels beyond effects on CL. First, we observed a decrease of total PC and PE in the PGC-1αβ−/− heart. This could be explained, in part, by the observed decrease in phosphate cytidylyltransferase 2, ethanolamine (Pcyt2) expression, a rate-limiting step in the Kennedy pathway. Second, we observed that n-3 polyunsaturated species (22:6n-3 and 22:5n-3) were reduced in the PGC-1αβ−/− heart. This pattern suggested the reduction of the stability of some species, perhaps by peroxidation secondary to reactive oxygen species that is known to damage polyunsaturated fatty acids (35). However, analysis of lipid peroxidation as measured by the formation of HNE did not demonstrate evidence of increased peroxidation damage. Interestingly, the reduction of n-3 PUFA species in PC and PE, as well as CL, was paralleled with a concomitant increase of n-6 PUFA species (20:3n-6 and 20:4n-6), among other saturated and monounsaturated FA. The association of decreased n-3 PUFA and increased n-6/n-3 ratio with impaired mitochondrial function has been reported previously (41). It has been suggested that the mitochondrion is a site for de novo biosynthesis of n-3 PUFA through a carnitine-dependent enzymatic pathway that has not been fully delineated (42, 43). Consistent with this notion, genetic defects in mitochondrial FA β-oxidation are associated with lower levels of 22:6n-3 (44, 45). Thus, down-regulation of PGC-1 target genes in the mitochondrial FA β-oxidation pathway (9) may contribute to the observed decrease in n-3 PUFA-containing PE and PC species in the PGC-1αβ−/− heart. This may also explain the decrease in total PC and PE levels, given that the 22–6n-3-containing species constitute a major component of PC and PE. Finally, the high levels of 22:6n-3 containing PL in the mitochondria suggest that this species is critical in maintaining high level respiration rates in tissues such as the heart. Taken together, these results suggest that the PGC-1 coactivators likely regulate PL biosynthesis via effects on several nodal points, including CDS-1 and additional mitochondrial FA metabolic pathways.
Evidence is emerging that altered PGC-1 signaling may be involved in several human diseases, including diabetes, neurodegeneration, skeletal muscle disorders, and heart failure. The importance of maintaining normal mitochondrial PL lipid levels in cardiac mitochondria is exemplified by the cardiomyopathy phenotype of human Barth syndrome. It is tempting to speculate that altered mitochondrial PL synthesis is relevant to the mitochondrial dysfunction known to occur in other inborn and common acquired diseases in which activity of PGC-1 coactivators is known to be reduced (46).
Acknowledgments
We thank Lorenzo Thomas for assistance in preparation of the manuscript; John Shelley at the Sanford-Burnham Medical Research Institute Histology Core at Lake Nona; William Kraft and Karen Green in the Pathology Electron Microscopy Core at Washington University in Saint Louis; Qi Zhang at the Materials Characterization Facility at the University of Central Florida; Denis M. Medeiros at Kansas State University for assistance with electron microscopy, and Hua Cheng for expert technical assistance with the lipidomics studies. Special thanks to Juliet Zechner, Deanna Collia, Corin Riggs, Beatrice Alvarado, Orlando Rodriguez, and Lauren Ashley Gabriel for assistance with the animal studies.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK045416 (to D. P. K.), R01 HL58493 (to D. P. K.), and R01 HL101189 (to D. P. K.).
This article was selected as a Paper of the Week.
- PL
- phospholipid
- CDS
- CDP-diacylglycerol synthase
- CL
- cardiolipin
- DAG
- diacylglycerol
- ERR
- estrogen-related receptor
- FA
- fatty acid
- HNE
- 4-hydroxynonenal
- MLCL
- monolysocardiolipin
- NRCM
- neonatal rat cardiac myocytes
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PGC-1
- peroxisome proliferator-activated receptor γ coactivator 1
- PUFA
- polyunsaturated fatty acyl
- TAM
- tamoxifen
- ERRE
- ERR-response element
- qRT-PCR
- quantitative real-time PCR.
REFERENCES
- 1. Zambrano F., Fleischer S., Fleischer B. (1975) Lipid composition of the Golgi apparatus of rat kidney and liver in comparison with other subcellular organelles. Biochim. Biophys. Acta 380, 357–369 [DOI] [PubMed] [Google Scholar]
- 2. Bissler J. J., Tsoras M., Göring H. H., Hug P., Chuck G., Tombragel E., McGraw C., Schlotman J., Ralston M. A., Hug G. (2002) Infantile dilated X-linked cardiomyopathy, G4.5 mutations, altered lipids, and ultrastructural malformations of mitochondria in heart, liver, and skeletal muscle. Lab. Invest. 82, 335–344 [DOI] [PubMed] [Google Scholar]
- 3. Hallman M. (1971) Changes in mitochondrial respiratory chain proteins during perinatal development. Evidence of the importance of environmental oxygen tension. Biochim. Biophys. Acta 253, 360–372 [DOI] [PubMed] [Google Scholar]
- 4. Smolich J. J., Walker A. M., Campbell G. R., Adamson T. M. (1989) Left and right ventricular myocardial morphometry in fetal, neonatal, and adult sheep. Am. J. Physiol. 257, H1–H9 [DOI] [PubMed] [Google Scholar]
- 5. Marin-Garcia J., Ananthakrishnan R., Goldenthal M. J. (2000) Heart mitochondrial DNA and enzyme changes during early human development. Mol. Cell. Biochem. 210, 47–52 [DOI] [PubMed] [Google Scholar]
- 6. Scarpulla R. C., Vega R. B., Kelly D. P. (2012) Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 23, 459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin J., Handschin C., Spiegelman B. M. (2005) Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370 [DOI] [PubMed] [Google Scholar]
- 8. Lehman J. J., Barger P. M., Kovacs A., Saffitz J. E., Medeiros D. M., Kelly D. P. (2000) Peroxisome proliferator-activated receptor γ coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 106, 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lai L., Leone T. C., Zechner C., Schaeffer P. J., Kelly S. M., Flanagan D. P., Medeiros D. M., Kovacs A., Kelly D. P. (2008) Transcriptional coactivators PGC-1α and PGC-lβ control overlapping programs required for perinatal maturation of the heart. Genes Dev. 22, 1948–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leone T. C., Lehman J. J., Finck B. N., Schaeffer P. J., Wende A. R., Boudina S., Courtois M., Wozniak D. F., Sambandam N., Bernal-Mizrachi C., Chen Z., Holloszy J. O., Medeiros D. M., Schmidt R. E., Saffitz J. E., Abel E. D., Semenkovich C. F., Kelly D. P. (2005) PGC-1α deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin J., Wu P. H., Tarr P. T., Lindenberg K. S., St-Pierre J., Zhang C. Y., Mootha V. K., Jäger S., Vianna C. R., Reznick R. M., Cui L., Manieri M., Donovan M. X., Wu Z., Cooper M. P., Fan M. C., Rohas L. M., Zavacki A. M., Cinti S., Shulman G. I., Lowell B. B., Krainc D., Spiegelman B. M. (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119, 121–135 [DOI] [PubMed] [Google Scholar]
- 12. Lelliott C. J., Medina-Gomez G., Petrovic N., Kis A., Feldmann H. M., Bjursell M., Parker N., Curtis K., Campbell M., Hu P., Zhang D., Litwin S. E., Zaha V. G., Fountain K. T., Boudina S., Jimenez-Linan M., Blount M., Lopez M., Meirhaeghe A., Bohlooly-Y M., Storlien L., Strömstedt M., Snaith M., Oresic M., Abel E. D., Cannon B., Vidal-Puig A. (2006) Ablation of PGC-1β results in defective mitochondrial activity, thermogenesis, hepatic function, and cardiac performance. PLoS Biol. 4, e369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sonoda J., Mehl I. R., Chong L. W., Nofsinger R. R., Evans R. M. (2007) PGC-1β controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proc. Natl. Acad. Sci. U.S.A. 104, 5223–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soriano F. X., Liesa M., Bach D., Chan D. C., Palacín M., Zorzano A. (2006) Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-γ coactivator-1α, estrogen-related receptor-α, and mitofusin 2. Diabetes 55, 1783–1791 [DOI] [PubMed] [Google Scholar]
- 15. Sihag S., Cresci S., Li A. Y., Sucharov C. C., Lehman J. J. (2009) PGC-1α and ERRα target gene downregulation is a signature of the failing human heart. J. Mol. Cell. Cardiol. 46, 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mootha V. K., Handschin C., Arlow D., Xie X., St Pierre J., Sihag S., Yang W., Altshuler D., Puigserver P., Patterson N., Willy P. J., Schulman I. G., Heyman R. A., Lander E. S., Spiegelman B. M. (2004) Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. U.S.A. 101, 6570–6575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Christie W. W., Han X. (2010) Lipid Analysis: Isolation, Separation, Identification and Lipidomic Analysis, 4th Ed., pp. 55–66 The Oily Press, Bridgewater, UK [Google Scholar]
- 18. Han X., Yang K., Gross R. W. (2008) Microfluidics-based electrospray ionization enhances the intrasource separation of lipid classes and extends identification of individual molecular species through multi-dimensional mass spectrometry: development of an automated high-throughput platform for shotgun lipidomics. Rapid Commun. Mass Spectrom. 22, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang K., Cheng H., Gross R. W., Han X. (2009) Automated lipid identification and quantification by multidimensional mass spectrometry-based shotgun lipidomics. Anal. Chem. 81, 4356–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Han X., Yang K., Yang J., Cheng H., Gross R. W. (2006) Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J. Lipid Res. 47, 864–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang K., Han X. (2011) Accurate quantification of lipid species by electrospray ionization mass spectrometry–Meet a key challenge in lipidomics. Metabolites 1, 21–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang M., Fang H., Han X. (2012) Shotgun lipidomics analysis of 4-hydroxyalkenal species directly from lipid extracts after one-step in situ derivatization. Anal. Chem. 84, 4580–4586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koo S. H., Satoh H., Herzig S., Lee C. H., Hedrick S., Kulkarni R., Evans R. M., Olefsky J., Montminy M. (2004) PGC-1 promotes insulin resistance in liver through PPAR-α-dependent induction of TRB-3. Nat. Med. 10, 530–534 [DOI] [PubMed] [Google Scholar]
- 24. Lin J., Yang R., Tarr P. T., Wu P. H., Handschin C., Li S., Yang W., Pei L., Uldry M., Tontonoz P., Newgard C. B., Spiegelman B. M. (2005) Hyperlipidemic effects of dietary saturated fats mediated through PGC-1β coactivation of SREBP. Cell 120, 261–273 [DOI] [PubMed] [Google Scholar]
- 25. Huss J. M., Torra I. P., Staels B., Giguère V., Kelly D. P. (2004) Estrogen-related receptor α directs peroxisome proliferator-activated receptor α signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol. 24, 9079–9091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vega R. B., Huss J. M., Kelly D. P. (2000) The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Mol. Cell. Biol. 20, 1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huss J. M., Kopp R. P., Kelly D. P. (2002) Peroxisome proliferator-activated receptor coactivator-1α (PGC-1α) coactivates the cardiac-enriched nuclear receptors estrogen-related receptor-α and -γ. Identification of novel leucine-rich interaction motif within PGC-1α. J. Biol. Chem. 277, 40265–40274 [DOI] [PubMed] [Google Scholar]
- 28. Lin J., Puigserver P., Donovan J., Tarr P., Spiegelman B. M. (2002) Peroxisome proliferator-activated receptor γ coactivator 1β (PGC-1β), a novel PGC-1-related transcription coactivator associated with host cell factor. J. Biol. Chem. 277, 1645–1648 [DOI] [PubMed] [Google Scholar]
- 29. Wende A. R., Huss J. M., Schaeffer P. J., Giguère V., Kelly D. P. (2005) PGC-1α coactivates PDK4 gene expression via the orphan nuclear receptor ERRα: a mechanism for transcriptional control of muscle glucose metabolism. Mol. Cell. Biol. 25, 10684–10694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang J., Williams R. S., Kelly D. P. (2009) Bcl3 interacts cooperatively with peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α to coactivate nuclear receptors estrogen-related receptor α and PPARα. Mol. Cell. Biol. 29, 4091–4102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schlame M., Rua D., Greenberg M. L. (2000) The biosynthesis and functional role of cardiolipin. Prog. Lipid Res. 39, 257–288 [DOI] [PubMed] [Google Scholar]
- 32. van Werkhoven M. A., Thorburn D. R., Gedeon A. K., Pitt J. J. (2006) Monolysocardiolipin in cultured fibroblasts is a sensitive and specific marker for Barth syndrome. J. Lipid Res. 47, 2346–2351 [DOI] [PubMed] [Google Scholar]
- 33. Houtkooper R. H., Rodenburg R. J., Thiels C., van Lenthe H., Stet F., Poll-The B. T., Stone J. E., Steward C. G., Wanders R. J., Smeitink J., Kulik W., Vaz F. M. (2009) Cardiolipin and monolysocardiolipin analysis in fibroblasts, lymphocytes, and tissues using high-performance liquid chromatography-mass spectrometry as a diagnostic test for Barth syndrome. Anal. Biochem. 387, 230–237 [DOI] [PubMed] [Google Scholar]
- 34. Han X., Yang K., Gross R. W. (2012) Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom. Rev. 31, 134–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sun D., Gilboe D. D. (1994) Effect of the platelet-activating factor antagonist BN 50739 and its diluents on mitochondrial respiration and membrane lipids during and following cerebral ischemia. J. Neurochem. 62, 1929–1938 [DOI] [PubMed] [Google Scholar]
- 36. Lu Z., Xu X., Hu X., Fassett J., Zhu G., Tao Y., Li J., Huang Y., Zhang P., Zhao B., Chen Y. (2010) PGC-1α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxid. Redox Signal. 13, 1011–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dufour C. R., Wilson B. J., Huss J. M., Kelly D. P., Alaynick W. A., Downes M., Evans R. M., Blanchette M., Giguère V. (2007) Genome-wide orchestration of cardiac functions by the orphan nuclear receptors ERRα and γ. Cell Metab. 5, 345–356 [DOI] [PubMed] [Google Scholar]
- 38. Huss J. M., Kelly D. P. (2004) Nuclear receptor signaling and cardiac energetics. Circ. Res. 95, 568–578 [DOI] [PubMed] [Google Scholar]
- 39. Finck B. N., Kelly D. P. (2006) PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J. Clin. Invest. 116, 615–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tamura Y., Harada Y., Nishikawa S., Yamano K., Kamiya M., Shiota T., Kuroda T., Kuge O., Sesaki H., Imai K., Tomii K., Endo T. (2013) Tam41 is a CDP-diacylglycerol synthase required for cardiolipin biosynthesis in mitochondria. Cell Metab. 17, 709–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ovide-Bordeaux S., Bescond-Jacquet A., Grynberg A. (2005) Cardiac mitochondrial alterations induced by insulin deficiency and hyperinsulinaemia in rats: targeting membrane homeostasis with trimetazidine. Clin. Exp. Pharmacol. Physiol. 32, 1061–1070 [DOI] [PubMed] [Google Scholar]
- 42. Infante J. P., Huszagh V. A. (1997) On the molecular etiology of decreased arachidonic (20:4n-6), docosapentaenoic (22:5n-6), and docosahexaenoic (22:6n-3) acids in Zellweger syndrome and other peroxisomal disorders. Mol. Cell. Biochem. 168, 101–115 [DOI] [PubMed] [Google Scholar]
- 43. Infante J. P., Huszagh V. A. (2001) Impaired arachidonic (20:4n-6) and docosahexaenoic (22:6n-3) acid synthesis by phenylalanine metabolites as etiological factors in the neuropathology of phenylketonuria. Mol. Genet. Metab. 72, 185–198 [DOI] [PubMed] [Google Scholar]
- 44. Harding C. O., Gillingham M. B., van Calcar S. C., Wolff J. A., Verhoeve J. N., Mills M. D. (1999) Docosahexaenoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 22, 276–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gillingham M., Van Calcar S., Ney D., Wolff J., Harding C. (1999) Dietary management of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). A case report and survey. J. Inherit. Metab. Dis. 22, 123–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sparagna G. C., Chicco A. J., Murphy R. C., Bristow M. R., Johnson C. A., Rees M. L., Maxey M. L., McCune S. A., Moore R. L. (2007) Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J. Lipid Res. 48, 1559–1570 [DOI] [PubMed] [Google Scholar]