

Background: Whether the mitochondrial or cytoplasmic protein synthesis inhibition predominates to aminoglycoside-induced ototoxicity is unclear.
Results: The ototoxicity of an aminoglycoside correlates primarily with its ability to block mitochondrial rather than cytoplasmic protein synthesis.
Conclusion: Designer aminoglycosides that selectively inhibit cytoplasmic rather than mitochondrial ribosome show decreased ototoxicity.
Significance: The results are beneficial for development of aminoglycoside-based drugs to treat human genetic diseases.
Keywords: Antibiotics Action, Genetic Diseases, Mitochondrial Apoptosis, Oxidative Stress, Ribosome Function, Aminoglycosides, Cytoplasmic Protein Synthesis Inhibition, Mitochondrial Protein Synthesis Inhibition, Ototoxicity
Abstract
There is compelling evidence that aminoglycoside (AG) antibiotics can induce the mammalian ribosome to suppress disease-causing nonsense mutations and partially restore the expression of functional proteins. However, prolonged AG treatment can cause detrimental side effects in patients, including most prominently, ototoxicity. Recent mechanistic discussions have considered the relative contributions of mitochondrial and cytoplasmic protein synthesis inhibition to AG-induced ototoxicity. We show that AGs inhibit mitochondrial protein synthesis in mammalian cells and perturb cell respiration, leading to a time- and dose-dependent increase in superoxide overproduction and accumulation of free ferrous iron in mitochondria caused by oxidative damage of mitochondrial aconitase, ultimately leading to cell apoptosis via the Fenton reaction. These deleterious effects increase with the increased potency of AG to inhibit the mitochondrial rather than cytoplasmic protein synthesis, which in turn correlates with their ototoxic potential in both murine cochlear explants and the guinea pig in vivo. The deleterious effects of AGs were alleviated in synthetic derivatives specially designed for the treatment of genetic diseases caused by nonsense mutations and possessing low affinity toward mitochondrial ribosomes. This work highlights the benefit of a mechanism-based drug redesign strategy that can maximize the translational value of “readthrough therapy” while mitigating drug-induced side effects. This approach holds promise for patients suffering from genetic diseases caused by nonsense mutations.
Introduction
Aminoglycosides (AGs)2 are a powerful class of bactericidal antibiotics, acting against a wide spectrum of different microorganisms, including Gram-positive and Gram-negative bacteria, and mycobacteria (1). These antibiotics target the 16 S rRNA of the 30 S ribosomal subunit and interfere with translational fidelity and the translocation step of protein synthesis, eventually resulting in bacterial cell death (2). Although a prokaryotic selectivity of action is crucial to the therapeutic utility of AGs as antibiotics, they are not entirely selective for the bacterial ribosome; they also bind to some extent to the eukaryotic cytoplasmic rRNA (3) and promote mistranslation (4). In fact, this lack of selectivity has been exploited as a treatment for genetic diseases that result from nonsense mutations (5). Recent studies in tissue culture and in animal models and clinical trials confirm the ability of AGs to allow mammalian cytoplasmic ribosomes to read through disease-causing premature termination codon (PTC) mutations and generate full-length functional proteins in several genetic disorders (6).
However, a major drawback that limits the potential of AGs for suppression therapy is their ototoxicity, i.e., their propensity to cause irreversible hearing loss (7). In addition, the low specificity of clinical AGs to the mammalian cytoplasmic ribosome (8) necessitates their use in higher doses than usually employed in antibiotic treatment, which in turn causes more deleterious toxic effects and hence largely limits their therapeutic utility against genetic diseases (9). Nevertheless, despite its established ototoxicity and the reduced readthrough efficiency at subtoxic doses (10), the clinical AG antibiotic gentamicin was and is frequently used for proof of concept experiments in various diseases models and in clinical trials, and no systematic study has been performed to design AG structures for better readthrough activity and low toxicity.
Recently, two different mechanisms have been proposed to explain the AG-induced ototoxicity. One model suggests that AGs exert their ototoxicity by inhibiting mitochondrial protein synthesis, followed by mitochondrial dysfunction caused by oxidative stress, which ultimately leads to cell death (11, 12). In contrast, an alternative model considers cytoplasmic protein synthesis inhibition as a potential trigger of a cellular pathway, similar to ribotoxic stress response, leading to hair cell apoptosis (13). Although in principle the inhibition of either the mitochondrial or cytoplasmic protein synthesis can contribute to ototoxicity, it needs to be determined which mechanism operates or predominates in vivo.
Given the proposed bacterial origin of mitochondria (14), we hypothesized that AG derivatives exhibiting a greater selectivity for the cytoplasmic versus the mitochondrial rRNA can improve functional PTC suppression at low dosages, thereby decreasing deleterious effects on mitochondrial protein synthesis and reducing ototoxic potential. Such compounds would be excellent candidates for the treatment of human genetic diseases (9). By addressing the need for such compounds, we have systematically developed compound NB74 and compound NB84 (15) as novel pseudo-trisaccharide derivatives of the extraordinary cytotoxic natural aminoglycoside G418 (16) (see Fig. 1). Both NB74 and NB84 showed markedly higher PTC suppression and cytoplasmic ribosome inhibition activities while exhibiting significantly lower cytotoxicity than gentamicin. However, both NB74 and NB84 also exhibited distinctly decreased bacterial and human mitochondrial ribosome specificity in comparison to those of gentamicin and the parent drug G418 (15, 17) and hence did not show significant antibacterial activity (in both Gram-negative and Gram-positive bacteria). These observations left unanswered the question of whether the lower affinity to mitochondrial or cytoplasmic ribosomes was responsible for the lower ototoxicity.
FIGURE 1.

Chemical structures of a series of standard (G418 and gentamicin) and designer (NB74 and NB84) aminoglycosides that were investigated in this study.
Here, we focus on characterizing the ototoxic potential of a series of standard and designer AGs, both in vitro and in vivo. Particular emphasis was placed on the question of whether mitochondrial or cytoplasmic protein synthesis is a major cause in AG ototoxicity. We show that AGs that inhibit mitochondrial protein synthesis will perturb cell respiration, leading to a time- and dose-dependent increase in superoxide overproduction and accumulation of free ferrous iron in mitochondria caused by oxidative damage of mitochondrial aconitase, ultimately leading to cell apoptosis via the Fenton reaction. We find that these deleterious effects increase with the increased inhibition potency of AG on mitochondrial protein synthesis, which in turn correlates with the ototoxic potential of the tested compounds both in cochlear explants in culture and in the guinea pig in vivo. These results validate the power of rational design strategy to combat the adverse side effects of AGs and are therefore beneficial for further research in two directions: the design of new AG-based structures for the treatment of human genetic diseases and the design of new AG-based antibiotics with diminished deleterious effects on humans.
EXPERIMENTAL PROCEDURES
Materials
Compounds NB74 and NB84 were prepared as described previously by us (15). Geneticin (G418) was purchased from Apollo Scientific Ltd. as sulfate salt. Gentamicin sulfate was purchased from Molekula. Neomycin sulfate was purchased from Sigma. All other chemicals and biochemicals, unless otherwise stated, were obtained from Merck or Sigma. In all biological tests, all the tested aminoglycosides were in their sulfate salt forms (Mw g/mol of the sulfate salts were as follows: gentamicin, 653.2; G418, 692.7; neomycin, 908.8; NB74, 564.3; and NB84, 695.5).
Cell Toxicity Assays
For the cytotoxicity assays, HeLa cells were grown in 96-well plates (5,000 cells/well) in DMEM (Sigma) containing 10% FBS and 1% glutamine (90 μl; Biological Industries) at 37 °C and 5% CO2. Following overnight incubation, different concentrations of the tested AGs were added (10 μl/well), and the cells were incubated for an additional 48 h. A cell proliferation assay (XTT-based colorimetric assay; Biological Industries) was performed by using a 3-h incubation protocol, according to the manufacturer's instructions. Optical density was measured using an ELISA plate reader. Cell viability was calculated as the ratio between the numbers of living cells in cultures grown in the presence of the tested compounds and those in cultures grown under the identical protocol without the tested compound. The half-maximal lethal concentration (LC50) values were obtained from fitting concentration-response curves to the data of at least three independent experiments, using GraFit 5 software.
Mitochondrial Protein Synthesis Inhibition Assay
HeLa cells were grown in 10-cm dishes in DMEM supplemented with 10% fetal bovine serum (Sigma) until cells reached 80% confluence. Approximately 2 × 107 cells were incubated with different concentration of AGs for 24 h. Cells were washed with PBS flowed by addition of a fresh DMEM without l-methionine (Sigma). Preincubation was carried out at 37 °C for 60 min, followed by addition of emetine (10 μm, an inhibitor of 80 S ribosome; Sigma) for 10 min and then addition of [35S]methionine (150 μCi) for 60 min. Cells were washed twice with PBS, trypsinized (Biological Industries), and centrifuged at 500 × g for 10 min. HeLa cells pellets were resuspended in 60 μl of lysis buffer (2% sodium dodecyl sulfate, 2 mm EDTA, 0.2% (v/v) 2-mercaptoethanol, 0.05 m Tris, pH 6.8, and 10% (v/v) glycerol) and heated for 2 min at 90 °C. The resulted mixture was then either stored in a freezer or used immediately for electrophoresis or scintillation measurements (18). Protein concentration was determined by the method of Bradford using bovine serum albumin as standard.
Autoradiography
Radioactivity was measured by acid precipitation of the labeled proteins: the lysed mixture from the above (15 μl) was added with trichloroacetic acid (15%), methionine (1 mm), and BSA (50 μg/ml) to a total volume of 1.9 ml. The resulted mixture was incubated on ice for 60 min. The precipitated proteins were harvested onto filter paper disks (Whatman 3 mm, 2.3 cm) using a Tomtec harvester and washed twice with 2 ml of 5% trichloroacetic acid, and the filters were dried at 60 °C for 30 min. The filters were then inserted into the scintillation vials containing 5 ml of scintillation solution: toluene (1 liter), Triton X-100 (0.5 liter), 2,2′-p-phenylene-bis(5-phenyloxazole) (0.3 g), and 2,5-diphenyloxazole (3 g), followed by counting on a scintillation counter. The half-maximal inhibitory concentration (IC50Mito) values of mitochondrial protein synthesis were determined by Grafit5 software. Resolution of labeled mitochondrial proteins was also carried out by electrophoresis on 15% acrylamide gel. After electrophoresis the gel was fixed, stained by Coomassie Blue, dried, and visualized by autoradiography.
Cell Respiration Inhibition Measurements
Oxygen utilization was measured polarographically with a thermostatically controlled (37 °C) Clark oxygen electrode (Strathkelvin 782 Oxygen System; Strathkelvin Instrument Ltd.). Freshly prepared HeLa cells were treated with G418 (1 and 2 mm), NB84 (1 and 2 mm) for 24 h and with rotenone (10 μm) for 1 h. Rotenone experiments were used as positive controls. Cells were washed with PBS, trypsinized (Biological Industries), and centrifuged at 500 × g for 10 min. HeLa cells pellets were resuspended in serum-free DMEM low glucose medium without phenol red (Invitrogen), and 10 μl were taken for cell counting (hemocytometer cell counting chamber; Hausser Scientific). Cell concentrations were normalized to 2 × 106 cells/ml, and then 1 ml of cell suspension was added to a water-jacketed chamber. Respiration was recorded for 10 min and calculated as rate of change in the oxygen concentration. Cell respiration was converted to a percentage of control.
Superoxide Radical Measurements
Superoxide radical in whole cells were determined by using redox-sensitive probes, dihydroethidium (DHE; Sigma) (19) and MitoSOX Red (Molecular Probes) (20), for cellular and mitochondrial compartments, respectively. Freshly prepared HeLa cells were treated with different AGs for 24 h. Cells were washed with PBS, trypsinized (Biological Industries), and centrifuged at 500 × g for 10 min; washed again in Hanks' balanced salt solution containing NaCl (135 mm), HEPES (20 mm), KCl (4 mm), Na2HPO4 (1 mm), CaCl2 (2 mm), MgCl2 (1 mm), and glucose (10 mm), pH 7.3. Cells were then resuspended in Hanks' balanced salt solution containing DHE (1 μm) or MitoSOX Red (1 μm) and incubated at 37 °C for 25 min. An aliquot of 10 μl was taken for cell counting (hemocytometer cell counting chamber, Hausser Scientific); cell concentrations were normalized to 2 × 106 cells/ml and transferred to a stirred thermostatted cuvette, and the fluorescence measurements were performed at 37 °C; DHE fluorescence (excitation, 518 nm; emission, 605 nm; slits, 10 nm) and MitoSOX Red fluorescence (excitation, 485 nm; emission, 590 nm; slits, 10 nm) were measured in a Varian Cary Eclipse fluorescence spectrophotometer with a temperature-controlled cuvette holder and magnetic stirrer.
Aconitase Activity Assay
HeLa cells were grown in 10-cm dishes in DMEM supplemented with 10% fetal bovine serum without the addition of penicillin/streptomycin (Sigma) to 80% confluence. Approximately 7 × 106 cells were incubated with different concentrations of AGs for 24 h. Cells were washed with PBS followed by lyses (lyses buffer: Tris-Cl (25 mm), pH 8.0, glycerol (10%, v/v), and bromphenol blue (0.025% w/v)). Cell lysates (5 mg protein/ml) were loaded on the aconitase activity gels as follow: a separating gel contained acrylamide (8%), Tris base (132 mm), borate (132 mm), citrate (3.6 mm), and a stacking gel contained acrylamide (4%), Tris base (67 mm), borate (67 mm), and citrate (3.6 mm). The running buffer contained Tris (25 mm), pH 8.3, glycine (192 mm), and citrate (3.6 mm). Electrophoresis was carried out at 180 V at 4 °C. Aconitase activity was assayed by incubating the gel in the dark at 25 °C in 100 mm Tris, pH 8.0, 1 mm NADP, 2.5 mm cis-aconitic acid, 5 mm MgCl2, 1.2 mm 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 0.3 mm phenazine methosulfate, and 5 units/ml isocitrate dehydrogenase, and quantitation was performed using gel analyzer image software.
Intracellular Free Iron Measurements
Freshly prepared HeLa cells were incubated with different concentration of aminoglycosides for 5 h, followed by addition of rhodamine B-[(1,10)phenanthrolin-5-yl) aminocarbonyl]benzyl ester (RPA; 1 μm, Squarix) (31). Cells were kept at 37 °C for 25 min. Excess RPA was rinsed by HEPES buffer (20 mm, pH 7.3), centrifuged, and resuspended in HEPES buffer (2 ml, 20 mm, pH 7.3). Cell concentrations were normalized to 2 × 106 cells/ml, and the fluorescence measurements were performed at 37 °C. RPA fluorescence (excitation, 543 nm; emission, 584 nm; slits, 10 nm) was measured in a Varian Cary Eclipse Fluorescence spectrophotometer with a temperature-controlled cuvette holder and magnetic stirrer. Following the stabilization of base line, an excess of fast permeating chelator Deferiprone (L1, 200 μmol/liter; Sigma), was added; the following increase in fluorescence was measured as reactive mitochondrial iron value.
EPR Measurements
The pool of intracellular chelatable iron was detected by established EPR method (21). Freshly prepared HeLa cells were incubated with different AGs at a final concentration of 4 mm for 5 h. Cells were centrifuged at 10,000 rpm for 5 min at 4 °C, and the pellets were resuspended in DMEM (900 μl), added with desferrioxamine mesylate (DFO, Sigma; 100 μl of 0.2 m solution in Tris-HCl, pH 7.4) and incubated for 10 min with shaking at 37 °C. Cells were centrifuged (10,000 rpm for 5 min), washed with ice cold Tris-HCl buffer (20 mm, pH 7.4), and resuspended in 200 μl of the same buffer containing 10% (v/v) glycerol. An aliquot of 10 μl was taken for cell counting, and the cell concentration was normalized to 5 × 106 cells/ml. Samples were loaded into 4-mm quartz EPR tubes and immediately frozen in liquid nitrogen. EPR spectra were recorded at 150 K on a Bruker EMX-10/12 X-band (ν = 9.3 GHz) digital EPR spectrometer equipped with a Bruker N2 temperature controller. EPR spectra were recorded at microwave power of 200 milliwatts, magnetic field of 100 kHz, modulation of 20.0 G amplitude, sweep time of 83.9 s, conversion time of 40.96 ms, time constant of 81.9 ms, and receiver gain of 4.48 × 104. The digital field resolution was 2048 points/spectrum.
Assays of Ototoxic Potential
For the evaluation of ototoxic potential, explants of the organ of Corti were obtained by dissection of the cochlea from postnatal day 2 mice. The tissue was positioned as a flat surface preparation on a collagen-coated incubation dish in 1 ml of serum-free Basal medium Eagle (BME) plus serum-free supplement (Sigma), 1% bovine serum albumin, 2 mm glutamine, 5 mg/ml glucose, and 10 units/ml penicillin and incubated for 4–5 h (37 °C, 5% CO2). Then an additional 1.5 ml of the culture medium were added, and the preincubation was continued for 40–48 h. Then the medium was exchanged for a new medium containing a specific concentration of drug and further incubated for 72 h. For hair cell counts, organ cultures were fixed overnight in 4% paraformaldehyde at 4 °C and then permeabilized for 30 min with 3% Triton X-100 in PBS. The specimens were washed three times with PBS for 10 min each time at room temperature, followed by incubation at room temperature for 60 min for fluorescent visualization of F-actin with rhodamine-phalloidin or Alexa 488-phalloidin (Molecular Probes). After several rinses in PBS, the specimens were mounted on a slide with fluoromount-G (Southern Biotech).
Hair cells in the surface preparations were counted using a 50× oil immersion objective on a Leitz Orthoplan microscope equipped for epifluorescence. The right objective had a calibrated scale (0.19 mm) superimposed on the field as a reference guide. The single row of inner hair cells and all three rows of outer hair cells were oriented longitudinally within each 0.19-mm frame. The presence or absence of hair cells was evaluated for each row beginning at the apex and moving down to the base, assessing each successive 0.19-mm field. Counts were entered into a computer program, which calculated the percentage of missing hair cells and plotted the result as a function of distance from the apical end of the cochlea.
In Vivo Tests
Albino male Hartley guinea pigs of initially ∼200 g (Charles River Breeding Laboratories) had free access to water and food and were acclimated for 1 week before experiments. AGs were administered once daily subcutaneously for 14 days at the concentration as indicated in the figure legend (see Fig. 7C); saline injections of the same volume served as controls. Auditory function was measured as auditory brainstem response at 12 and 32 kHz under anesthesia with an intramuscular injection of 40 mg of ketamine, 2 mg of xylazine, and 1 mg of acepromazine/kg of body weight as described previously (22). For each animal, the thresholds were determined before the study and at 3 weeks after drug treatment.
FIGURE 7.

Aminoglycoside-induced hair cell loss in cochlear explants and loss of auditory function in vivo. A, explants of the mouse organ of Corti were incubated for 72 h with a different AGs (as indicated) at concentrations of 3.3 μm and stained for actin as described under “Experimental Procedures.” Following treatment with NB84 and NB74, morphology remains essentially normal, gentamicin causes considerable destruction of hair cells, and cell loss is almost complete with G418. Sections of the basal part are shown. OHC, outer hair cells; IHC, inner hair cells. Calibration bar, 25 μm. B, dose-response curves of missing outer hair cells as a function of AG concentration. Hair cell loss was quantified along the entire length of cochlear explants, and the concentration at 50% loss of hair cells (LC50Coch) was determined by Grafit5 software. The data represent means ± S.E., n = 3–8/point. C, effect of chronic AG treatment (once daily injections at indicated concentrations for 14 days) in vivo on auditory thresholds. Threshold shift is the difference in threshold before and 3 weeks after the end of treatment. The data represent means ± S.D., n = 3/point. Note that dB is a logarithmic scale, i.e., a 10-dB difference indicates a 1 log10 difference in sound intensity. Cont, control; Gent, gentamicin; Neo, neomycin.
Cytocochleograms showing quantitative evaluation of hair cell loss were generated from surface preparations of the organ of Corti (see Fig. 8). The temporal bones were removed from the guinea pigs immediately following euthanasia and were perfused through the cochlear scalae with 4% paraformaldehyde in PBS, pH 7.4, and kept in this medium overnight at 4 °C. After rinsing in PBS, the cochleae were decalcified in 5% sodium EDTA (adjusted with HCl to pH 7.4) for 24 h at room temperature. Subsequently, the softened bony capsule and extraneous tissues (stria vascularis, Reissner's membrane, and tectorial membrane) were removed. The remaining cochlear structure was permeabilized with 0.3% Triton X-100 in PBS for 30 min and stained for actin with a 1:100 dilution of rhodamine phalloidin for 1 h at room temperature. After several rinses with PBS, the epithelium of the organ of Corti was removed from the modiolus and mounted on a slide with anti-fade mounting media. Hair cells were counted as described above for the evaluation of cochlear explants.
FIGURE 8.
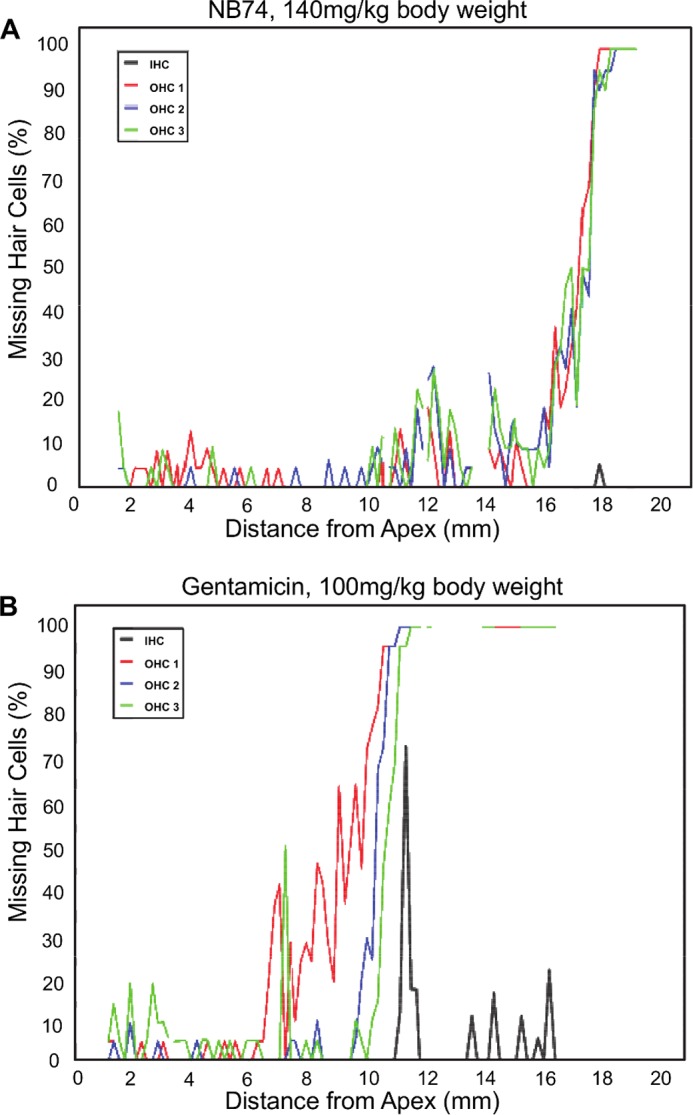
Cytocochleograms showing quantitative evaluation of hair cell loss. Surface preparations of guinea pig cochleae were evaluated quantitatively by counting the presence or absence of hair cells along the entire length of the cochlea. A, after treatment of guinea pigs with NB74 (140 mg/kg of body weight for 14 days), only minimal loss of outer hair cells is observed at the extreme base of the cochlea. B, after treatment with gentamicin (100 mg/kg of body weight for 14 days), loss of outer hair cells (OHC) already begins in the upper middle turn of the cochlea and increases steeply with a total loss in the lower middle and basal turns of the cochlea. Inner hair cells (IHC) show scattered loss toward the base. Representative examples of treatment with gentamicin and NB74 are shown.
RESULTS
Synthetic AGs with Significantly Reduced Inhibition of Mitochondrial Protein Synthesis Show Reduced Impairment of Mitochondrial Respiration in Mammalian Cells
As a first step for our comparative study, we especially selected G418, gentamicin, NB74, and NB84 (Fig. 1) because they greatly diverge in their abilities to block cytoplasmic and the mitochondrial protein synthesis (15, 17) (Table 1): a 30-fold difference in their cytoplasmic protein synthesis inhibition (IC50Cyto values of 2.0, 2.8, 17, and 62 μm for G418, NB84, NB74, and gentamicin, respectively) and a 74-fold difference in their mitochondrial protein synthesis inhibition (IC50Mito values of 13, 26, 404, and 965 μm for G418, gentamicin, NB84, and NB74, respectively). An additional advantage in choosing these particular structures was that their relative inhibition tendencies in two systems are different; for example, whereas both G418 and NB84 exhibit a similar potency for cytoplasmic protein synthesis inhibition, G418 is ∼31-fold more potent in blocking mitochondrial protein synthesis compared with NB84.
TABLE 1.
Comparative toxicity and inhibition of protein translation in cytoplasmic and mitochondrial systems of G418, gentamicin, and the designer structures NB84 and NB74
| AG | Ototoxicity |
Cell toxicity (LC50) |
Translation inhibitiond |
||||
|---|---|---|---|---|---|---|---|
| Cochleotoxicity (LC50Coch)a | In vivob | HEK-293c | HeLa | In vitro IC50Cyto | Ex vivo IC50Mito | In vitro IC50Mito | |
| μm | mm | mm | μm | mm | μm | ||
| G418 | 0.7 | ++++ | 1.3 ± 0.1 | 2.1 ± 0.1 | 2.0 ± 0.3 | 2.3 ± 0.2 | 13 ± 1 |
| Gentamicin | 3.5 | ++ | 2.5 ± 0.3 | 2.9 ± 0. 2 | 62.0 ± 9.0 | 13.3 ± 0.4 | 26 ± 2 |
| NB84 | 20.0 | NDe | 5.8 ± 0.7 | 8.1 ± 1.3 | 2.8 ± 0.3 | >16 | 404 ± 26 |
| NB74 | 140.0 | + | 22.2 ± 1.1 | 27.4 ± 1.5 | 17.0 ± 0.6 | >16 | 965 ± 155 |
a LC50 values in human embryonic kidney (HEK-293) cells were previously reported (17).
b The cytoplasmic in vitro translation inhibition (IC50Cyto) and the mitochondrial in vitro translation inhibition (IC50Mito) were previously reported (17). The mitochondrial translation inhibition (IC50Mit) values ex vivo are derived from the data in Fig. 2B.
c Cochleotoxicity curves are presented in Fig. 7B.
d The relative ototoxicity in vivo are derived from the data in Fig. 7C.
e ND, not determined.
To address whether the previously observed inhibition of protein synthesis in mitochondria isolated from HeLa cells (17) correlates with their ability to penetrate the cells, we first examined the direct impact of AGs on mitochondrial protein synthesis in HeLa cells at the whole cell level. The mitoribosome in mammalian cells is responsible for synthesis of 13 mtDNA-encoded proteins, which are integral parts of the mitochondrial respiratory chain complexes. We exposed HeLa cells to selected AGs, and the inhibition levels were quantified by using a radioactive assay (Fig. 2) (18). Proteins synthesized in mitochondria were selectively labeled by including the cytoribosome inhibitor emetine into the incubation.
FIGURE 2.

Dose-response effects of AGs on mitochondrial protein synthesis as determined in whole cell assay. HeLa cells were incubated with varied concentrations of G418, neomycin, gentamicin, NB74, and NB84 as indicated. A, after lyses, equal amounts of protein (Coomassie staining) were fractionated on SDS-PAGE, and [35S]methionine-labeled mitochondrial protein levels were determined by densitometry. Tentative identities of labeled mitochondrial proteins were determined as reported (23): COX1 (57 kDa), COX2 (26 kDa), COX3 (29 kDa), subunits I, II, and III of cytochrome c oxidase; cytochrome b (44 kDa) apocytochrome b of the bc1 complex; ATPase 6 (25 kDa), and ATPase 8 (10 kDa) subunits 6 and 8 of the F0 portion of ATP synthase; ND1 (35 kDa), ND2 (39 kDa), ND3 (13 kDa), ND4 (52 kDa), ND5 (67 kDa), ND6 (18 kDa), and ND4L (8 kDa), subunits of NADH dehydrogenase. B, semilogarithmic plots of scintillation counting as a function of AG concentration. Radioactivity was measured on the lysed mixture after acid precipitation as described under “Experimental Procedures.” The 50% inhibitory concentrations (IC50Mit) were determined by Grafit5 software. The data represent means ± S.D., n = 3/point.
As a representative example, the data in Fig. 2A show the comparative effects of the standard AG, neomycin, and the designer structure NB74 on mitochondrial protein synthesis after 24 h of incubation as determined by SDS-PAGE; a typical electrophoretic pattern of the organelle-specific translation products was observed (23). Neomycin-treated cells showed a clear dose-dependent effect with the tendency toward a lower rate of protein labeling relative to NB74. Note that even though we did not include it into all our experiments regularly, neomycin is well characterized and one of the most ototoxic aminoglycoside reported to date (9). Here we show that G418 is actually even more ototoxic than neomycin (see below). Protein levels were quantified by direct scintillation counting of the radiolabeled proteins after acid precipitation (Fig. 2B; for more details see “Experimental Procedures”). The antibiotic G418 is the most potent inhibitor of the mitoribosome (IC50Mito = 2.3 mm) followed by neomycin (IC50Mito = 6.3 mm) and gentamicin (IC50Mito = 13.3 mm), whereas the synthetic derivatives NB74 and NB84 exhibit significantly lower potency of inhibition (IC50Mito > 16 mm). Notably, the observed trend in HeLa cells is very similar to that observed previously by us in isolated mitochondria (17), with the only difference that the IC50Mito values in the cell line are in the millimolar range, whereas in intact mitochondria these values are in micromolar range, probably because of the poor mammalian cell permeability of AGs (9).
We next tested whether the inhibition of mitochondrial protein synthesis is associated with the mitochondrial dysfunction, in particular with the inhibition of mitochondrial respiration. To directly test the mitochondrial respiratory capacity of AG-treated cells, we measured changes in the oxygen consumption rate of intact HeLa cells using Clark oxygen electrode (Strathkelvin Instrument Ltd.; Fig. 3A). Compared with untreated cells, G418-treated cells exhibited a significant reduction in respiration of 31 and 41% at 1 and 2 mm concentration, respectively (Fig. 3B). Cells treated with NB84 at the same concentrations showed a significantly lesser change in respiratory capacity: only 6 and 10% reduction at concentrations of 1 and 2 mm, respectively. To ensure the accuracy of the observed data, we used rotenone, a mitochondrial complex I inhibitor, as a positive control in all our experiments. The cell respiration inhibition by rotenone (10 μm) was 60%, as previously reported (24). These data demonstrate that AG-induced inhibition of mitoribosome is associated with the impairment of the function of mitochondria and that the increased inhibition of mitoribosome correlates with the increased dysfunction of mitochondria, reflected by the impairment of the overall respiratory capacity of the cell.
FIGURE 3.

Inhibitory effects of G418, NB84, and rotenone (Rot) on mitochondrial respiration in HeLa cells. A, representative tracings of oxygen consumption as recorded by using Clark oxygen electrode. Rates for oxygen consumption in control (Cont, untreated), NB84-, G418-, and rotenone-treated cells were 11.3, 9.6, 6.5, and 4.4 μmole/liter/min, respectively. B, mitochondrial respiration inhibition induced by the indicated concentrations of G418, NB84, and rotenone. Respiration was calculated as a percentage of control. The data represent means ± S.D., n = 3/point.
Synthetic AGs Induce Significantly Reduced Oxidative Stress and Damage in Mammalian Cells
We next examined whether the observed AG-induced impairment of the overall respiratory capacity triggers oxidative stress and damage to mammalian cells. In mammalian cells, the mitochondrial electron transport chain complexes are the major source of reactive oxygen species (ROS) during normal metabolism because of leakage of electrons (25). Superoxide (O2⨪), a reactive oxygen by-product, is the primary radical formed because of incomplete reduction of oxygen to water in a one-electron transfer (26). The superoxide radical is a precursor to other forms of ROS, including H2O2, in a process that is catalyzed by superoxide dismutase. In addition, the superoxide radical can directly interact with cellular components, resulting in cell damage. We first measured superoxide production in HeLa cells by using two different fluorescence probes; DHE that detects the total superoxide in the cell (19) and MitoSOX Red, recently characterized as a highly selective detection probe of mitochondrial superoxide production in live cells (20) (Fig. 4).
FIGURE 4.

Effect of AGs on superoxide radical production. HeLa cells were incubated with a series of AGs as indicated, and the superoxide radical formation was detected by using two different fluorescence probes. A, DHE (1 μm) probe, measuring fluorescence at 518/605 nm. The data represent means ± S.D., n = 3/point. B, rate changes (as a percentage of control) of superoxide radical production under DHE treatment. The data represent means ± S.D., n = 3/point. C, MitoSox Red (1 μm) probe, measuring fluorescence at 510/580 nm. The data represent means ± S.D., n = 3/point. D, plot of the superoxide radical levels as measured by DHE fluorescence (data from A) against the levels obtained from MitoSox Red (data from C) (linearity R2 = 0.9546). E and F, dose-dependent effect of AGs on superoxide production. Following the treatment with AG (24 h), cells were incubated with DHE (1 μm, E) or MitoSox Red (1 μm, F) for an additional 170 min, 37 °C, and the fluorescence was measured as above. The data represent means ± S.D., n = 3/point. Cont, control; Gent, gentamicin; Neo, neomycin.
Exposure of HeLa cells to G418 and gentamicin at 2 mm concentration resulted in ∼2- and 1.6-fold increases of superoxide production over that of untreated cells, as detected by the DHE probe (Fig. 4A). In contrast, the treatment with NB74 at the same concentration had only little effect. For the positive control, we used antimycin A and performed various control experiments by following the fluorescence change over the time (170 min; data not shown). In the AG-treated and untreated cells, we determined the relative rates of superoxide radical production: the changes were 140, 70, and 20% for G418, gentamicin, and NB74, respectively (Fig. 4B), indicating a similar trend on superoxide overproduction: G418 > gentamicin > NB74.
To determine whether such increases in superoxide production might originate from mitochondria, MitoSOX Red was then employed as a probe. HeLa cells exposed to a series of standard and synthetic AGs at 2 mm concentrations generated a significant increase in superoxide radicals by the mitochondria (Fig. 4C). Treatment with G418, neomycin, and gentamicin resulted in superoxide over expression by 92, 71, and 62%, relative to untreated cells, whereas the effects of NB84 and NB74 were significantly milder, resulting in a 33 and 18% increase. We also examined the dose-dependent effect of AGs on superoxide radical production, using both DHE (Fig. 4E) and MitoSOX Red (Fig. 4F) probes, and found that the increased concentration of each AG tested resulted in increased level of superoxide produced. The DHE fluorescence data correlated well with the MitoSOX Red response (Fig. 4D) (R2 = 0.9546), suggesting that the electron leakage from the mitochondrial electron transport chain is a major source of the superoxide radicals produced because of the AG treatment.
Superoxide can directly interact with various cellular components and cause cell damage. Among other cellular components, particular attention has focused on reactions with the members of [4Fe-4S]-containing dehydratases, including the citric acid cycle enzyme aconitase. This reaction occurs rapidly (with estimated second order rate constants in the range of 107 m−l s−1) (27) and is considered the strongest oxidative damage to aconitase, resulting in the release of Fe2+ from [4Fe-4S] clusters and H2O2 accumulation (28). To characterize AG-induced cell damage, we monitored the dose-dependent impact of AGs on mitochondrial and cytoplasmic aconitase activity by using a nondenaturing gel assay, an aconitase activity assay (29) (Fig. 5). Such an in-gel activity assay allowed us a clear and convenient separation between the effects on the cytoplasmic and mitochondrial aconitases (c-aconitase and m-aconitase, respectively). Among the AGs tested, the largest effect was observed for G418, which caused a strong dose-dependent inactivation of both aconitases, with a significant preference for the mitochondrial versus the cytoplasmic protein: at 4 mm concentration, only ∼7% of m-aconitase activity but 51% of c-aconitase activity were retained. The residual activities of both aconitases caused by the exposure of cells to gentamicin were considerably higher than for G418, whereas the effect of NB74 was almost negligible. These data are consistent with the mitoribosome inhibition, cell respiration, and mitochondrial superoxide overexpression, highlighting that the oxidative stress and cell damage induced by the synthetic AGs is significantly milder than that of the standard AGs tested.
FIGURE 5.

In-gel activity assay of aconitase enzyme from mitochondrial and cytoplasmic origin (m-aconitase and c-aconitase), in HeLa cells Triton lysate as a function of AG concentration as indicated. HeLa cells were incubated with different AGs for 24 h. After lysis, the lysates (5 mg protein/ml) were subjected to electrophoresis, followed by activity quantization (29) as described under “Experimental Procedures.”
To further substantiate the cellular damage caused by inactivation of the mitochondrial aconitase, we measured the accumulation of intracellular free iron (Fe2+) in mitochondria. Intracellular free iron is of additional interest because of its role in both DNA and protein damage and lipid peroxidation. The primary experimental approach was to block these forms of oxidative damage by the presence of cell-permeable iron chelators (30). To monitor the levels of free Fe2+ selectively in mitochondria, we used the fluorescence probe RPA (31). We measured mitochondrial Fe2+ in HeLa cells and found that both standard and synthetic AGs induced a dose-dependent increase in Fe2+ accumulation (Fig. 6, A and B): at concentrations of 4 mm, G418 and gentamicin elevated free Fe2+ accumulation 8.7- and 4.2-fold, respectively, whereas the effects of NB84 and NB74 were significantly lower, 2.4- and 1.7-fold, respectively (Fig. 6B).
FIGURE 6.

Effect of AGs on intracellular free iron formation monitored by fluorescence and by EPR spectroscopy. A, HeLa cells were incubated with different AGs as indicated for 5 h, followed by addition of the florescence probe RPA (1 μm). Fluorescence was monitored at 543/584 nm, and after ∼10 min of base-line stabilization, Deferiprone (L1, 200 μm) was added. The time course was monitored for 140 min at 543/585 nm. The data represent means ± S.D., n = 3/point. B, dose-response effects of AGs at concentrations as indicated. The experiments were performed as in A, and the differences of fluorescence relative to untreated cells (ΔFluorescence) at indicated concentration of AG after 120 min are plotted as a function of AG concentration. The data represent means ± S.D., n = 3/point. C, iron EPR signals (g = 4.3) from whole HeLa cells. HeLa cells were incubated with different AGs (4 mm) for 5 h. The samples were incubated with DFO (20 mm) for 10 min and then frozen in liquid nitrogen. The EPR spectra were recorded at 150 K as described under “Experimental Procedures.” D, the data from C are presented as intracellular free iron fold change in exposure to different AGs as indicated versus untreated cells (control). The data represent means ± S.D., n = 3/point. Cont, control; Gent, gentamicin.
These data were further validated by using whole cell EPR method (21), frequently used for quantitative analysis of free iron levels both in bacteria (32) and yeast. This method is based on the incubation of cells, after exposure to AG, with the penetrating iron chelator DFO, which gives an easily detectable sharp EPR signal at g = 4.3. Because DFO completely blocks iron-mediated cell damage, it appears that the iron-DFO signal includes all iron that participates in oxidative chemistry in the cell. Furthermore, DFO failed to extract iron from a variety of iron-containing enzymes, indicating that the signal does not represent iron that had been integrated into proteins. First, we tested the adequacy of this method for mammalian cells by performing a series of control experiments with and without the presence of DFO (data not shown) and found it to be accurate and valid for the purpose. The spectra in Fig. 6C were recorded after incubation of HeLa cells without (control) and with 4 mm AG, followed by incubation with DFO. The observed peak amplitudes were normalized against the control cells (without the AG), and the changes are presented in Fig. 6D. The observed relative impact of different tested AGs on the free iron production into whole cells detected by EPR method correlates well with that detected by using fluorescence probe RPA (Fig. 6B).
Synthetic AGs Exhibit Significantly Lower Ototoxic Potential than Standard AG Drugs
Organ cultures of the early postnatal mouse are frequently used in research on ototoxic agents and their mechanisms. The pattern of toxicity is similar to the in vivo action of aminoglycosides: preferential loss of outer hair cells in a base to apex fashion that is characteristic of aminoglycoside damage to the cochlea in both human and experimental animals (7). For the assessment of a comparative ototoxicity potential, we tested G418, gentamicin, NB84, and NB74 at 3.3 μm concentrations with cochlear explants (Fig. 7A). Staining for F-actin revealed the organized outline of the array of three rows of outer hair cells and one row of inner hair cells in segments from the basal part of the cochlea, the region most sensitive to aminoglycoside damage. Hair cells are almost complete absent with G418, show considerable destruction with gentamicin, but show no significant pathology with NB84 and NB74.
The comparative dose-response curves (Fig. 7B) show that 50% loss of hair cells (LC50Coch) was observed at concentrations of 20 μm for NB84 and 140 μm for NB74. These values compare favorably to G418 (LC50Coch = 0.7 μm) and gentamicin (LC50Coch = 3.5 μm), demonstrating that both NB84 and NB74 have significantly lower ototoxic potential than G418 and gentamicin, with a rank order of G418 > gentamicin ≫ NB84 ≫ NB74. Notably, the observed ∼5-fold higher ototoxic potential of G418 over that of gentamicin correlates with their capacity to inhibit ex vivo mitochondrial protein synthesis (IC50Mito values of 2.3 and 13.3 mm for G418 and gentamicin, respectively; Fig. 2B and Table 1). Because of the solubility limit, the IC50Mito values for NB84 and NB74 could not be determined precisely in ex vivo experiments (for both IC50Mito > 16 mm); consequently, the complete correlation for all the tested AGs cannot be clearly depicted visually (Fig. 9A). However, by plotting the IC50Mito data measured in vitro in isolated mitochondria from HeLa cells (17) against the cochlear toxicity data (LC50Coch), we observed that the increased specificity of action toward mitochondrial ribosome correlates with the increased cochlear toxicity of all the tested AGs (Fig. 9B). A similar correlation was previously observed by us between the IC50Mito data (measured in vitro on the isolated mitochondria from HeLa cells) (17) and the cytotoxicity data of AGs, suggesting that increased inhibition of mitochondrial protein synthesis is associated with the increased cytotoxicity and ototoxicity potential.
FIGURE 9.

Relationship between the ability of a series of AGs (as indicated) to block mitochondrial protein synthesis (IC50Mito values; Table 1) and their ex vivo cochleotoxicity (LC50Coch values; Table 1). A, the relationship with IC50Mito values measured in vitro on intact mitochondria isolated from HeLa cells. B, the relationship with IC50Mito values measured ex vivo in HeLa cells. Genta, gentamicin.
To further substantiate the observed cochlear toxicity data in vivo, as representative examples, we tested the comparative toxicity of G418, neomycin, gentamicin, and NB74 in guinea pigs in vivo (Fig. 7C). AG-induced effects on auditory thresholds were determined by auditory brain stem evoked responses, and pathology was assessed by hair cell counts, analogous to the ex vivo results above. Particularly indicative of ototoxic potential are threshold shifts at 32 kHz, which assess the sensitive base of the cochlea. Gentamicin-induced ototoxicity rises sharply when the animals were exposed for 14 days to 100, 120, and 140 mg of gentamicin/kg of body weight, with a profound threshold shift of 64 dB at 140 mg/kg. In contrast, treatment of animals with NB74 at 140 mg/kg of body weight showed essentially no effect on auditory thresholds, whereas the treatment with only 50 mg of G418/kg of body weight resulted in the death of all animals. The quantitative evaluation of hair cells along the entire length of the cochlea confirmed significantly less damage by NB74 (Fig. 8A) than by gentamicin (Fig. 8B). This in vivo ototoxicity correlates with the ex vivo cochlear toxicity data and thus highlights the role of AG-induced mitochondrial protein synthesis inhibition in the production of oxidative cell and tissue damage, in this case causing irreversible hearing loss.
In addition, the in vivo treatment with NB74 did not cause any apparent systemic toxicity. Parameters of renal function, taken after the final auditory brain stem evoked response measurement, remained normal: albumin, 3.0 + 0.1 g/dl for NB74 treatment versus 3.0 + 0.1 g/dl for saline animals; creatinine, 0.46 + 0.03 mg/dl versus 0.51 + 0.05 mg/dl; and blood urea nitrogen, 17 + 3 mg/dl versus 18 + 3 mg/dl.
One can argue that the observed alleviated ototoxicity of NB84 and NB74 in cochlear explants and that of NB74 in vivo in guinea pigs might be due to a relatively decreased uptake into the cells and hence a decreased accessibility to mitochondrial and/or cytoplasmic ribosomes as compared with G418 and gentamicin. However, the similar trends in the relative inhibition potencies, both in the isolated mitochondria and in the whole cell based assays (Table 1), effectively rules out the possibility of significant differences in accessibility of mitochondrial protein synthesis to these tested compounds. Furthermore, in our previous studies (15, 17), we performed a comparative readthrough activity tests of this set of compounds (G418, gentamicin, NB74, and NB84) in both cell-free and cell-based luciferase assays and found a similar activity trend in both assays: G418 ≥ NB84 > NB74 > gentamicin, which is in accordance with and correlates to their relative inhibition of cytoplasmic protein synthesis (Table 1). Because the readthrough activity is a result of the interaction of the tested compounds with the cytoplasmic ribosomes, these data effectively rule out the possibility of differences in their accessibility of cytoplasmic protein synthesis.
DISCUSSION
The ototoxic side effects of AGs have been known for a long time, and ROS appear causally involved (33). However, the mechanism(s) by which these antibiotics cause ROS formation and subsequent hair cell death are still unclear. Early genetic analysis of patients hypersensitive to AGs had shown that a particular mitochondrial DNA mutation, A1555G, can account for this rare trait, and pointed to a mechanism in which an interference with mitochondrial protein synthesis might be central in AG-induced hair cell death (34). Because the mitochondria are evolved from bacteria, the mitochondrial ribosomes share more structural similarities to bacterial ribosomes than do eukaryotic cytoplasmic ribosomes; this fact further supported the involvement of mitochondrial ribosomes as one of the primary target sites in AGs ototoxicity (11, 35, 36).
Extensive research has shown that exposure to AGs may lead to impairment of RNA translation and inhibition of protein synthesis within mitochondria, resulting in increased formation of ROS, which then promote apoptotic cell death (11, 37–39). This mechanistic postulate is further supported by a recent finding by Matt et al. (12) that apramycin, an AG with reduced ability to inhibit mitochondrial protein synthesis, also shows signs for a reduced ROS accumulation and exhibits reduced ototoxicity.
In contrast, the most recent work by Francis et al. (13) suggests that AGs ototoxicity correlates closely with cytoplasmic protein synthesis inhibition. Using the bioorthogonal noncanonical amino acid tagging method, this group investigated the role of cytoplasmic protein synthesis inhibition in hair cells and demonstrated that the ability of a particular AG to block cytoplasmic protein synthesis and to activate the JNK pathway correlates with its ototoxic potential. Particularly, it was shown that the reduced ototoxicity of apramycin in comparison to that of gentamicin correlates equally well with its relatively reduced ability to block cytoplasmic protein synthesis and JNK activity in hair cells, leaving the question open as to whether mitochondrial or cytoplasmic protein synthesis inhibition predominates to AG-induced ototoxicity.
In the present work, we demonstrate that ototoxicity exerted by a particular AG in cochlear explants and in guinea pigs in vivo correlates primarily with its ability to block mitochondrial rather than cytoplasmic protein synthesis. The impact of a particular AG on mitochondrial protein synthesis was first demonstrated at the whole cell level using HeLa cells (Fig. 2 and Table 1). G418 is the most potent inhibitor of the mitoribosome, followed by gentamicin, and the synthetic derivatives NB84 and NB74 exhibited significantly reduced potency of inhibition. Consistent with their relative ability in blocking mitochondrial protein synthesis, G418 caused a significant reduction in the overall respiratory capacity, whereas NB84 induced significantly less change (Fig. 3). These data demonstrate that: (i) AGs impair the function of mitochondria and, consequently, the overall respiratory capacity of the cell and (ii) increased inhibition of mitoribosome correlates with the increased dysfunction of mitochondria.
The presence of AGs within hair cells leads to increased formation of ROS (7, 40), and the mitigating effect of antioxidants on ototoxicity is perhaps the strongest argument for a causal relationship (41, 42) between ROS formation and auditory sensory cell damage. A common mechanism for the formation of lethal ROS is the Fenton reaction (43):
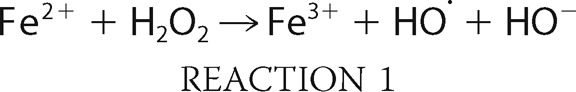 |
Execution of this reaction requires the presence of excess Fe2+ and H2O2 in mitochondria, which can be produced by a direct oxidative damage of [4Fe-4S]-containing dehydratases, including the mitochondrial aconitase, promoted by an excess of superoxide radical (28). For this, the required excess superoxide can be formed via disturbance of energy metabolism caused by AG-induced blockage of mitochondrial protein synthesis and accumulation of abnormal respiratory complexes (11). This scenario is a potential mechanism for an AG-induced oxidative stress and cell apoptosis, and some of its particular steps have previously been independently demonstrated in different model systems (12, 35, 37). To our knowledge, however, a comprehensive study comparing the ability of different AG derivatives in promoting all these steps in one system has not been carried out.
Here, we first show that the AG-induced disturbance of energy metabolism (as reflected by impairment of cell respiration) indeed triggers the production of excess superoxide radical and that the extent of superoxide production correlates directly with the extent of the inhibitory effect of AGs on mitochondrial protein synthesis (Fig. 4); the trend for the induction of superoxide overproduction is G418 > gentamicin > NB84 > NB74 and is essentially identical to that observed for the inhibition of mitochondrial protein synthesis in mammalian cells (Fig. 2), as well as for the impairment of cell respiratory capacity (Fig. 3). Next, by using in-gel activity assay, we show that the elevated superoxide levels cause oxidative damage to cellular aconitases, preferentially to the one of the mitochondrial origin and that the extent of this damage again correlates directly with the extent of the inhibitory effect of AGs on mitochondrial protein synthesis (Fig. 5). Finally, because the reaction of superoxide with the [4F-4S] cluster in the aconitase enzyme results in the formation of free Fe2+ and H2O2 (28), we monitored the accumulation of free Fe2+ in intact human cells (Fig. 6). The exposure of cells to a particular AG induced a dose-dependent accumulation a free Fe2+, the extent of which is in a direct correlation with the inhibitory capacity on mitochondrial protein synthesis with a rank: G418 > gentamicin > NB84 > NB74.
Importantly, our comparative data further indicate that the ability of a particular AG to induce oxidative stress and accumulation of lethal ROS within the mammalian cell correlates with its ototoxicity potential (Fig. 7). First, we show here that the exposure (72 h) of murine organ of Corti explants to clinically relevant levels (3.3 μm) of a particular AG (Fig. 7A) (44) causes a significantly different organ pathology; the basal cochlea shows almost complete destruction with G418, considerable destruction with gentamicin, and no significant pathology with NB84 and NB74. The LC50Coch values derived from dose-response curves covering a 200-fold concentration range were: 0.7, 3.5, 20, and 140 μm for G418, gentamicin, NB84, and NB74, respectively (Fig. 7B and Table 1). These ex vivo data were further corroborated in vivo in the guinea pig (Fig. 7C). The auditory threshold shifts at 32 kHz indicated that the treatment with gentamicin (140 mg of drug/kg of body weight for 14 days) causes a profound hearing loss of 64 dB. In contrast, treatment with NB74 at the same dosing regimen caused essentially no significant effect on auditory thresholds compared with untreated animals. G418 was so highly toxic that a dose of 50 mg of drug/kg of body weight resulted in early death of all three experimental animals. The sum of these results demonstrated that susceptibility of mitochondrial ribosomes to AGs both in isolated mitochondria (Fig. 9B) and at the whole cell level (Fig. 9A), correlates with their relative cochleotoxicity (Table 1). This correlation is further supported by the in vivo ototoxicity results, reinforcing the conclusion that AG-induced cochleotoxicity is mainly determined by the AG activity against mitochondrial ribosomes rather than against cytoplasmic ribosomes. This notion is also in accordance with the recent report on hybrid ribosomes by Böttger and co-workers (12, 45). Furthermore, our findings here are consistent with our previous work in which we showed that the increased specificity of AG toward the mitochondrial ribosome correlates with its increased cytotoxicity (17).
Our studies demonstrate properties of the designer structures NB74 and NB84 that can be exploited in the treatment of human genetic diseases caused by nonsense mutations. In this context, we have recently demonstrated the ability of some of our recent lead compounds of the NB series, including NB74 and NB84, to partially restore protein function in various clinically relevant cellular and animal models of genetic diseases, including cystic fibrosis (46), Rett syndrome (47, 48), Usher syndrome (49–51), and Hurler syndrome (52, 53). These observations, together with the relatively low toxicity of NB74 and NB84, encourage the further testing of these novel designer AGs in animal models and human subjects to maximize the translational impact of our work.
Acknowledgments
We thank Marianna Hainrichson of our group for assistance at the initial steps of mitochondrial protein synthesis assays. We thank Gadi Schuster (Technion) for helping us in mitochondrial protein synthesis assays and Boris Tumanskii (Technion) for assistance in performing EPR experiments. V. B. acknowledges the Ministry of Science and Technology, Israel (Kamea Program).
This work was supported, in whole or in part, by National Institutes of Health Grant 1 R01 GM094792-01 A1 (to T. B. and J. S.). This work was also supported by German-Israeli Foundation Grant G-1048-95.5/2009 (to T. B.).
- AG
- aminoglycoside
- DHE
- dihydroethidium
- RPA
- rhodamine B-[(1,10-phenanthrolin-5-yl)aminocarbonyl]benzyl ester
- DFO
- desferrioxamine mesylate
- PTC
- premature termination codon
- ROS
- reactive oxygen species.
REFERENCES
- 1. Davis B. D. (1987) Mechanism of bactericidal action of aminoglycosides. Microbiol. Rev. 51, 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moazed D., Noller H. F. (1987) Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 327, 389–394 [DOI] [PubMed] [Google Scholar]
- 3. Böttger E. C., Springer B., Prammananan T., Kidan Y., Sander P. (2001) Structural basis for selectivity and toxicity of ribosomal antibiotics. EMBO Rep. 2, 318–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eustice D. C., Wilhelm J. M. (1984) Fidelity of the eukaryotic codon-anticodon interaction. Interference by aminoglycoside antibiotics. Biochemistry 23, 1462–1467 [DOI] [PubMed] [Google Scholar]
- 5. Burke J. F., Mogg A. E. (1985) Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 13, 6265–6272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Keeling K. M., Wang D., Conard S. E., Bedwell D. M. (2012) Suppression of premature termination codons as a therapeutic approach. Crit. Rev. Biochem. Mol. Biol. 47, 444–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Forge A., Schacht J. (2000) Aminoglycoside antibiotics. Audiol. Neurootol. 5, 3–22 [DOI] [PubMed] [Google Scholar]
- 8. Kondo J., Hainrichson M., Nudelman I., Shallom-Shezifi D., Barbieri C. M., Pilch D. S., Westhof E., Baasov T. (2007) Differential selectivity of natural and synthetic aminoglycosides towards the eukaryotic and prokaryotic decoding A sites. ChemBioChem 8, 1700–1709 [DOI] [PubMed] [Google Scholar]
- 9. Hainrichson M., Nudelman I., Baasov T. (2008) Designer aminoglycosides. The race to develop improved antibiotics and compounds for the treatment of human genetic diseases. Org. Biomol. Chem. 6, 227–239 [DOI] [PubMed] [Google Scholar]
- 10. Malik V., Rodino-Klapac L. R., Viollet L., Wall C., King W., Al-Dahhak R., Lewis S., Shilling C. J., Kota J., Serrano-Munuera C., Hayes J., Mahan J. D., Campbell K. J., Banwell B., Dasouki M., Watts V., Sivakumar K., Bien-Willner R., Flanigan K. M., Sahenk Z., Barohn R. J., Walker C. M., Mendell J. R. (2010) Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 67, 771–780 [DOI] [PubMed] [Google Scholar]
- 11. Hobbie S. N., Akshay S., Kalapala S. K., Bruell C. M., Shcherbakov D., Böttger E. C. (2008) Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 20888–20893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matt T., Ng C. L., Lang K., Sha S. H., Akbergenov R., Shcherbakov D., Meyer M., Duscha S., Xie J., Dubbaka S. R., Perez-Fernandez D., Vasella A., Ramakrishnan V., Schacht J., Böttger E. C. (2012) Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-monosubstituted 2-deoxystreptamine apramycin. Proc. Natl. Acad. Sci. U.S.A. 109, 10984–10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Francis S. P., Katz J., Fanning K. D., Harris K. A., Nicholas B. D., Lacy M., Pagana J., Agris P. F., Shin J. B. (2013) A novel role of cytosolic protein synthesis inhibition in aminoglycoside ototoxicity. J. Neurosci. 33, 3079–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gray M. W., Burger G., Lang B. F. (1999) Mitochondrial evolution. Science 283, 1476–1481 [DOI] [PubMed] [Google Scholar]
- 15. Nudelman I., Glikin D., Smolkin B., Hainrichson M., Belakhov V., Baasov T. (2010) Repairing faulty genes by aminoglycosides. Development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg. Med. Chem. 18, 3735–3746 [DOI] [PubMed] [Google Scholar]
- 16. Chernikov V. G., Terekhov S. M., Krokhina T. B., Shishkin S. S., Smirnova T. D., Kalashnikova E. A., Adnoral N. V., Rebrov L. B., Denisov-Nikol'skii Y. I., Bykov V. A. (2003) Comparison of cytotoxicity of aminoglycoside antibiotics using a panel cellular biotest system. Bull. Exp. Biol. Med. 135, 103–105 [DOI] [PubMed] [Google Scholar]
- 17. Kandasamy J., Atia-Glikin D., Shulman E., Shapira K., Shavit M., Belakhov V., Baasov T. (2012) Increased selectivity toward cytoplasmic versus mitochondrial ribosome confers improved efficiency of synthetic aminoglycosides in fixing damaged genes. A strategy for treatment of genetic diseases caused by nonsense mutations. J. Med. Chem. 55, 10630–10643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McKee E. E., Ferguson M., Bentley A. T., Marks T. A. (2006) Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob. Agents Chemother. 50, 2042–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bindokas V. P., Jordán J., Lee C. C., Miller R. J. (1996) Superoxide production in rat hippocampal neurons. Selective imaging with hydroethidine. J. Neurosci. 16, 1324–1336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mukhopadhyay P., Rajesh M., Yoshihiro K., Haskó G., Pacher P. (2007) Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem. Biophys. Res. Commun. 358, 203–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woodmansee A. N., Imlay J. A. (2002) Quantitation of intracellular free iron by electron paramagnetic resonance spectroscopy. Methods Enzymol. 349, 3–9 [DOI] [PubMed] [Google Scholar]
- 22. Lautermann J., McLaren J., Schacht J. (1995) Glutathione protection against gentamicin ototoxicity depends on nutritional status. Hear. Res. 86, 15–24 [DOI] [PubMed] [Google Scholar]
- 23. Chomyn A., Cleeter M. W., Ragan C. I., Riley M., Doolittle R. F., Attardi G. (1986) URF6, last unidentified reading frame of human mtDNA, codes for an NADH dehydrogenase subunit. Science 234, 614–618 [DOI] [PubMed] [Google Scholar]
- 24. Chen Q., Vazquez E. J., Moghaddas S., Hoppel C. L., Lesnefsky E. J. (2003) Production of reactive oxygen species by mitochondria. Central role of complex III. J. Biol. Chem. 278, 36027–36031 [DOI] [PubMed] [Google Scholar]
- 25. Dröse S., Brandt U. (2012) Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 748, 145–169 [DOI] [PubMed] [Google Scholar]
- 26. Turrens J. F. (2003) Mitochondrial formation of reactive oxygen species. J. Physiol. 552, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hausladen A., Fridovich I. (1994) Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem. 269, 29405–29408 [PubMed] [Google Scholar]
- 28. Cantu D., Schaack J., Patel M. (2009) Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS One 4, e7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tong W. H., Rouault T. A. (2006) Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 3, 199–210 [DOI] [PubMed] [Google Scholar]
- 30. Imlay J. A., Chin S. M., Linn S. (1988) Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro. Science 240, 640–642 [DOI] [PubMed] [Google Scholar]
- 31. Petrat F., Weisheit D., Lensen M., de Groot H., Sustmann R., Rauen U. (2002) Selective determination of mitochondrial chelatable iron in viable cells with a new fluorescent sensor. Biochem. J. 362, 137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Y., Imlay J. A. (2013) Cell death from antibiotics without the involvement of reactive oxygen species. Science 339, 1210–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xie J., Talaska A. E., Schacht J. (2011) New developments in aminoglycoside therapy and ototoxicity. Hear. Res. 281, 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hutchin T., Cortopassi G. (1994) Proposed molecular and cellular mechanism for aminoglycoside ototoxicity. Antimicrob. Agents Chemother. 38, 2517–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kalghatgi S., Spina C. S., Costello J. C., Liesa M., Morones-Ramirez J. R., Slomovic S., Molina A., Shirihai O. S., Collins J. J. (2013) Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci. Transl. Med. 5, 192ra185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prezant T. R., Agapian J. V., Bohlman M. C., Bu X., Oztas S., Qiu W. Q., Arnos K. S., Cortopassi G. A., Jaber L., Rotter J. I., Shohat M., Fischelghodsian N. (1993) Mitochondrial ribosomal-RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat. Genet. 4, 289–294 [DOI] [PubMed] [Google Scholar]
- 37. Böttger E. C., Schacht J. (2013) The mitochondrion. A perpetrator of acquired hearing loss. Hear. Res. 303, 12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huth M. E., Ricci A. J., Cheng A. G. (2011) Mechanisms of aminoglycoside ototoxicity and targets of hair cell protection. Int. J. Otolaryngol. 2011, 937861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kurtz D. I. (1974) Fidelity of protein synthesis with chicken embryo mitochondrial and cytoplasmic ribosomes. Biochemistry 13, 572–577 [DOI] [PubMed] [Google Scholar]
- 40. Clerici W. J., Yang L. (1996) Direct effects of intraperilymphatic reactive oxygen species generation on cochlear function. Hear. Res. 101, 14–22 [DOI] [PubMed] [Google Scholar]
- 41. Sha S. H., Qiu J. H., Schacht J. (2006) Aspirin to prevent gentamicin-induced hearing loss. N. Engl. J. Med. 354, 1856–1857 [DOI] [PubMed] [Google Scholar]
- 42. Feldman L., Efrati S., Eviatar E., Abramsohn R., Yarovoy I., Gersch E., Averbukh Z., Weissgarten J. (2007) Gentamicin-induced ototoxicity in hemodialysis patients is ameliorated by N-acetylcysteine. Kidney Int. 72, 359–363 [DOI] [PubMed] [Google Scholar]
- 43. Thomas C., Mackey M. M., Diaz A. A., Cox D. P. (2009) Hydroxyl radical is produced via the Fenton reaction in submitochondrial particles under oxidative stress. Implications for diseases associated with iron accumulation. Redox Rep. 14, 102–108 [DOI] [PubMed] [Google Scholar]
- 44. Shalev M., Kandasamy J., Skalka N., Belakhov V., Rosin-Arbesfeld R., Baasov T. (2013) Development of generic immunoassay for the detection of a series of aminoglycosides with 6′-OH group for the treatment of genetic diseases in biological samples. J. Pharm. Biomed. Anal. 75, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hobbie S. N., Bruell C. M., Akshay S., Kalapala S. K., Shcherbakov D., Böttger E. C. (2008) Mitochondrial deafness alleles confer misreading of the genetic code. Proc. Natl. Acad. Sci. U.S.A. 105, 3244–3249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rowe S. M., Sloane P., Tang L. P., Backer K., Mazur M., Buckley-Lanier J., Nudelman I., Belakhov V., Bebok Z., Schwiebert E., Baasov T., Bedwell D. M. (2011) Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J. Mol. Med. 89, 1149–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brendel C., Belakhov V., Werner H., Wegener E., Gärtner J., Nudelman I., Baasov T., Huppke P. (2011) Readthrough of nonsense mutations in Rett syndrome. Evaluation of novel aminoglycosides and generation of a new mouse model. J. Mol. Med. 89, 389–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vecsler M., Ben Zeev B., Nudelman I., Anikster Y., Simon A. J., Amariglio N., Rechavi G., Baasov T., Gak E. (2011) Ex vivo treatment with a novel synthetic aminoglycoside NB54 in primary fibroblasts from Rett syndrome patients suppresses MECP2 nonsense mutations. PLoS One 6, e20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nudelman I., Rebibo-Sabbah A., Cherniavsky M., Belakhov V., Hainrichson M., Chen F., Schacht J., Pilch D. S., Ben-Yosef T., Baasov T. (2009) Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 52, 2836–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Goldmann T., Rebibo-Sabbah A., Overlack N., Nudelman I., Belakhov V., Baasov T., Ben-Yosef T., Wolfrum U., Nagel-Wolfrum K. (2010) Beneficial read-through of a USH1C nonsense mutation by designed aminoglycoside NB30 in the retina. Invest. Ophthalmol. Vis. Sci. 51, 6671–6680 [DOI] [PubMed] [Google Scholar]
- 51. Goldmann T., Overlack N., Möller F., Belakhov V., van Wyk M., Baasov T., Wolfrum U., Nagel-Wolfrum K. (2012) A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 4, 1186–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang D., Belakhov V., Kandasamy J., Baasov T., Li S. C., Li Y. T., Bedwell D. M., Keeling K. M. (2012) The designer aminoglycoside NB84 significantly reduces glycosaminoglycan accumulation associated with MPS I-H in the Idua-W392X mouse. Mol. Genet. Metab. 105, 116–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Keeling K. M., Wang D., Dai Y., Murugesan S., Chenna B., Clark J., Belakhov V., Kandasamy J., Velu S. E., Baasov T., Bedwell D. M. (2013) Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS One 8, e60478. [DOI] [PMC free article] [PubMed] [Google Scholar]