

Background: During thrombus formation, platelet integrin αIIbβ3 binds fibrin; however, the mechanism of this interaction is unclear.
Results: Mutations of discontinuous negatively charged and aromatic residues in the αIIb β-propeller domain impair fibrin clot retraction and cell adhesion.
Conclusion: Integrin αIIbβ3 has multiple binding sites for fibrin.
Significance: Uncovered recognition specificity of αIIbβ3 for fibrin may be used to select inhibitors of this interaction.
Keywords: Adhesion, Fibrin, Fibrinogen, Integrin, Platelets, Clot Retraction
Abstract
The currently available antithrombotic agents target the interaction of platelet integrin αIIbβ3 (GPIIb-IIIa) with fibrinogen during platelet aggregation. Platelets also bind fibrin formed early during thrombus growth. It was proposed that inhibition of platelet-fibrin interactions may be a necessary and important property of αIIbβ3 antagonists; however, the mechanisms by which αIIbβ3 binds fibrin are uncertain. We have previously identified the γ370–381 sequence (P3) in the γC domain of fibrinogen as the fibrin-specific binding site for αIIbβ3 involved in platelet adhesion and platelet-mediated fibrin clot retraction. In the present study, we have demonstrated that P3 can bind to several discontinuous segments within the αIIb β-propeller domain of αIIbβ3 enriched with negatively charged and aromatic residues. By screening peptide libraries spanning the sequence of the αIIb β-propeller, several sequences were identified as candidate contact sites for P3. Synthetic peptides duplicating these segments inhibited platelet adhesion and clot retraction but not platelet aggregation, supporting the role of these regions in fibrin recognition. Mutant αIIbβ3 receptors in which residues identified as critical for P3 binding were substituted for homologous residues in the I-less integrin αMβ2 exhibited reduced cell adhesion and clot retraction. These residues are different from those that are involved in the coordination of the fibrinogen γ404–411 sequence and from auxiliary sites implicated in binding of soluble fibrinogen. These results map the binding of fibrin to multiple sites in the αIIb β-propeller and further indicate that recognition specificity of αIIbβ3 for fibrin differs from that for soluble fibrinogen.
Introduction
Integrin αIIbβ3, a major membrane protein expressed on the surface of platelets, plays central roles in normal hemostasis and pathological thrombosis. On stimulated platelets, αIIbβ3 serves as a specific receptor for the plasma protein fibrinogen. Fibrinogen binding to activated αIIbβ3 induces platelet aggregation, the essential cellular event in the formation of the primary hemostatic plug. Furthermore, platelets bind fibrin, a product of the enzymatic transformation of soluble fibrinogen into insoluble fibrin, which is formed early and dominates the entire process of thrombus growth (1–3). Because the same molecular pathways mediate pathological thrombus formation, the interaction between αIIbβ3 and fibrinogen has been targeted for antithrombotic therapy (for a review, see Ref. 4). It has also been proposed that inhibition of platelet interactions with fibrin may be a necessary and important property of αIIbβ3 antagonists (5). The initial interaction of soluble fibrinogen with αIIbβ3 occurs via the COOH-terminal sequence in the globular γC domains of fibrinogen with γ404GAKQAGDV411 (γC peptide) providing critical coordination residues that bind to the interface between the αIIb β-propeller domain of the α integrin subunit and the β3 I domain of the β subunit (6). The γC sequence is unique to fibrinogen, and binding of fibrinogen to αIIbβ3 through γC is highly specific (7, 8). Although four integrin recognition RGD sequences are present in fibrinogen and the RGD peptide inhibits αIIbβ3 adhesive reactions and can bind within the same pocket that is occupied by γGAKQAGDV, none of the RGDs in fibrinogen are required for platelet aggregation (9).
Fibrinogen binding to αIIbβ3 is a multistep process: initial reversible contact is followed by irreversible binding such that the bound ligand no longer readily dissociates (10, 11). The binding of fibrinogen to the receptor is accompanied by the alteration of fibrinogen conformation and leads to unmasking of cryptic sequences that potentially can serve as new αIIbβ3-binding sites (12, 13). Also, as the thrombus formation proceeds, the interaction of αIIbβ3 with fibrin engages new contacts that lead to clot retraction. Thus, the overall process of thrombus formation in vivo involves the interactions of αIIbβ3 with different forms of fibrinogen: soluble fibrinogen and an insoluble fibrin(ogen) matrix. The evidence accumulated so far suggests that these interactions involve differential recognition specificity. In contrast to platelet aggregation, the γC sequence is not absolutely required for adhesion to immobilized fibrinogen and fibrin clot retraction (14, 15). Furthermore, RGDs do not contribute to αIIbβ3-mediated clot retraction. Recombinant human fibrinogen in which all RGDs in the Aα chains were mutated and γ408AGDV411 in the γC domains were truncated exhibits delayed but otherwise normal clot retraction (16). Also, neither RGD nor γC peptides inhibit clot tension development during retraction (17), and some anti-αIIbβ3 mAbs inhibit clot retraction but not fibrinogen binding and vice versa (17–19). In addition, fibrinogen from mice in which the γC domain was targeted to delete γ407QAGDV411 does not support platelet aggregation but still mediates normal clot retraction (20). Finally, some αIIbβ3 antagonists have different efficacies in inhibiting clot retraction despite the equivalent antiaggregatory potency (21). Taken together, these data indicate that the site(s) involved in the initial binding of fibrinogen to αIIbβ3 during platelet aggregation is different from those that participate in the interaction of platelets with the insoluble fibrin(ogen) matrix during thrombus growth and clot retraction.
The existence of alternative binding sites in addition to γC and RGD that are involved in binding of fibrinogen to αIIbβ3 was first suggested by Parise et al. (22). They found that αIIbβ3 binding to fibrinogen immobilized on agarose was not inhibited by either RGD or γ400HHLGGAKQAGDV411 (named the H12 peptide). The subsequent studies have localized two sites in the γC domain that may mediate the interaction of αIIbβ3 with insoluble forms of fibrin(ogen). The mutations within the γ316–322 sequence of recombinant fibrinogen diminished platelet aggregation and platelet adhesion under flow (23, 24). We have previously identified the sequence γ370ATWKTRWYSMKK381 (termed P3) as the binding site for αIIbβ3 in adhesion and clot retraction (15, 25). We further found that the mechanism by which αIIbβ3 binds P3 is distinct from the γC recognition. First, P3-mediated adhesion of platelets to fibrinogen fragments lacking the γC residues 406KQAGDV411 does not require their prior stimulation, whereas the engagement of γC by αIIbβ3 is activation-dependent (15). Second, P3 is fibrin-specific in that it is poorly exposed on the surface of intact soluble fibrinogen but becomes fully available after the transformation of fibrinogen to fibrin or after deposition of fibrinogen on various surfaces, including aggregated platelets (12, 26). Third, P3 binding to αIIbβ3 depends on its positively charged residues (25). Because P3 contains no sequences resembling the γ404GAKQAGDV411 or RGD motif, it is reasonable to assume that the binding site(s) for P3 in αIIbβ3 is unlike that utilized by RGD or γC. Here, we performed the binding analyses to demonstrate that αIIbβ3 contains multiple binding sites for P3. Furthermore, using synthetic peptide libraries and mutational analyses, we have localized these sites in the αIIb β-propeller domain of the receptor.
EXPERIMENTAL PROCEDURES
Proteins, Peptides, and Monoclonal Antibodies
Human fibrinogen, thrombin, and plasmin were obtained from Enzyme Research Laboratories (South Bend, IN). The D98 fragment of fibrinogen (98 kDa) was prepared by digestion of fibrinogen with plasmin and purified as described (27). The DD dimer fragment was purified from the cross-linked fibrin as described (28). Recombinant fibrinogen with the binding site for αIIbβ3 in the γC domain of fibrinogen γ408–411 (Fg γ407)2 deleted was produced as described previously (14). The platelet integrin αIIbβ3 was isolated from outdated human platelets (The Blood Center, Hammond, LA) using an affinity chromatography with concanavalin A-agarose (25). The peptides corresponding to the αIIb sequences (αIIb 64–78, αIIb 94–108, αIIb 153–162, αIIb 229–237, αIIb 241–255, αIIb 361–375, αIIb 421–435, and αIIb 10–20) were synthesized by Peptide 2.0 (Chantilly, VA). The peptide duplicating the fibrinogen sequence γ370–381 (P3) was synthesized and labeled with 125I using IODO-GEN (Thermo Scientific Pierce Protein Research Products, Rockford, IL).
The 9-fluorenylmethoxycarbonyl (Fmoc)-protected and pentafluorophenyl-activated amino acids for synthesis of the peptide libraries were purchased from Bachem (King of Prussia, PA). Pentafluorophenyl-activated Trp was obtained from Novabiochem. mAb AP3 against the β3 integrin subunit was isolated from hybridoma cells obtained from ATCC (Manassas, VA). The anti-αIIb mAb sc-51654 was from Santa Cruz Biotechnology (Dallas, TX), mAb LM609 was from Millipore (Billerica, MA), and mAb 7E3 was a gift from Dr. B. Coller. The plasmids pcDNA3.1/Neo and pcDNA3.1/Hygro containing the full-length cDNA encoding the human αIIb and β3 integrin subunits, respectively, were provided by Dr. T. O'Toole. The primers for mutagenesis were obtained from Integrated DNA Technologies, Inc. (Coralville, IA). PfuTurbo DNA polymerase was from Agilent (Santa Clara, CA), and Lipofectamine 2000 was from Invitrogen.
Surface Plasmon Resonance (SPR)
The interaction of αIIbβ3 with P3 was examined using a BIAcore 2000 SPR-based biosensor (Biacore AB, Uppsala, Sweden). Purified αIIbβ3 was coupled to a CM5 sensor chip (Biacore) using the amine coupling kit according to the manufacturer's protocol. The sensor chip was coated to achieve ∼1500 response units, which correspond to ∼62 μm αIIbβ3. Different concentrations of P3 in HBSP buffer (10 mm HEPES buffer with pH 7.4, 150 mm NaCl, and 0.005% v/v Surfactant P20) (Biacore) containing 1 mm CaCl2 and 1 mm MgCl2 were passed over the flow cell at 10 μl/min, and the association between immobilized protein and peptide was detected as the change in the SPR response. All data were corrected for the response obtained using a blank reference flow cell that was activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide and then blocked with ethanolamine. The chip surface was regenerated with 2 m NaCl and 50 mm NaOH followed by re-equilibration with the binding buffer. Experimental data were analyzed using the BIAevaluation 4.1 program supplied with the instrument. The data for the construction of Scatchard plots were obtained from the equilibrium portions of SPR sensorgrams, and the dissociation equilibrium constant (Kd) was estimated by analysis of the binding curve using the steady-state affinity model provided by the same software.
Synthesis of Cellulose-bound Peptide Libraries and Screening for P3 Binding
The αIIb β-propeller (residues 1–451)-derived peptide libraries were prepared by parallel spot synthesis as described (25, 29). Peptides were COOH-terminally attached to cellulose via a (β-Ala)2 spacer and were acetylated NH2-terminally. The cellulose membranes with covalently coupled peptides were incubated for 1 min in methanol and then washed with TBS buffer. After blocking with 1% BSA for 2 h at 22 °C, the membranes were incubated with 10 μg/ml 125I-labeled P3 (105 cpm/ml) in phosphate-buffered saline (PBS) for 3 h at 22 °C. After washing with TBS containing 0.05% Tween 20, the membranes were dried, and P3 binding was detected by autoradiography and analyzed by densitometry as described (25).
Cells and Stable Transfection of Integrin Subunit Constructs
Platelets were collected from fresh aspirin-free human blood in the presence of 2.8 μm prostaglandin E1 and isolated by differential centrifugation followed by gel filtration on Sepharose 2B in divalent cation-free Tyrode's buffer, pH 7.2 containing 0.1% BSA. Human embryonic kidney 293 (HEK293) cells were stably transfected with pcDNA3.1 plasmids with inserted wild-type (WT) αIIb and β3 or mutant αIIb and WT β3 using Lipofectamine 2000 reagent (Invitrogen). After 48 h at 37 °C in 5% CO2, cells were harvested and cultured in medium with 0.5 mg/ml G418 (Invitrogen) and 0.25 mg/ml hygromycin (Invitrogen). After 14 days, surviving cells were collected and sorted. The expression of αIIbβ3 on the surface of the cells was evaluated by FACS analyses using anti-β3 mAb AP3 (10 μg/ml) and a FACSCalibur flow cytometer (BD Biosciences). The αIIbβ3-expressing HEK293 cells were maintained in DMEM/F-12 (Invitrogen) supplemented with 10% FBS, 2 mm glutamine, 15 mm HEPES, 0.25 mg/ml G418, and 0.1 mg/ml hygromycin.
Immunoprecipitation
Cells (5 × 106) were labeled with 100 μg Immunopure Sulfo-NHS-LC-Biotin (Thermo Scientific, Rochester, NY) in 200 μl of PBS for 30 min at 22 °C. The cells were solubilized with a lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1 mm CaCl2, 1 mm PMSF, 100 μg/ml leupeptin, 10 mm benzamidine) for 30 min at 22 °C. The lysates were incubated with 10 μg of normal mouse IgG (Sigma) and 50 μl of Zysorbin-G (Zymed Laboratories Inc., San Francisco, CA) for 2 h at 4 °C. After centrifugation, the supernatant was incubated with anti-αIIb mAb sc-51654 (10 μg) for 2 h at 4 °C. The integrin-mAb complex was captured by incubating with 50 μl of protein A-Sepharose (Amersham Biosciences) for 2 h at 4 °C. The immunoprecipitated proteins were eluted with SDS-polyacrylamide gel electrophoresis loading buffer and analyzed by Western blotting. The Immobilon-P membranes (Millipore, New Bedford, MA) were incubated with streptavidin conjugated to horseradish peroxidase and developed using enhanced SuperSignal chemiluminescent substrate (Pierce).
Fibrin Clot Retraction
Clot retraction assays using isolated platelets were performed as described previously (15). Briefly, the reaction mixture (total volume, 1.0 ml) consisted of 3 × 108 platelets in isotonic HEPES buffer (20 mm HEPES, pH 7.3, 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2, 3.3 mm NaH2PO4 containing 35 mg/ml BSA and 1 mg/ml glucose), 0.25 mg/ml fibrinogen, and 1 mm CaCl2 in glass tubes coated with Sigmacote (Sigma). For clot retraction assays with recombinant Fg γ407, the volume of the reaction mixture was reduced to 0.25 ml. Clot retraction was initiated by adding of 1 unit/ml thrombin at 22 °C. Clot retraction mediated by wild-type HEK293 cells and generated mutant cell lines was performed as described (30). The reaction mixture consisted of 2 × 106 cells in 1 ml containing 10 mm tranexamic acid, 0.25 mg/ml fibrinogen, and 2 mm CaCl2. Clot retraction was initiated by adding of 1 unit/ml thrombin at 37 °C. To block the effect of αvβ3, cells were first preincubated with mAb LM609 (10 μg/ml) for 10 min at 22 °C.
Clot retraction triggered by platelets and HEK293 cells was monitored by taking photographs of clots at several time intervals using a digital camera. The images were scanned, and the areas occupied by clots were calculated using NIH ImageJ software. The effect of the αIIb-derived peptides on platelet-mediated clot retraction was evaluated by determining several parameters (lag phase and IC50) obtained from the kinetic curves of retraction as described previously (15). The value of IC50 is defined as a concentration of the inhibitor that produces 50% of maximal inhibition. The lag phase is defined as the time spanned from the onset of the process until the first visible changes in clot morphology are observed.
Adhesion Assays
The wells of 96-well tissue culture plates (Costar, Cambridge, MA) were coated with the fibrinogen fragment D98 for 3 h at 37 °C or overnight at 4 °C. The wells were postcoated with 1% BSA inactivated at 75 °C for platelet adhesion assays or 1% polyvinyl alcohol for HEK293 cells. Cells were labeled with 10 μm calcein AM (Molecular Probes, Inc., Eugene, OR) for 30 min at 37 °C. Platelets were washed in isotonic HEPES buffer and resuspended at 1 × 108/ml in the same buffer supplemented with 1% BSA, 1 mm MgCl2, and 1 mm CaCl2. Calcein-labeled wild-type and αIIbβ3-expressing HEK293 cells were washed and resuspended in DMEM/F-12 medium at 1 × 105 cells/ml. Aliquots (100 μl) of cells were added to the wells and incubated at 37 °C for 30 min. The nonadherent cells were removed by two washes with PBS, and fluorescence was measured in a fluorescence plate reader (Applied Biosystems, Framingham, MA). The number of adherent cells was determined using the fluorescence of aliquots with a known number of labeled cells.
Platelet Aggregation
Platelet aggregation studies were performed using isolated platelets as described previously (13). 1 × 108/ml platelets were incubated with different concentrations of the αIIb-derived peptides or RGDV peptide for 10 min before the initiation of aggregation. Platelet aggregation with 0.25 mg/ml fibrinogen in the presence of 10 μm ADP and 10 μm epinephrine was measured in a platelet aggregometer (Chronolog Corp., Haverton, PA) at 37 °C with continuous stirring at 1200 rpm. The maximal aggregation, achieved within 5 min after addition of agonists, was determined and expressed as a percentage of aggregation in the absence of peptides. All aggregation assays were conducted within 3 h after venipuncture.
RESULTS
Multiple Binding Sites for the P3 Peptide in αIIbβ3
To characterize the interaction between αIIbβ3 and the P3 peptide, binding studies using SPR were performed. For these analyses, the isolated αIIbβ3 was coupled to the CM5 sensor surface, and the SPR profiles across a range of P3 concentrations flowed over protein surfaces were examined. Fig. 1A demonstrates the sensorgrams for the binding of P3 in the range of 0–45 μm. The maximal responses achieved in the equilibrium portions of the sensorgrams for each P3 concentration were determined, and binding data were used to construct Scatchard plots and calculate the number of binding sites and the equilibrium dissociation constants (Kd). The binding of P3 to αIIbβ3 was saturable at 45 μm (maximal testable concentration) and occurred with Kd of 19.4 ± 2 μm (Fig. 1B). The stoichiometry of P3 binding to αIIbβ3 obtained from extrapolation of the linear parts of Scatchard plots was found to be 7 ± 0.6:1 (Fig. 1B, inset), indicating that the binding of P3 to αIIbβ3 may occur at several sites.
FIGURE 1.

The interaction of αIIbβ3 with the P3 peptide analyzed by SPR. A, representative profiles of the SPR responses for P3 peptide concentrations ranging from 0 to 45 μm binding to purified αIIbβ3 coupled onto a CM5 sensor chip. RU, response units. B, saturable binding curve and Scatchard plot (inset) of P3 binding to αIIbβ3. Req is the response at equilibrium. The abscissa in the Scatchard plot is the ratio of the number of P3 molecules bound per molecule of αIIbβ3. The ratio of bound to free peptide is given on the ordinate.
Screening of the αIIb Propeller-derived Peptide Libraries for P3 Binding
Previous studies demonstrated that, in addition to a well defined set of amino acid residues in the αIIb β-propeller and the β3 I-like domain involved in ligation of RGD and γ404GAKQAGDV411, numerous residues scattered throughout the αIIb β-propeller domain provide contact sites for soluble fibrinogen (31–33). Therefore, we focused on this region of the receptor. To localize the binding sites for P3 in our initial analyses, we screened the cellulose-bound peptide library representing the complete sequence of the αIIb β-propeller. The library consisting of 9-mer peptides with a 3-residue offset spanning the entire αIIb β-propeller (residues 1–451) (Fig. 2A) was synthesized, and the membrane with covalently attached peptides was probed with 125I-labeled P3 peptide. The results of autoradiography revealed that several peptides bound P3 (Fig. 2, B and C). The majority of the P3-binding spots formed clusters; i.e. they were formed by the stretches of overlapping peptides. The eight P3-binding clusters correspond to the following sequences: αIIb 64–78 (cluster 1), αIIb 94–108 (cluster 2), αIIb 151–174 (cluster 3), αIIb 223–243 (cluster 4), αIIb 241–254 (cluster 5), αIIb 259–276 (cluster 6), αIIb 361–375 (cluster 7), and αIIb 421–435 (cluster 8). The observation that the central spots within clusters display the highest intensity suggests that common sequences in these peptides may be responsible for their activities. For example, within cluster 1 formed by three overlapping peptides (spots 22–24), the central spot has the highest P3 binding activity (Fig. 2C). The overlapping parts of these peptides contain the sequence FDL, which potentially may be responsible for P3 binding. In cluster 2, the overlapping peptides contain the common WSD sequence. Other clusters with more than three spots (clusters 4 and 6) may contain more extended recognition sites.
FIGURE 2.

The binding of P3 to the peptide library spanning the sequence of the αIIb β-propeller. A, the amino acid sequence of the of αIIb β-propeller (residues 1–451). The β-strands from the seven blades are marked and underlined. The numbering of residues is shown with a dot, which marks every 10th residue. B, a peptide library consisting of 9-mer peptides derived from the αIIb β-propeller (residues 1–451) was screened for P3 binding. The membrane with covalently attached peptides was incubated with 125I-labeled P3 and subjected to autoradiography. C, clusters of the overlapping sequences selected based on the highest P3 binding activity are shown. The numbers of peptides correspond to the numbering of spots in B.
To ensure that these types of analyses detect integrin-ligand recognition events reported previously (33), we have independently re-examined the binding of the DD fragment to the αIIb β-propeller. This fibrin-derived fragment contains both the H12 and P3 sequences, and thus, its binding to αIIbβ3 may potentially involve both fibrinogen- and fibrin-specific sites. Screening membranes with 125I-DD revealed that, among 16 binders, 10 corresponded to the segments identified by Kamata et al. (33) as critical for binding of soluble fibrinogen to αIIbβ3 (supplemental Fig. 1S). Furthermore, among 40 individual amino acid residues identified as critical in that study (33), 36 were present in the DD binders (supplemental Fig. 1SB). Several residues, including Tyr143, Pro145, Asp163, Leu183, and Thr207, mutations of which were identified in Glanzmann thrombasthenia (34),3 were also found in the DD binders. In addition, in agreement with previous data (33) indicating that mutations in the αIIb 298–304 region (a predicted binding site for fibrinogen (36)) do not affect fibrinogen binding, no interaction of the DD fragment with this segment was detected. Among other DD-binding regions, five segments (αIIb 25–33, αIIb 52–60, αIIb 349–363, αIIb 379–393, and αIIb 442–451) were not previously found to be important for fibrinogen binding, and two (αIIb 400–408 and αIIb 415–423) were not analyzed (33). Thus, the screening experiments with the DD fragment largely supported conclusions drawn from previous mutational analyses using soluble fibrinogen (33) and, thus, substantiated the validity of this mapping strategy. However, they also revealed the differences between the binding of the DD fragment and P3 to the αIIb-derived peptide libraries; i.e. only three regions, αIIb 109–117, αIIb 228–239, and αIIb 256–267, bound both ligands.
The αIIb β-Propeller-derived Peptides Inhibit Platelet-mediated Clot Retraction and Adhesion but Not Platelet Aggregation
To assess whether the P3-binding peptides identified in the screening experiments above can mirror the effect of P3 in functional analyses, the peptides duplicating the sequences of the P3 binders, including αIIb 64–78, αIIb 94–108, αIIb 153–162, αIIb 229–237, αIIb 241–255, αIIb 361–375, and αIIb 421–435, were synthesized and examined for their ability to inhibit clot retraction and platelet adhesion. The peptide duplicating cluster 6 was not synthesized because only 1 residue (Trp262) in its most active part (αIIb 259–271, spots 87–89) is exposed on the surface. Fig. 3 shows the effect of αIIb 241VGEFDGDLNTTEYVV255 as an example. The increasing concentrations of peptide progressively blocked clot retraction (Fig. 3, A and B), and at 200 μm, fibrin clots did not retract after 4 h. The IC50 value calculated from the progress curves of retraction was 72 ± 6 μm (Fig. 3C and Table 1). All other peptides, except αIIb 361–375, were capable of inhibiting clot retraction albeit with different efficiencies. The IC50 value and lag phase determined for the concentration of each peptide allowed the comparison of their potency (Table 1). The αIIb 241–255 was most active followed by αIIb 94–108, αIIb 64–78, αIIb 421–435, αIIb 153–162, and αIIb 229–237. In additional experiments, we examined the effect of selected αIIb-derived peptides used in combinations. As shown in supplemental Fig. 3S, the mixtures of two or four peptides added to platelets at the concentrations to achieve a final concentration equal to that used with each individual peptide did not produce an additive effect in inhibition of clot retraction. This observation is consistent with a model in which the identified αIIb-derived peptides bind the same P3 site in fibrinogen.
FIGURE 3.
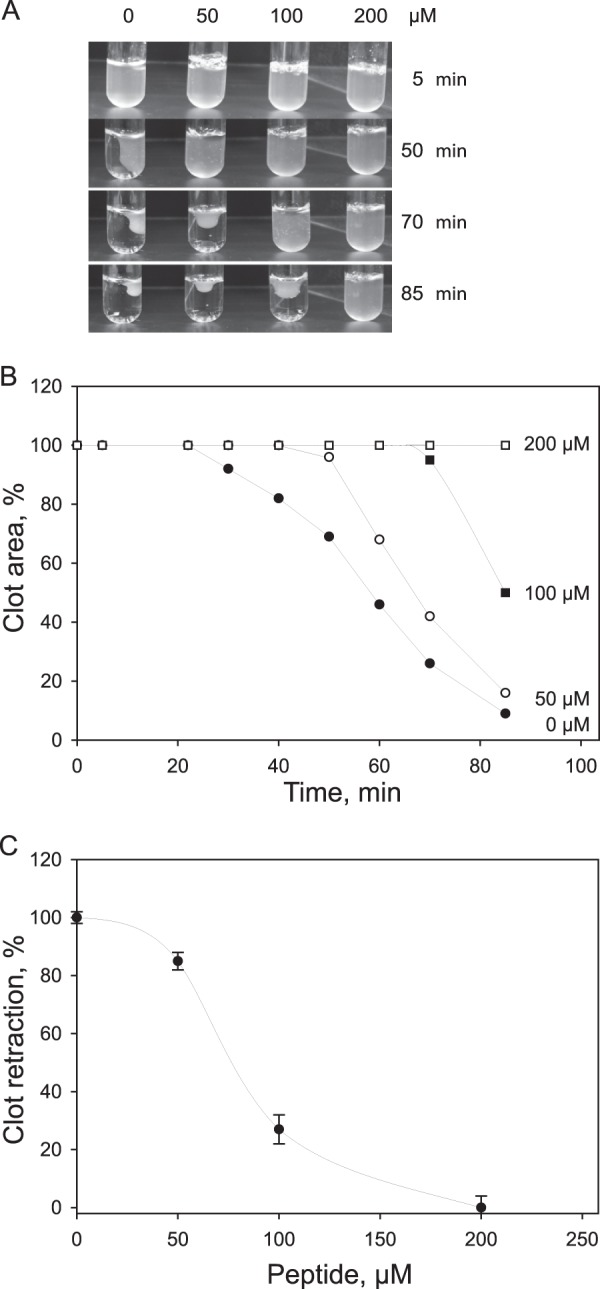
Effect of the αIIb 241–255 peptide on platelet-mediated fibrin clot retraction. A, platelets were mixed with 0.25 mg/ml fibrinogen in isotonic HEPES buffer containing 1 mm CaCl2 and different concentrations of the αIIb 241–255 peptide (0–200 μm), and fibrin clots were formed by adding 1 unit/ml thrombin at 22 °C. Clot retraction was observed by taking photographs at different times (0–85 min). The left lane of tubes (0) shows clot retraction in the absence of peptide. A representative experiment is shown. B, clot areas in each tube were measured from images in A, and a percentage of clot retraction was calculated. Kinetic curves of retraction in the absence (●) or presence of 50 (○), 100 (■), and 200 (□) μm αIIb 241–255 were generated by plotting clot areas versus time. C, dose-dependent inhibition of clot retraction by αIIb 241–255 is shown. Clot retraction in the presence of selected concentrations of the peptide was determined at 85 min. At this time, clot retraction in the absence of peptide was complete (100% retraction), and clot retraction in the presence of each concentration of peptide was at different stages of completion. At 200 μm, no clot retraction was observed. The data shown are means and S.D. (error bars) from three experiments.
TABLE 1.
Effect of the αIIb β-propeller-derived peptides on platelet-mediated fibrin clot retraction and platelet adhesion to fibrinogen or its D98 fragment
The potency of each peptide in adhesion and clot retraction assays was determined as described under “Experimental Procedures.”
| Peptide | Sequence | Adhesion to D98a | Adhesion to Fga | Clot retraction |
|
|---|---|---|---|---|---|
| IC50 | Lag phaseb | ||||
| % | % | μm | min | ||
| Control, no peptide | 100 | 100 | 23 ± 0.5 | ||
| αIIb 64–78 | QCPSLLFDLRDETRN | 57 ± 19 | 74 ± 5.1 | 144 ± 6 | 35 ± 2.0 |
| αIIb 94–108 | GASVVSWSDVIVACA | 43 ± 5 | 81 ± 5.2 | 116 ± 9 | 38 ± 2.0 |
| αIIb 153–162 | RIYVENDFSW | 67 ± 15 | 90 ± 2.2 | 200 ± 6 | 25 ± 1.0 |
| αIIb 229–237 | EYFDGYWGY | 60 ± 20 | 78 ± 2.4 | 262 ± 10 | 26 ± 0.5 |
| αIIb 241–255 | VGEFDGDLNTTEYVV | 71 ± 2 | 68 ± 9.0 | 72 ± 6 | 40 ± 4.0 |
| αIIb 361–375 | APLGDLDRDGYNDIA | 55 ± 6 | 105 ± 9.7 | No inhibition | 23 ± 0.5 |
| αIIb 421–435 | LRGAVDIDDNGYPDL | 55 ± 14 | 73 ± 3.8 | 165 ± 7 | 35 ± 2.0 |
| αIIb 10–20 | FYAGPNGSQFG | 100 | 100 | No inhibition | 23 ± 0.5 |
a The potency of the αIIb-derived peptides expressed as the maximal platelet adhesion to D98 or Fg attained in the presence of 250 μm each peptide.
b Lag phase of clot retraction in the presence of 50 μm each peptide was determined as the time spanned from the onset of the process until the first visible changes in clot morphology are observed.
Previous studies demonstrated that, although mutation of each of the RGD sequences in the Aα chains of recombinant fibrinogens did not affect clot retraction, deletion of the γC sequence 408–411 resulted in the slight delay of retraction when compared with normal fibrinogen (16). However, after the delay, retraction rates and the final size of clots for both mutant Fg γ407 and normal fibrinogen were similar, consistent with a mechanism in which γC may contribute to an initial phase of clot retraction but not to a subsequent step. We compared the potency of two representative αIIb β-propeller-derived peptides, 94–108 and 241–255, to inhibit retraction of clots formed by mutant γ407 and normal fibrinogens. In agreement with published data (16), the retraction rates for both fibrinogens after 20 min were similar (Fig. 4, A and B). The inhibitory effect of peptides on Fg γ407-mediated clot retraction was stronger than that on retraction of clots generated from normal fibrinogen (Fig. 4 and supplemental Fig. 2S; shown for αIIb 241–255), suggesting that peptides mainly block the γC-independent step of clot retraction.
FIGURE 4.
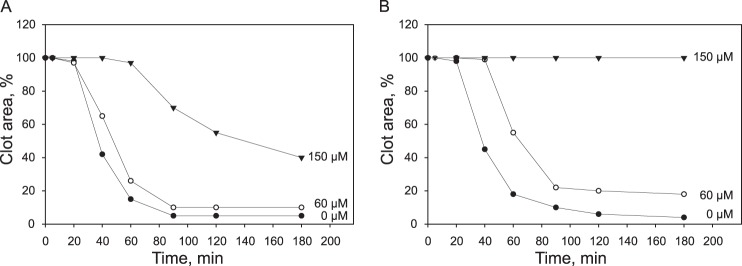
Effect of the αIIb 241–255 peptide on platelet-mediated retraction of clots formed from mutant γ407 and normal recombinant fibrinogens. Platelets (2 × 108/ml) were mixed with 0.25 mg/ml normal fibrinogen (A) and Fg γ407 (B) in isotonic HEPES buffer containing 1 mm CaCl2 and 60 μm (○) or 150 μm (▾) αIIb 241–255 (final volume, 0.25 ml). Fibrin clots were formed by adding 1 unit/ml thrombin at 22 °C, and clot retraction was observed by taking photographs at different times. Kinetic curves of retraction of clots formed from normal fibrinogen in the absence (●) or presence of αIIb 241–255 were generated by plotting clot areas versus time.
The effect of αIIb-derived peptides on platelet adhesion was tested using the immobilized D98 fragment. This fibrinogen fragment lacks the COOH-terminal γ404–411 sequence, and therefore, αIIbβ3-dependent platelet adhesion is mediated solely by P3 (15). The effect of peptide αIIb 94–108 is shown as an example (Fig. 5A). The peptides inhibited adhesion of resting platelets in a dose-dependent manner and at 250 μm produced ∼40–60% inhibition (Fig. 5B and Table 1). The inhibition was specific as αIIb-derived peptide αIIb 10–20 had no activity. As anticipated, the inhibitory effect of peptides on platelet adhesion to intact fibrinogen was less potent apparently due to the presence of strong γ404–411-binding sites (Table 1). In agreement with the clot retraction data, the equimolar mixtures of peptides did not produce additional inhibition of platelet adhesion to D98 (supplemental Fig. 4S).
FIGURE 5.
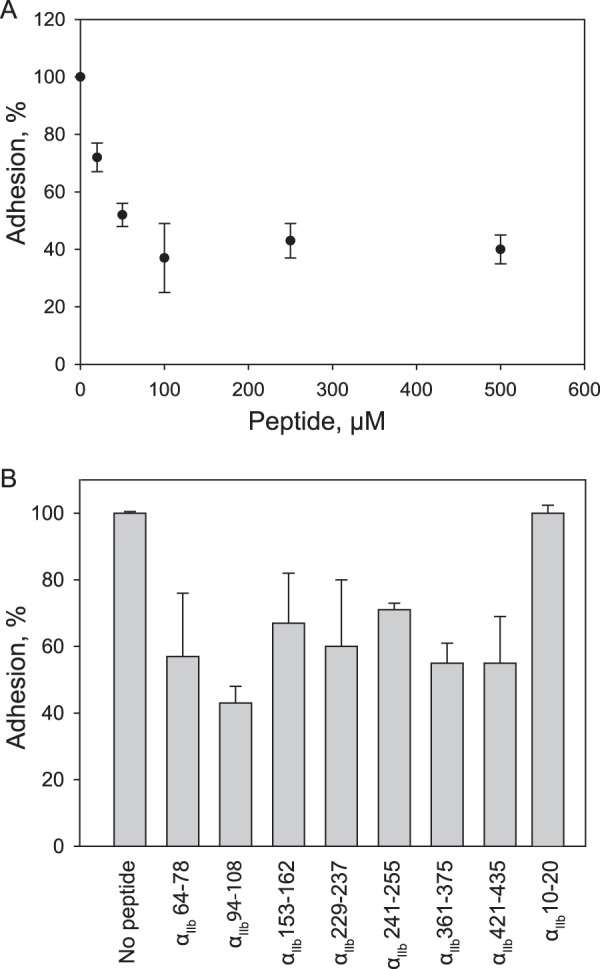
Effect of the αIIb β-propeller-derived peptides on platelet adhesion. Microtiter wells were coated with 10 μg/ml D98 fragment and postcoated with 1% BSA. 50-μl aliquots containing different concentrations of the αIIb-derived peptides in isotonic HEPES buffer were added to the wells for 15 min at 37 °C followed by suspensions of calcein-labeled platelets (1 × 107/50 μl). After 30 min at 37 °C, nonadherent cells were removed, and adhesion was determined. A, a dose-dependent inhibition of adhesion by αIIb 94–108 is shown. B, inhibition of platelet adhesion by each peptide used at 250 μm is shown. The data are expressed as the percentage of adhesion in the absence of peptides. Error bars represent S.D. The results shown are the average of triplicate measurements at each experimental point and are representative of three to five experiments.
To examine whether the αIIb-derived peptides were able to inhibit binding of soluble fibrinogen, we tested their effect on platelet aggregation. Isolated platelets were preincubated with different concentrations of synthetic peptides or RGDV (positive control) for 10 min before the initiation of aggregation. Among selected peptides, αIIb 64–78 inhibited platelet aggregation by ∼30% at 200 μm (supplemental Fig. 5S). No inhibition of aggregation by other peptides was detected. As expected, RGDV blocked aggregation in a dose-dependent manner and completely inhibited it at 100 μm. These observations lend further support to the idea that the sites responsible for binding of soluble fibrinogen are different from those involved in the interaction of αIIbβ3 with insoluble forms of fibrin(ogen).
Localization of Critical Amino Acid Residues in the P3-binding Clusters
With the above data indicating that the αIIb-derived peptides are able to inhibit platelet-mediated clot retraction, we sought to identify critical residues for P3 binding. Additional peptide libraries in which each residue in the identified clusters was mutated to Ala were synthesized and examined for P3 binding (supplemental Fig. 6S). On the basis of densitometry analyses, mutation of Leu69, Phe70, Asp71, Asp74, Glu75 (cluster 1), Trp100, Asp102 (cluster 2), Glu157, Asp159, Glu168, Trp162 (cluster 3), Trp235, Glu243 (cluster 4), Asp247, Glu252, Tyr253 (cluster 5), Asp369 (cluster 7), Asp426, Asp428, and Asp429 (cluster 8) to Ala resulted in a >70% loss of P3 binding (Table 2). Furthermore, mutations of Leu68, Trp162, Tyr166, Asp224, Glu229, Tyr230, Phe231, Tyr237, Asp365, Asp367, Tyr371, Asp373, Gly423, and Asp434 reduced P3 binding by ∼40–70% (Table 2). The finding that the majority of critical residues in the αIIb β-propeller are negatively charged is consistent with the high positive charge of P3. In addition, hydrophobic residues (Leu and Phe) and aromatic residues (Trp and Tyr) were found to contribute to binding. The role of these residues is also highlighted by the finding that not all negatively charged peptides in the αIIb β-propeller scan bound P3 (e.g. 43LGPSQEETG51, 115VLEKTEEAE123, and 295VTDVNGDGR303).
TABLE 2.
Densitometry analyses of P3 binding to the substitutional peptide libraries derived from the active αIIb β-propeller clusters in which each residue was mutated to Ala
| Amino acid | Reactivitya |
|---|---|
| % | |
| Leu68 | 50 |
| Leu69 | 81 |
| Phe70 | 89 |
| Asp71 | 94 |
| Asp74 | 80 |
| Glu75 | 85 |
| Trp100 | 89 |
| Asp102 | 95 |
| Glu157 | 92 |
| Asp159 | 93 |
| Trp162 | 64 |
| Tyr166 | 40 |
| Glu168 | 90 |
| Asp224 | 40 |
| Glu229 | 67 |
| Tyr230 | 36 |
| Phe231 | 41 |
| Trp235 | 72 |
| Tyr237 | 43 |
| Val241 | 31 |
| Glu243 | 89 |
| Asp247 | 70 |
| Glu252 | 89 |
| Tyr 253 | 70 |
| Asp365 | 52 |
| Asp367 | 53 |
| Asp369 | 82 |
| Tyr371 | 43 |
| Asp373 | 65 |
| Gly423 | 43 |
| Asp426 | 75 |
| Asp428 | 70 |
| Asp429 | 73 |
a The loss of reactivity is shown as a percentage of binding to wild-type peptides. Mutations that resulted in the loss of ≥70% binding are in boldface.
The analyses of the three-dimensional structure of the αIIb β-propeller (Protein Data Bank code 2VDO (6)) indicated that not all residues in the P3 binders are exposed on the surface (supplemental Fig. 6S). The following residues that are exposed on the surface of the αIIb β-propeller and when mutated to Ala exhibited significant loss of P3 binding were selected as initial candidates for subsequent analyses: Leu68, Leu69, Phe70, Asp71, Asp74, Glu75 (cluster 1), Trp100, Asp102 (cluster 2), Glu157, Asp159, Trp162 (cluster 3), Asp224, Glu229, Phe231, Trp235 (cluster 4), Glu243, Asp247, Glu252 (cluster 5), Asp365, Asp367, Asp369, Tyr371 (cluster 7), Asp426, Asp428, and Asp429 (cluster 8).
HEK293 Cells Expressing Mutant αIIbβ3 Receptors Support Reduced Cell Adhesion and Clot Retraction
To determine whether the residues identified as critical in the experiments above are involved in the αIIbβ3 function in adhesion and clot retraction, we replaced them with corresponding residues of the I-less αM β-propeller of integrin αMβ2. The rationale for this strategy is based upon the observations that, although both the αIIb and I-less αM β-propeller domains have relatively high homology (30% identical residues and 48% conservative substitutions) and both αIIbβ3 and αMβ2 bind fibrinogen, it is the αMI domain inserted between the second and third repeats of the αM propeller that is responsible for the binding of fibrinogen by αMβ2. The deletion of the αMI domain generates the I-less integrin, which supports ∼10–15% adhesion to fibrinogen compared with wild-type αMβ2 (37). Because of the considerable homology between the two propellers, substitutions of individual residues in αIIb propeller with corresponding αM residues are not expected to alter the conformation of the mutant receptor. The sequence alignment of two domains (supplemental Fig. 7S) revealed that many residues identified as candidates for P3 binding, especially those that are negatively charged, are not conserved between the two domains. For example, the 69LFDLRDE75 sequence (cluster 1; critical residues are underlined) in the αIIb β-propeller is replaced with RLQVPVE in the αM β-propeller. Thus, Leu69, Asp70, and Asp74 were substituted with homologous residues in αM. Glu75, which is identical in both propellers, remained unchanged. In another example, Trp and Asp in αIIb 100WSD102 were replaced with Ser and Pro present in the homologous αM SPP sequence. Residues Asp224, Glu229, and Phe231 in cluster 4 were excluded from the analyses because Asp224 and Phe231 coordinate γLys406 in soluble fibrinogen (6); therefore, it is unlikely that residues in this segment bind P3. In addition, in view of high homology between the αIIb residues in cluster 7 and those in αM, they were left unchanged. The residues that have been mutated are boxed in supplemental Fig. 7S. Wild-type and mutant αIIb subunits were transfected into the HEK293 cells together with wild-type β3 subunit, and stable cell lines carrying mutant integrins were established. Heterodimer association of mutants was evaluated by immunoprecipitation of detergent-lysed surface-labeled cells, and mutant cell lines were sorted to select the expressors with similar levels of integrins (supplemental Fig. 8S).
The effect of point mutations on the αIIbβ3 function was explored using adhesion and clot retraction assays. Fig. 6A shows adhesion of wild-type αIIbβ3-expressing HEK293 cells and W100S/D102P mutant as an example to the increasing concentrations of D98. To exclude the potential effect of αvβ3 on cell adhesion to D98 (38), cells were preincubated with anti-αvβ3 mAb LM609. Adhesion of wild-type and mutants cells reached a maximal level at 10 μg/ml D98, and the effect of mutations was expressed as a percentage of maximal adhesion attained with wild-type expressing cells. As shown in Fig. 6B, adhesion of cells expressing W100S/D102P, E157A/D159S/W162F, W235L, E243L/D247A/E252S, and D248T/D429M mutants was reduced by ∼40–70% of WT cells. Adhesion of cells expressing the L69R/D71Q/D74V triple mutant was not impaired (not shown), and that of cells expressing the quadruple L69R/F70L/D71Q/D74V mutant was reduced by ∼20%. A mutant in which L69R, D71Q, D74V, W100S, D102P, W235L, E243L, E252S, D428T, and D429M (10-residue mutant) were simultaneously mutated supported ∼20% adhesion (Fig. 6B).
FIGURE 6.
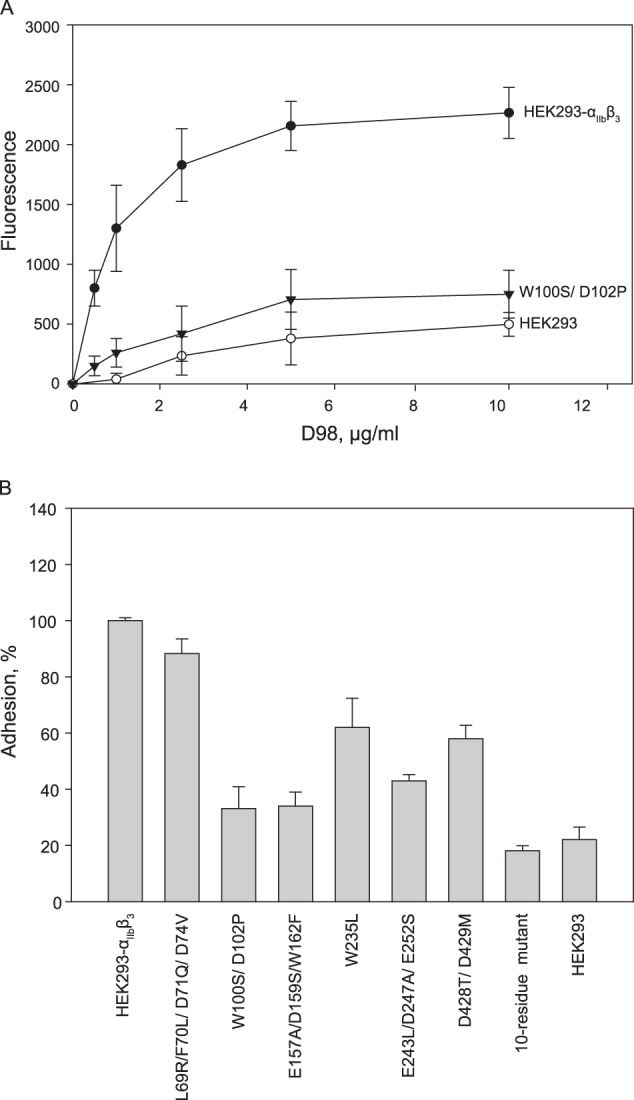
Adhesion of HEK293 cells expressing wild-type and mutant αIIbβ3 receptors. A, adhesion of HEK293 cells expressing the αIIb β-propeller double point mutant W100S/D102P is shown. Microtiter wells were coated with different concentrations of the D98 fragment (0–10 μg/ml). Calcein-labeled HEK293 cells expressing WT (●) or mutant (W100S/D102P) integrin αIIbβ3 (▾) were added to the wells (5 × 104 cells/well) and allowed to adhere for 30 min at 37 °C. After washing of non-adherent cells, cell adhesion was assessed by measuring fluorescence. Adhesion on WT HEK293 cells (○) was also determined. B, adhesion of each mutant to different concentrations of D98 was performed as described in A. Adhesion of each mutant reached the maximal level at 10 μg/ml D98 and is shown as a percentage of maximal adhesion attained with HEK293 cells expressing WT αIIbβ3. The data are means ± S.D. (error bars) of triplicate determinations at experimental data point and are representative of 3–5 experiments.
Further evidence for the role of selected residues in αIIbβ3 function was obtained in clot retraction experiments. These analyses were performed in the presence of mAb LM609 to block αvβ3, which is known to support clot retraction of HEK293 cells transfected with the β3 integrin subunit (30). While cells expressing wild-type αIIbβ3 supported clot retraction, retraction of cells expressing the 10-residue mutant was significantly delayed. Compared with cells bearing wild-type integrins that began to retract clots after ∼38 min, the lag phase for mutant cells was prolonged to ∼100 min (Fig. 7). Furthermore, the final clot size retracted by mutant cells after 4 h was ∼70% compared with 45% retracted by cells expressing wild-type αIIbβ3. Clot retraction of cells expressing E157A/D159S/W162F, E243L/D247A/E252S, and D428T/D429M mutants was also delayed (Table 3); however, final clot size was not significantly different from that retracted by cells expressing wild-type receptor. No significant change in clot retraction by the L69R/F70L/D71Q/D74V mutant cell line was detected, suggesting that the loss of one site may be compensated by another site(s). The activity of the W100S/D102P cell line was not tested. These data indicate that mutations of selected negatively charged and aromatic residues in αIIbβ3 impair the interaction of the receptor with fibrin required for the development of contractile force during clot retraction.
FIGURE 7.

Clot retraction mediated by HEK293 cells expressing mutant αIIbβ3. A, aliquots (2 × 106 cells/ml) of HEK293 cells stably expressing WT (left panel) or mutant αIIbβ3 (right panel)in Tyrode's/HEPES buffer were incubated with 10 mm tranexamic acid, 0.25 mg/ml fibrinogen, and 2 mm CaCl2 in siliconized glass tubes for 5 min at 37 °C. mAb LM609 (10 μg/ml) was added to block αvβ3-mediated clot retraction. Fibrin polymerization and clot retraction were initiated by adding 1 unit/ml thrombin to cell suspensions, and fibrin gels were incubated at 37 °C for the next 220 min. Clot retraction was monitored by taking digital photographs. Representative photographs of retracted gels are shown. B, kinetic curves of retraction by WT αIIbβ3 (●) or mutant receptor carrying 10 point mutations (○) were generated by plotting clot areas versus time. HEK293 cells (▾) do not support clot retraction. The data are expressed as percentage of clot retraction as described under “Experimental Procedures.” Results are representative of three separate experiments.
TABLE 3.
HEK293 cells expressing mutant αIIbβ3 receptors support delayed clot retraction
| Cell line | Lag phase | Final clot volume |
|---|---|---|
| min | % | |
| WT αIIbβ3 | 38 ± 3 | 42 ± 6 |
| E157A/D159S/W162F | 50 ± 2 | 43 ± 3 |
| E243L/D247A/E252S | 90 ± 5 | 50 ± 3 |
| D428T/D429M | 42 ± 3 | 46 ± 5 |
| 10-Residue mutant | 110 ± 8 | 70 ± 4 |
DISCUSSION
In this study, we characterized the interaction of integrin αIIbβ3 with the fibrin-specific peptide P3 (γ370ATWKTRWYSMKK381) and identified residues critical for P3 binding in the αIIb β-propeller domain of the receptor. The analyses of the binding data obtained by SPR indicate that P3 can bind to multiple sites in αIIbβ3. In agreement with this finding, P3 bound to various peptides in the peptide library spanning the sequence of the αIIb β-propeller. The peptides duplicating the P3-binding sequences inhibit clot retraction and platelet adhesion but not platelet aggregation. A common feature of the peptides is their enrichment with negatively charged and aromatic residues. Indeed, substitutions of Leu 69, Phe70, Asp71, Asp74, Trp100, Asp102, Glu157, Asp159, Trp162, Trp235, Glu243, Asp247, Glu252, Asp428, and Asp429 in the αIIb β-propeller blocked adhesion and clot retraction mediated by HEK293 cells expressing mutant receptors. These amino acid residues potentially represent the sites through which αIIbβ3 contacts fibrin fibers during clot retraction.
The amino acid residues identified as critical for P3 binding in the αIIb β-propeller are largely different from those that coordinate the fibrinogen recognition peptide γ404GAKQAGDV411 and situated at the interface between the αIIb β-propeller and β3 I domains (6). One notable exception is αIIb Asp224 and αIIb Phe231, mutation of which in peptides constituting the substitutional peptide library (supplemental Fig. 6S) modestly reduced P3 binding. However, the fact that synthetic peptide 229EYFDGYWGY237, which contains Phe231, does not inhibit platelet aggregation, although it efficiently blocks clot retraction and platelet adhesion, suggests that its activity depends on other residues. Indeed, mutations of Glu229 and Trp235 strongly reduced P3 binding and decreased adhesion of HEK293 cells expressing αIIbβ3 carrying the W235L mutation. The P3-binding residues are also distinct from 40 discontinuous residues in the αIIb β-propeller identified by Takada and co-workers (32, 33) as critical for binding of soluble fibrinogen. Those residues have been mapped to the loops in repeats 2–4 and at the boundary between repeats 4 and 5 of the αIIb β-propeller. The crystal structure of αIIbβ3 in complex with the γC peptide (6) has subsequently revealed that among these residues not only are those that coordinate γC but many residues that form the αIIb cap subdomain, the region of the αIIb β-propeller where epitopes for several function-blocking antibodies were identified (33, 39). In the three-dimensional structure of the αIIb β-propeller, the P3-binding residues Leu69, Phe70, Asp71, Asp74, Trp100, and Asp102 surround Insert 1 of the cap subdomain; Glu157, Asp159, and Trp162 are present within Insert 3 of the cap subdomain; and Trp235 is adjacent to Insert 4 of the cap (Fig. 8). Other residues, including Glu243, Glu252, Asp428, and Asp429, are found within or in the vicinity of the Ca2+-binding sites located in blades W4 and W6 of the β-propeller.
FIGURE 8.
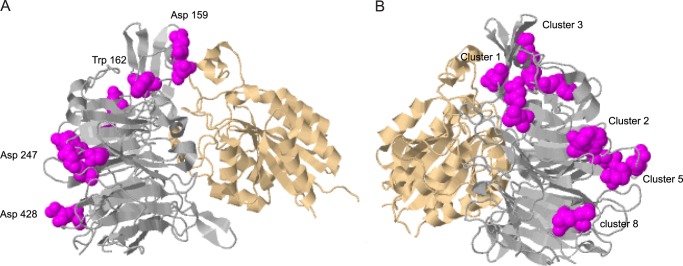
The ribbon model of the αIIbβ3 headpiece based on the crystal structure (Protein Data Bank code 2VDO). The αIIb subunit is shown in gray, and β3 is shown in tan. Amino acid residues identified as critical for the binding of fibrin-specific peptide P3 in the αIIb β-propeller are shown in magenta (selected residues are labeled). Two views (A and B) are rotated relative to each other by 180° about the vertical axis.
Identification of distinct binding sites for the γC peptide, whole fibrinogen, and the P3 peptide is consistent with a mechanism where αIIbβ3 exhibits differential recognition specificity for soluble fibrinogen and the insoluble fibrin matrix in platelet aggregation and clot retraction. The binding of soluble fibrinogen to αIIbβ3 during platelet aggregation was described as a two-step process with potential engagement of different amino acid residues at each step. Accordingly, the binding of soluble fibrinogen to agonist-activated platelets results in the formation of platelet aggregates that can dissociate under certain conditions (10, 40). The underlying mechanism for this phenomenon appears to be a reversible binding of fibrinogen to platelets (40). That initial binding of fibrinogen to αIIbβ3 is mediated by γC 408AGDV411 has been documented in numerous studies that showed that recombinant fibrinogen lacking this sequence does not support platelet aggregation (9, 14) and was recently confirmed with isolated receptor (41). Reversible platelet aggregation is followed by an irreversible step that was proposed to result from the progressive stabilization of the complex between αIIbβ3 and fibrinogen (10, 11). Indeed, biophysical studies revealed the two-step binding mechanism of fibrinogen binding to isolated αIIbβ3 in which fast weak binding is followed by slow strong complex formation (42, 43). Although conclusive data are not available and other mechanisms may account for the irreversible step, it is reasonable to assume that stabilization of the complex between αIIbβ3 and fibrinogen involves the amino acid residues in the αIIb β-propeller cap domain (33, 39). These residues together with those that coordinate the γC sequences may constitute the complete integrin-fibrinogen binding interface formed during platelet aggregation.
In contrast to platelet aggregation, the interaction of αIIbβ3 with fibrin is strongly associated with the development of platelet contractile activity. A requirement for clot retraction is the spatially even distribution of platelets within fibrin because preaggregated platelets do not retract clots (44). On the basis of electron microscopic studies of clots retracting under isometric tension, it has been proposed that close apposition of platelet pseudopods to long fibrin fibers is the major mechanism for the transmission of contractile force (45). As pseudopods crawl and pull on bound fibrin, the fibers become stretched and aligned in the direction of tension. The contacts between the platelet surface and fibrin strands in retracting clots were observed across a ∼15-nm space established by the structures that initially were called “stubs” and presumably represent integrins (46, 47). In contrast, the interplatelet bridges in aggregating platelets span the space of ∼50 nm (48, 49). Another distinction between platelet-fibrin and platelet-fibrinogen interactions is their sensitivity to EDTA: i.e. although EDTA-treated and then washed platelets do not aggregate in response to agonists, they support normal clot retraction (50). From the comparison of these characteristics of platelet aggregation and clot retraction, it is clear that αIIbβ3 is involved in two separable processes utilizing different mechanisms. Therefore, as a receptor for polymerizing fibrin, αIIbβ3 may engage the binding sites that are different from those required for platelet aggregation. Indeed, clot retraction does not absolutely depend on the γC-binding site because recombinant fibrinogen with γ408AGDV411 missing or fibrinogen from mice in which the γC domain was targeted to delete γ407QAGDV411 supports substantial clot retraction (16, 20). The proposal that P3 serves as the binding site for αIIbβ3 in fibrin was put forward based upon the ability of mAb 2G5 directed against P3 and synthetic peptides duplicating the P3 sequence to inhibit platelet-mediated clot retraction (15). It has also been shown that natural and recombinant fibrinogens with mutations in the P3 sequence exhibit delayed clot retraction (25). The direct interaction between P3 and αIIbβ3 was confirmed by affinity chromatography using a P3-agarose affinity matrix (15). Further evidence that αIIbβ3 utilizes distinct recognition specificity toward fibrin comes from the fact that P3 is poorly exposed in soluble fibrinogen and becomes available after its conversion to fibrin (12). Because during clot retraction platelet-fibrin clumps stretch and align fibrin in the direction of tensile force, it is tempting to speculate that P3 may become fully exposed under tension. It should be noted that even though the γC sequence may be exposed on the surface of fibrin fibers it is unlikely that αIIbβ3 is capable of utilizing it. In fibrin, γLys406, one of the amino acid residues that coordinate several residues in αIIb (6), is cross-linked by Factor XIIIa to γGln399/398 on the neighboring molecule and may not be available for αIIbβ3 binding.
The identification of several P3-binding segments in the αIIb β-propeller suggests a model in which a single receptor may form multiple low affinity contacts with a fibrin polymer. The P3-αIIbβ3 interactions are largely electrostatic; i.e. positively charged P3 binds negatively charged amino acid residues in the αIIb β-propeller, although aromatic residues are also involved. The relatively limited requirements for P3 recognition by the αIIb β-propeller domain imply that other sequences in fibrin(ogen) that contain analogous combinations of amino acid residues or have similar physicochemical properties could function as the αIIbβ3-binding sites in fibrin. For example, the second homologous domain in the D region of fibrinogen, βC, contains a sequence highly homologous to P3 (β438MNWKGSWYSMRK449). In contrast to platelet aggregation where each αIIbβ3 binds a single fibrinogen molecule, during clot retraction, the receptor makes contacts with already preformed fibrin. In fibrin polymer, the formation of fibers through the lateral association of protofibrils may cluster the P3 and β438–449 sequences in the interacting γC and βC domains. Although the lateral association of protofibrils that gives rise to mature fibrin has not been defined at the level of atomic resolution, the packing of human and chicken fibrinogens in crystals revealed several interactions that have been proposed to be good candidates for those that occur in fiber formation, including the γC-γC and βC-βC interfaces (51, 52). It is not impossible that each αIIbβ3 molecule may establish multiple contacts with P3 and/or the βC sequences brought together in fibrin. However, an alternative possibility is that P3 and P3-like sequences may indiscriminately engage one of the negatively charged clusters in αIIb β-propeller.
Among seven P3-binding sites, three have been identified in the segments that span the Ca2+-binding sites in blades W4, W6, and W7. Within these peptides, for example in αIIb 241–255 that spans Ca2+-binding site 1 in W4 and includes flanking residues, mutations of several negatively charged residues, including Glu243, Asp247, and Glu252, resulted in the loss of P3 binding (Table 2 and supplemental Fig. 6S). Among these residues, Glu243 and Asp247 provide metal-coordinating side-chain oxygen atoms, whereas Glu252 is outside of the 9-residue Ca2+-coordinating segment (Ref. 53 and references therein). Likewise, although mutations of both Asp428 and Asp429 in site 4 reduced P3 binding, Asp429 is not involved in metal coordination. Mutations of these residues introduced into the whole receptors reduced cell adhesion and clot retraction (Fig. 6 and Table 3). At first glance, this finding might seem to indicate that the loss of Ca2+ resulted in the alteration of the receptor conformation, providing support for previous reports that Ca2+-binding sites are essential for αIIb folding and αIIbβ3 heterodimer formation (53). However, receptors carrying triple and double mutations in the Ca2+-binding sites were assembled normally and expressed on the cell surface at levels comparable with that of WT integrin, suggesting that even though the local conformation may be distorted it does not hamper the biogenesis of integrin. Furthermore, clot retraction is only marginally sensitive to Ca2+ (50).4 Although the dependence on the αIIb negatively charged residues is compatible with the overall P3 cationic nature, other residues in the vicinity of or within the Ca2+-binding sites, including Tyr253, Tyr371, and Gly423, may contribute to binding (supplemental Fig. 6S). Thus, although coordination of calcium by oxygen atoms from side chains of Asp and Glu reduces the negative surface electrostatic potential of this region, other negatively charged and aromatic residues in the vicinity may be involved in P3 binding. One puzzling observation is that no binding of either P3 or the DD fragment was detected to peptides duplicating or overlapping Ca2+-binding site 2 in W5. The lack of DD binding to this region is consistent with previous studies (33) showing that swapping the Ca2+-binding loop W5:1–2 did not affect the binding of soluble fibrinogen; however, the reason for the absence of P3 binding is not clear. Like others, the Ca2+-binding site in W5 contains negatively charged residues forming the consensus motif. The only difference between the residues that constitute this Ca2+-binding site is the absence of aromatic residues present in sites 1, 3, and 4 that may impart additional specificity to P3 recognition. Previous studies showed that the β-propeller Ca2+-binding sites are involved in αIIbβ3-fibrinogen interactions and in binding α4β1 to several ligands (54, 55). Clarifying whether the Ca2+-binding sites also contribute to fibrin recognition and the role of Ca2+ in this process will require further efforts.
It has long been proposed that inhibition of platelet interactions with fibrin may be a necessary and important property of αIIbβ3 antagonists (5). Recent studies using intravital confocal microscopy as well as traditional histological methods have demonstrated the presence of fibrin in early thrombi (1–3), suggesting that αIIbβ3 may interact with fibrin not only in retracting clots that contain large masses of fibrin but also during platelet aggregation. The role of αIIbβ3-fibrin interactions in platelet aggregation is consistent with the finding of the unique GPIb-thrombin pathway that does not depend on the binding of fibrinogen to platelets but instead requires fibrin (56). The relatively simple complementarity between the cationic P3 site and negatively charged residues in the αIIb β-propeller may explain a well known ability of various positively charged compounds, including natural polyamines, to interact with platelets and modulate their responses (57–59). Likewise, negatively charged compounds may affect the αIIbβ3-fibrin interactions. Consistent with this proposal, polyphosphates released from activated platelets (35) inhibit clot retraction.5 Further studies may help to define the reagents that specifically target platelet-fibrin bonds in thrombus formation.
This work was supported, in whole or in part, by National Institutes of Health grants HL63199 and HL107539 (to T. P. U.). This work was also supported by American Heart Association Grant 0835257N (to N. P.).

This article contains supplemental Figs. 1S–8S.
Glanzmann Thrombasthenia Database (2013) sinaicentral.mssm.edu/intranet/research/glanzmann/play?page=nomenclature.
N. P. Podolnikova, unpublished data.
N. P. Podolnikova, T. P. Ugarova, and J. Morrissey, unpublished data.
- Fg
- fibrinogen
- SPR
- surface plasmon resonance.
REFERENCES
- 1. Falati S., Gross P., Merrill-Skoloff G., Furie B. C., Furie B. (2002) Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat. Med. 8, 1175–1181 [DOI] [PubMed] [Google Scholar]
- 2. Hayashi T., Mogami H., Murakami Y., Nakamura T., Kanayama N., Konno H., Urano T. (2008) Real-time analysis of platelet aggregation and procoagulant activity during thrombus formation in vivo. Pflugers Arch. 456, 1239–1251 [DOI] [PubMed] [Google Scholar]
- 3. Hechler B., Nonne C., Eckly A., Magnenat S., Rinckel J. Y., Denis C. V., Freund M., Cazenave J. P., Lanza F., Gachet C. (2010) Arterial thrombosis: relevance of a model with two levels of severity assessed by histologic, ultrastructural and functional characterization. J. Thromb. Haemost. 8, 173–184 [DOI] [PubMed] [Google Scholar]
- 4. Coller B. S., Shattil S. J. (2008) The GPIIb/IIIa (integrin αIIbβ3) odyssey: a technology-driven saga of a receptor with twists, turns, and even bend. Blood 112, 3011–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nurden A. T., Nurden P. (2003) GPIIb/IIIa Antagonists and other anti-integrins. Semin. Vasc. Med. 3, 123–130 [DOI] [PubMed] [Google Scholar]
- 6. Springer T. A., Zhu J., Xiao T. (2008) Structural basis for distinctive recognition of fibrinogen γC peptide by the platelet integrin αIIbβ3. J. Cell Biol. 182, 791–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kloczewiak M., Timmons S., Lukas T. J., Hawiger J. (1984) Platelet receptor recognition site on human fibrinogen. Synthesis and structure-function relationship of peptides corresponding to the carboxy-terminal segment of the γ chain. Biochemistry 23, 1767–1774 [DOI] [PubMed] [Google Scholar]
- 8. Kloczewiak M., Timmons S., Bednarek M. A., Sakon M., Hawiger J. (1989) Platelet receptor recognition domain on the gamma chain of human fibrinogen and its synthetic peptide analogues. Biochemistry 28, 2915–2919 [DOI] [PubMed] [Google Scholar]
- 9. Farrell D. H., Thiagarajan P., Chung D. W., Davie E. W. (1992) Role of fibrinogen α and γ chain sites in platelet aggregation. Proc. Natl. Acad. Sci. U.S.A. 89, 10729–10732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marguerie G. A., Edgington T. S., Plow E. F. (1980) Interaction of fibrinogen with its platelet receptor as part of a multistep reaction in ADP-induced platelet aggregation. J. Biol. Chem. 255, 154–161 [PubMed] [Google Scholar]
- 11. Peerschke E. I., Wainer J. A. (1985) Examination of irreversible platelet fibrinogen interactions. Am. J. Physiol. Cell Physiol. 248, C466–C472 [DOI] [PubMed] [Google Scholar]
- 12. Zamarron C., Ginsberg M. H., Plow E. F. (1991) A receptor-induced binding site in fibrinogen elicited by its interaction with platelet membrane glycoprotein IIb-IIIa. J. Biol. Chem. 266, 16193–16199 [PubMed] [Google Scholar]
- 13. Ugarova T. P., Budzynski A. Z., Shattil S. J., Ruggeri Z. M., Ginsberg M. H., Plow E. F. (1993) Conformational changes in fibrinogen elicited by its interaction with platelet membrane glycoprotein GPIIb-IIIa. J. Biol. Chem. 268, 21080–21087 [PubMed] [Google Scholar]
- 14. Rooney M. M., Parise L. V., Lord S. T. (1996) Dissecting clot retraction and platelet aggregation. J. Biol. Chem. 271, 8553–8555 [DOI] [PubMed] [Google Scholar]
- 15. Podolnikova N. P., Yakubenko V. P., Volkov G. L., Plow E. F., Ugarova T. P. (2003) Identification of a novel binding site for platelet integrins αIIbβ3(GPIIbIIIa) and α5β1 in the γC-domain of fibrinogen. J. Biol. Chem. 278, 32251–32258 [DOI] [PubMed] [Google Scholar]
- 16. Rooney M. M., Farrell D. H., van Hemel B. M., de Groot P. G., Lord S. T. (1998) The contribution of the three hypothesized integrin-binding sites in fibrinogen to platelet-mediated clot retraction. Blood 92, 2374–2381 [PubMed] [Google Scholar]
- 17. Cohen I., Burk D. L., White J. G. (1989) The effect of peptides and monoclonal antibodies that bind to platelet glycoprotein IIb-IIIa complex on the development of clot tension. Blood 73, 1880–1887 [PubMed] [Google Scholar]
- 18. Frelinger A. L., 3rd, Cohen I., Plow E. F., Smith M. A., Roberts J., Lam S. C., Ginsberg M. H. (1990) Selective inhibition of integrin function by antibodies specific for ligand-occupied receptor conformers. J. Biol. Chem. 265, 6346–6352 [PubMed] [Google Scholar]
- 19. Osdoit S., Rosa J.-P. (2001) Fibrin clot retraction by human platelets correlates with αIIbβ3 integrin-dependent protein tyrosine dephosphorylation. J. Biol. Chem. 276, 6703–6710 [DOI] [PubMed] [Google Scholar]
- 20. Holmbäck K., Danton M. J., Suh T. T., Daugherty C. C., Degen J. L. (1996) Impaired platelet aggregation and sustained bleeding in mice lacking the fibrinogen motif bound by integrin αIIbβ3. EMBO J. 15, 5760–5771 [PMC free article] [PubMed] [Google Scholar]
- 21. Hantgan R. R., Ramsamooj P., Taylor R. G., Lewis J. C. (1989) in Fibrinogen 3. Biochemistry, Biological Functions, Gene Regulation and Expression (Mosesson M. W., Amrani D. L., Siebenlist K. R., DiOrio J. P., eds) pp. 203–206, Elsevier Science Ltd., London [Google Scholar]
- 22. Parise L. V., Steiner B., Nannizzi L., Criss A. B., Phillips D. R. (1993) Evidence for novel binding sites on the platelet glycoprotein IIb and IIIa subunits and immobilized fibrinogen. Biochem. J. 289, 445–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hogan K. A., Gorkun O. V., Lounes K. C., Coates A. I., Weisel J. W., Hantgan R. R., Lord S. T. (2000) Recombinant fibrinogen Vlissingen/Frankfurt IV. The depletion of residues 319 and 320 from the γ chain of fibrinogen alters calcium binding, fibrin polymerization, cross-linking, and platelet aggregation. J. Biol. Chem. 275, 17778–17785 [DOI] [PubMed] [Google Scholar]
- 24. Remijn J. A., IJsseldijk M. J., van Hemel B. M., Galanakis D. K., Hogan K. A., Lounes K. C., Lord S. T., Sixma J. J., de Groot P. G. (2002) Reduced platelet adhesion in flowing blood to fibrinogen by alterations in segment γ 316–322, part of the fibrin-specific region. Br. J. Haematol. 117, 650–657 [DOI] [PubMed] [Google Scholar]
- 25. Podolnikova N. P., Gorkun O. V., Loreth R. M., Yee V. C., Lord S. T., Ugarova T. P. (2005) A cluster of basic amino acid residues in the γ370–381 sequence of fibrinogen comprises a binding site for platelet integrin αIIbβ3 (GPIIb/IIIa). Biochemistry 44, 16920–16930 [DOI] [PubMed] [Google Scholar]
- 26. Zamarron C., Ginsberg M. H., Plow E. F. (1990) Monoclonal antibodies specific for a conformationally altered state of fibrinogen. Thromb. Haemost. 64, 41–46 [PubMed] [Google Scholar]
- 27. Lishko V. K., Kudryk B., Yakubenko V. P., Yee V. C., Ugarova T. P. (2002) Regulated unmasking of the cryptic binding site for integrin αMβ2 in the γC-domain of fibrinogen. Biochemistry 41, 12942–12951 [DOI] [PubMed] [Google Scholar]
- 28. Yakovlev S., Makogonenko E., Kurochkina N., Nieuwenhuizen W., Ingham K., Medved L. (2000) Conversion of fibrinogen to fibrin: mechanism of exposure of t-PA and plasminogen binding sites. Biochemistry 39, 15730–15741 [DOI] [PubMed] [Google Scholar]
- 29. Lishko V. K., Podolnikova N. P., Yakubenko V. P., Yakovlev S., Medved L., Yadav S. P., Ugarova T. P. (2004) Multiple binding sites in fibrinogen for integrin αMβ2 (Mac-1). J. Biol. Chem. 279, 44897–44906 [DOI] [PubMed] [Google Scholar]
- 30. Katagiri Y., Hiroyama T., Akamatsu N., Suzuki H., Yamazaki H., Tanoue K. (1995) Involvement of αVβ3 integrin in mediating fibrin gel retraction. J. Biol. Chem. 270, 1785–1790 [DOI] [PubMed] [Google Scholar]
- 31. Kamata T., Irie A., Tokuhira M., Takada Y. (1996) Critical residues of integrin αIIb subunit for binding of αIIbβ3 (glycoprotein IIb-IIIa) to fibrinogen and ligand-mimetic antibodies (PAC-1, OP-G2, and LJ-CP3). J. Biol. Chem. 271, 18610–18615 [DOI] [PubMed] [Google Scholar]
- 32. Puzon-McLaughlin W., Kamata T., Takada Y. (2000) Multiple discontinuous ligand-mimetic antibody binding sites define a ligand binding pocket in integrin αIIbβ3. J. Biol. Chem. 275, 7795–7802 [DOI] [PubMed] [Google Scholar]
- 33. Kamata T., Tieu K. K., Irie A., Springer T. A., Takada Y. (2001) Amino acid residues in the αIIb subunit that are critical for ligand binding to integrin αIIbβ3 are clustered in the β-propeller model. J. Biol. Chem. 276, 44275–44283 [DOI] [PubMed] [Google Scholar]
- 34. Bennett J. S. (2001) Platelet-fibrinogen interactions. Ann. N.Y. Acad. Sci. 936, 340–354 [DOI] [PubMed] [Google Scholar]
- 35. Morrissey J. H. (2012) Polyphosphate multi-tasks. J. Thromb. Haemost. 10, 2313–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. D'Souza S. E., Ginsberg M. H., Matsueda G. R., Plow E. F. (1991) A discrete sequence in a platelet integrin is involved in ligand recognition. Nature 350, 66–68 [DOI] [PubMed] [Google Scholar]
- 37. Yalamanchili P., Lu C., Oxvig C., Springer T. A. (2000) Folding and function of I domain-deleted Mac-1 and lymphocyte function-associated antigen-1. J. Biol. Chem. 275, 21877–21882 [DOI] [PubMed] [Google Scholar]
- 38. Yokoyama K., Zhang X.-P., Medved L., Takada Y. (1999) Specific binding of integrin αVβ3 to the fibrinogen γ and αE chain C-terminal domains. Biochemistry 38, 5872–5877 [DOI] [PubMed] [Google Scholar]
- 39. Xiao T., Takagi J., Coller B. S., Wang J.-H., Springer T. A. (2004) Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 432, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mustard J. F., Packham M. A., Kinlough-Rathbone R. L., Perry D. W., Regoeczi E. (1978) Fibrinogen and ADP-induced platelet aggregation. Blood 52, 453–466 [PubMed] [Google Scholar]
- 41. Hantgan R. R., Stahle M. C., Lord S. T. (2010) Dynamic regulation of fibrinogen: integrin αIIbβ3 binding. Biochemistry 49, 9217–9225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Müller B., Zerwes H. G., Tangemann K., Peter J., Engel J. (1993) Two-step binding mechanism of fibrinogen to αIIbβ3 integrin reconstituted into planar lipid bilayers. J. Biol. Chem. 268, 6800–6808 [PubMed] [Google Scholar]
- 43. Huber W., Hurst J., Schlatter D., Barner R., Hübscher J., Kouns W. C., Steiner B. (1995) Determination of kinetic constants for the interaction between the platelet glycoprotein IIb-IIIa and fibrinogen by means of surface plasmon resonance. Eur. J. Biochem. 227, 647–656 [DOI] [PubMed] [Google Scholar]
- 44. De Gaetano G., Donati M. B., Vermylen J., Verstraete M. (1971) Inhibition of clot retraction by previous in vitro platelet aggregation. Thromb. Diath. Haemorrh. 26, 449–454 [PubMed] [Google Scholar]
- 45. Cohen I., Gerrard J. M., White J. G. (1982) Ultrastructure of clots during isometric contraction. J. Cell Biol. 93, 775–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Behnke O. (1976) in Contractile Systems in Non-Muscle Tissues: Proceedings of the International Symposium, Bressanone, Italy (Perry S. V., Margreth A., Adelstein R. S., eds) pp. 105–115, North-Holland Publishing Co., Amsterdam [Google Scholar]
- 47. Morgenstern E., Korell U., Richter J. (1984) Platelets and fibrin strands during clot retraction. Thromb. Res. 33, 617–623 [DOI] [PubMed] [Google Scholar]
- 48. Skaer R. J., Emmines J. P., Skaer H. B. (1979) The fine structure of cell contacts in platelet aggregation. J. Ultrastruct. Res. 69, 28–42 [DOI] [PubMed] [Google Scholar]
- 49. Morgenstern E., Reimers H. J., Miyashita C. (1984) Ultrastructural studies of the binding sites of fibrinogen on platelet surface during aggregation. Acta Histochem. Suppl. 29, 183–189 [PubMed] [Google Scholar]
- 50. White J. G. (2000) EDTA-induced changes in platelet structure and function: clot retraction. Platelets 11, 49–55 [DOI] [PubMed] [Google Scholar]
- 51. Yang Z., Mochalkin I., Doolittle R. F. (2000) A model of fibrin formation based on crystal structures of fibrinogen and fibrin fragments complexed with synthetic peptides. Proc. Natl. Acad. Sci. U.S.A. 97, 14156–14161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kollman J. M., Pandi L., Sawaya M. R., Riley M., Doolittle R. F. (2009) Crystal structure of human fibrinogen. Biochemistry 48, 3877–3886 [DOI] [PubMed] [Google Scholar]
- 53. Zhang K., Chen J. (2012) The regulation of integrin function by divalent cations. Cell Adh. Migr. 6, 20–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gulino D., Boudignon C., Zhang L. Y., Concord E., Rabiet M. J., Marguerie G. (1992) Ca2+-binding properties of the platelet glycoprotein IIb ligand-interacting domain. J. Biol. Chem. 267, 1001–1007 [PubMed] [Google Scholar]
- 55. Masumoto A., Hemler M. E. (1993) Mutation of putative divalent cation sites in the α4 subunit of the integrin VLA-4: distinct effects on adhesion to CS1/fibronectin, VCAM-1, and invasin. J. Cell Biol. 123, 245–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Soslau G., Class R., Morgan D. A., Foster C., Lord S. T., Marchese P., Ruggeri Z. M. (2001) Unique pathway of thrombin-induced platelet aggregation mediated by glycoprotein Ib. J. Biol. Chem. 276, 21173–21183 [DOI] [PubMed] [Google Scholar]
- 57. Jenkins C. S., Packham M. A., Kinlough-Rathbone R. L., Mustard J. F. (1971) Interactions of polylysine with platelets. Blood 37, 395–412 [PubMed] [Google Scholar]
- 58. Ganguly P., Bradford H. R. (1982) Inhibition of platelet aggregation by primary amines. Evidence for a possible role of membrane-associated calcium. Biochim. Biophys. Acta 714, 192–199 [DOI] [PubMed] [Google Scholar]
- 59. Houston D. S., Gerrard J. M., McCrea J., Glover S., Butler A. M. (1983) The influence of amines on various platelet responses. Biochim. Biophys. Acta 734, 267–273 [DOI] [PubMed] [Google Scholar]