

Background: Histone methylation plays important roles in development and embryonic stem cell (ESC) differentiation.
Results: Inhibition of the H3K9 demethylase JMJD1C directly down-regulated miR-302 and promoted neural differentiation of human ESCs (hESCs).
Conclusion: JMJD1C inhibits neural differentiation of hESCs at least partially by epigenetically sustaining miR-302 expression.
Significance: We provide novel evidence for epigenetic regulation of miR-302 to control neural differentiation of hESCs.
Keywords: Embryonic Stem Cell, Epigenetics, Histone Methylation, MicroRNA, Neurodifferentiation
Abstract
It has been recently reported that the regulatory circuitry formed by OCT4, miR-302, and NR2F2 controls both pluripotency and neural differentiation of human embryonic stem cells (hESCs). We show here that JMJD1C, a histone 3 lysine 9 (H3K9) demethylase expressed in hESCs, directly interacts with this circuitry. hESCs with stable knockdown of JMJD1C remain pluripotent while having reduced miR-302 expression, decreased BMP signaling, and enhanced TGFβ signaling. JMJD1C binds to the miR-302 promoter and reduces H3K9 methylation. Withdrawal of basic fibroblast growth factor (bFGF) from the culture induces neural differentiation of the knockdown, but not the control, cells within 3 days, accompanied by elevated NR2F2 expression. This can be attenuated with miR-302 mimics or an H3K9 methytransferase inhibitor. Together, our findings suggest that JMJD1C represses neural differentiation of hESCs at least partially by epigenetically sustaining miR-302 expression and that JMJD1C knockdown is sufficient to trigger neural differentiation upon withdrawal of exogenous bFGF.
Introduction
The development of early embryos and differentiation of embryonic stem cells (ESCs)4 are both subject to dynamic epigenetic regulation, including DNA methylation and histone modification (e.g. methylation and acetylation). Histone methylation is associated with either transcriptional activation (often in euchromatin) or repression (often in heterochromatin) and regulated by two groups of antagonistic enzymes, histone methyltransferases and histone demethylases. Hypermethylated histone 3 lysine 9 (H3K9) is generally a repressive mark on target genes and plays a pivotal role in embryogenesis, cell differentiation, carcinogenesis, etc. (1–3). For example, the H3K9 demethylases Jmjd1a and Jmjd2c can maintain mouse ESC pluripotency by regulating the expression of Tcl1 and Nanog, respectively, and their loss of function leads to inappropriate differentiation (4). It has been shown that Jmjd2c is required for expression of pluripotency genes, and depletion of Jmjd2c causes developmental arrest before the blastocyst stage (5). In addition, the H3K36 demethylase Jhdm1b has been shown to enhance somatic cell reprogramming in mice (6). However, little is known about the role of histone demethylation in pluripotency and differentiation of human ESCs (hESCs).
While mining our transcriptomic databases for hESCs, we noticed that expression of a histone demethylase, JMJD1C (also named TRIP8 or KDM3C), is highly correlated to the pluripotent state. JMJD1C is a H3K9 demethylase, which has been shown to control the balance of histone methylation status through interaction with histone methyltransferases and WHISTLE for transcriptional regulation (7). JMJD1C is abundantly expressed in hESCs and declines during hESC differentiation in response to either activation of BMP signaling (8) or inhibition of TGFβ and/or FGF signaling (9). Furthermore, comparative genomics analysis has shown that the JMJD1C promoter contains a putative OCT-binding site, suggesting that JMJD1C may play a role in the regulatory circuit of pluripotency as a downstream target gene of OCT4 (10).
Here we document that JMJD1C is indeed a part of the regulatory circuit that prevents hESCs from neural differentiation by demethylating H3K9 at the locus of a pluripotency-associated microRNA, the miR-302/367 gene cluster. hESCs with stable knockdown (KD) of JMJD1C remained pluripotent and could be expanded for a prolonged time. Upon withdrawal of exogenous bFGF (an inhibitor of neural initiation) from the cell culture, the JMJD1C KD hESCs differentiated into neural progenitors in 3 days, much faster than wild-type hESCs differentiated in similar conditions with inhibition of FGF signaling (11). It has been reported that both OCT4 and miR-302 act as neural repressors; during neural differentiation of hESCs, reduction of OCT4 and miR-302 triggers the transcription of the neural inducing gene NR2F2, and NR2F2 in turn represses OCT4 expression and sustains the activation of PAX6 and other neural genes (12). We demonstrate that this precocious differentiation of the JMJD1C KD cells is associated with down-regulation of miR-302 and that JMJD1C prevents neural induction in hESCs at least in part by epigenetic modification to promote expression of miR-302. In addition, it appears that JMJD1C also represses neural differentiation by modulating TGFβ and BMP signaling.
EXPERIMENTAL PROCEDURES
hESC Culture and Neural Differentiation
hESC lines H9 (13) and CT2 (14) cells (National Institutes of Health registration 0062 and 0069, respectively) were cultured in either the medium conditioned on mouse embryonic fibroblasts (15) or the defined medium mTeSR1 (T1) (16). For culture in the mouse embryonic fibroblast-conditioned medium (CM), hESCs were split and maintained on plates coated with Matrigel (BD Biosciences) in Dulbecco's modified Eagle's medium/F12 containing 20% knock-out serum replacer, 0.1 mm nonessential amino acids, 1 mm l-glutamine (all from Invitrogen), and 0.1 mm β-mercaptoethanol (Sigma-Aldrich), which had been conditioned on mouse embryonic fibroblasts and then supplemented with 4 ng/ml bFGF (Millipore). For culture in the T1 medium, hESCs were split and maintained on Matrigel-coated plates containing T1 medium. Neural differentiation was induced in hESCs through either embryoid body (three-dimensional) formation (17) or a monolayer (two-dimensional) culture in T1 depleted of bFGF (T1/F0) and supplemented with 10 μm SB431542 (Stemgent), an inhibitor of TGFβ signaling, and 0.5 μm dorsomorphin (Stemgent), an inhibitor of BMP signaling, similar to previously reported methods (11, 18).
The inhibitors were not added to T1/F0 for neural differentiation of the JMJD1C KD cells. The cells were cultured in the designated medium for up to 6 days with daily refreshing of the medium, followed by analyses of neural rosette formation and expression of the pluripotency marker OCT4 and neural markers NR2F2, PAX6, and MAP2.
Establishment of JMJD1C Knockdown Cell Lines Using Lentiviral shRNA
To knock down JMJD1C in hESCs, we used shRNAs delivered by a lentiviral vector (Thermo Scientific). A cassette with a U6 promoter-driven puromycin-resistant gene was contained in the vector for drug selection. The vector was packaged in 293T cells, and the viral particles were harvested at 48 h to transduce H9 and CT2 hESC lines cultured in CM. The cells were treated with 0.5 μm puromycin starting on day 3 post-transduction to select transduced clones. We generated stable JMJD1C KD cell clones from both H9 and CT2 hESC lines and also generated mismatch (MM) negative control clones using sequence-mismatched shRNA. KD was confirmed through RT-PCR and Western blotting. Handling of all recombinant DNA in this study followed National Institutes of Health guidelines.
RT-PCR and Quantitative PCR (qPCR)
RNA was isolated from cells using TRIzol reagent (Invitrogen), and cDNA was synthesized from the RNA using Superscript II (Invitrogen), according to the manufacturer's instructions. Gene expression was assessed through PCR with primers for specific genes (Table 2) under the following conditions: an initial 5-min denaturation at 95 °C; followed by 30 cycles of 45 s of denaturation at 95 °C, 45 s of annealing at 55 °C, and 45 s of extension at 72 °C; completed with a final extension at 72 °C for 10 min. The PCRs were spread on TBE gel via electrophoresis, and DNA bands were visualized via ethidium bromide staining.
TABLE 2.
Primers used in this study
| Primers |
||
|---|---|---|
| Forward | Reverse | |
| RT-PCR and -qPCR | ||
| JMJD1C | CCAAAATTGCCTGGAAAAGA | ACATCACTCTCTGGGCTGCT |
| OCT4 | GACAACAATGAGAATCTTCAGGAGA | CTGGCGCCGGTTACAGAACCA |
| NODAL | GGGCAAGAGGCACCGTCGACATCA | GGGACTCGGTGGGGCTGGTAACGTT |
| PAX6 | TGTCCAACGGATGTGTGAGT | TTTCCCAAGCAAAGATGGAC |
| NR2F2 | GCCATAGTCCTGTTCACCTCA | AATCTCGTCGGCTGGTTG |
| TGFBR2 | GCTACTCCATGGCTCTGGT | ATCTGGATGCCCTGGTGGTT |
| Pri-miR-302 | GGAGAACACGAATCTTTGGG | GAAAGAGTATTTGGGAAATGT |
| GAPDH | TCTTTTGCGTCGCCAGCCGAG | TCCCGTTCTCAGCCTTGACGGT |
| ChIP-PCR and -qPCR | ||
| Region 1 | GAAACACAATGCCTTTCTCG | TCTGCTTGTTTGATGGCATAG |
| Region 2 | TCTTGTGGTAATGGTTTTAGCTG | GGTGGGTCTGGATCCTTTTT |
| Region 3 | CAAAAGGCCCTTGTAATGATTC | GGGTAAAAGGCAGGGACTTC |
| Region 4 | GAACACGCTAACCTCATTTG | GACACCTCCACTGAAACATG |
| Region 5 | CTTGGAAGCTTCTGATGC | CAGCTCTGAAGATGCAGAATC |
For qPCR, the RT samples were analyzed by using Power SYBR® Green (Applied Biosystems) on the AB 7300 real-time PCR system (Applied Biosystems) using the same primers as for PCR (Table 2). The following conditions were used in qPCR: 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. GAPDH was tested as an endogenous reference to calculate the relative expression level of target genes. The results are displayed as relative mRNA levels.
Luciferase Reporter Assay
A JMJD1C promoter-responsive firefly luciferase reporter plasmid, pJMJD1C-Luc, was constructed by inserting 359-bp JMJD1C variant-2 promoter (including exon 1B) (10) into the pGL4.10 Basic vector (Promega). To test the role of OCT binding in regulation of the JMJD1C promoter activity, we generated a reporter vector with the predicted OCT binding site ATTTGCAT on the promoter deleted or mutated to CTGTTCCT, by using the Site-directed Mutagenesis Kit (Stratagene). All of the three constructed vectors were confirmed by sequencing.
The luciferase assay was performed by transfecting hESCs using FuGENE HD (Roche Applied Science) on day 1 with each of the reporter vectors together with a trace amount (one-twentieth of the DNA amount for the test vector) of pRL-tk plasmid (Promega) to express Renilla luciferase as an internal control. The cells were harvested on day 3, and the lysates were assayed for both the firefly and Renilla luciferase activities using the Dual-Luciferase Reporter Assay System (Promega) on a 3010 Luminometer (BD Biosciences). The ratio of relative luciferase/Renilla activities was displayed as relative luciferase activity.
Western Blotting
Protein samples were separated on SDS-polyacrylamide gels (Bio-Rad) and then transferred to PVDF membranes (Bio-Rad). The membranes were blocked with 5% nonfat milk and incubated with mouse or rabbit antibodies against SMAD2/3, phosphorylated SMAD1/5/8, phosphorylated SMAD2/3 (with phosphorylation at the two serine residuals in the C termini of these SMADs), β-ACTIN (Abcam), JMJD1C (Millipore), and SMAD1/5/8 (Cell Signaling) at 4 °C overnight, followed by washing with TBS-T solution three times. Membranes were incubated in horseradish peroxidase (HRP)-labeled donkey anti-mouse or anti-rabbit secondary antibody (Santa Cruz Biotechnology, Inc.) for 30 min and followed by TBS-T washing three times. Finally, target protein bands on the membranes were visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore).
Microarray and PluriTest
hESC lines H9 and CT2 and their derivative KD and MM cell lines were cultured on Matrigel-coated plates in CM as described above. Total RNA was isolated from each cell line (in duplicate) using the RNAqueous-4PCR kit (Invitrogen). The TotalPrepTM RNA amplification kit (Illumina) was used for generating biotinylated, amplified RNA for hybridization with Illumina Sentrix Arrays on the Illumina Human HT-12 v4 Beadchip. The results were analyzed using GenomeStudio software (Illumina). A PluriTest was conducted using the corresponding software available online (19).
Flow Cytometry Analysis
hESCs were cultured in T1 or T1/F0 with or without treatment for 4 days. Cell clumps were harvested by using Accutase (Innovative Cell Technologies Inc.) and were gently dissociated into single cells. The cells were washed with a FACS buffer, which contained PBS, 0.1% NaN3, and 2% FBS. After being fixed and permeabilized with ice-cold 0.1% paraformaldehyde for 10 min and 90% methanol for 30 min, the cells were incubated overnight with anti-PAX6 antibody at 1:1000 (DSHB) or a normal mouse IgG as a negative control. The cells were then washed and incubated with Alexa 488-conjugated goat anti-mouse IgG antibody. For OCT4 staining, the cells were further incubated with anti-OCT4 antibody conjugated with Alexa 647 at 1:200 (BD Bioscience). The cells were analyzed on a BD Biosciences FACS LSR-II instrument, and the PAX6+ cell ratio was calculated by using the Flowjo software (Tree Star).
Immunostaining
hESCs cultured in T1 or T1/F0 medium for 5 days were fixed with 4% paraformaldehyde for 10 min, followed by incubation in PBS containing 5% rabbit serum and 0.4% Triton X-100 for blocking and permeabilization, respectively. PBS containing 0.5% Tween 20 (PBS-T) was used to dilute antibodies and wash the cells in the following procedures. The cells were incubated with mouse antibodies against PAX6 (DSHB), OCT4 (Santa Cruz Biotechnology, Inc.), or NANOG (Abcam). The cell nuclei were counterstained with DAPI.
Northern Blotting for the miR-302 Isoforms
A non-radioactive method for detection of small RNAs (20) was used. Briefly, total RNA from hESCs was loaded onto 15% SequaGel (National Diagnostics), electrophoresed, and transferred to nylon membranes at 10–15 V (90 min) using a Trans-Blot SD semidry transfer cell (Bio-Rad). Membranes were cross-linked to the RNA at 60 °C for 1–2 h using freshly prepared cross-linking reagent Doc-S. Probes were labeled with the non-radioactive digoxigenin, using the End Tailing Kit (Roche Applied Science). We used miRCURY LNATM detection probes EXQON 38134-01, 38136-01, 38138-01, and 38140-01 to detect miR-302a, -b, -c, and -d, respectively, and EXQON 99002-01 to detect small nuclear RNA U6 as a loading control (all from Exiqon). Following hybridization, the membranes were washed at 37 °C twice using a low stringency buffer solution (2× SSC, 0.1% SDS) and a high stringency buffer solution (0.1× SSC, 0.1% SDS), for 5 and 10 min, respectively. Alkaline phosphatase-conjugated anti-digoxigenin antibody was used for visualization of the binding. Photoemissions were visualized by using an x-ray developer SRX-101A (Konica).
Chromatin Immunoprecipitation (ChIP)-PCR and -qPCR
ChIP was performed using the EZ-Magna ChIPTM G chromatin immunoprecipitation kit (Millipore) by following the manufacturer's instructions. The antibodies used included ChIP grade anti-JMJD1C antibody (Millipore), ChIPAb+ monomethyl-histone H3(Lys9), and ChIPAb+ dimethyl-histone H3(Lys9) (Millipore). DNA was released via decross-linking from chromatin fragments pulled down with each specific antibody and subjected to PCR or qPCR analysis for enrichment of regions around the transcription start site at the promoter of the miR-302/367 cluster using primer pairs listed in Table 2. The Power SYBR®Green (Applied Biosystems) was used for QPCR on the AB 7300 real-time PCR system. The following PCR conditions were used: 10 min at 95 °C and 40 cycles of 30 s at 95 °C and 1 min at 60 °C.
Transfection of hESCs with miR-302 Mimics and Inhibitors
To rescue miR-302 function in the KD hESCs, we transfected the KD cells with a mimic for miR-302b or miR-302c or a negative control mimic (Dharmacon) at 50 nm using DharmaFECT1 (Dharmacon) two times on days 0 and 2. To repress miR-302 function in MM hESCs, we transfected the MM hESCs with an inhibitor for miR-302b or miR-302c or a negative control inhibitor (Dharmacon) at 25 nm, also using DharmaFECT1 two times on days 0 and 2. The cells were then cultured in T1 or T1/F0 for 5 days for differentiation. The ratio of PAX6+ cells was measured via flow cytometry.
Statistical Analyses
Data are presented as mean ± S.D. Student's t test was used for statistical analysis. The percentage data were arcsine-transformed prior to analysis. p < 0.05 was considered significant, and p < 0.01 was considered highly significant.
RESULTS
JMJD1C Declines during Neural Differentiation of hESCs
As mentioned above, among the JMJD family members, JMJD1C has been shown to be expressed the highest in hESCs and decline concomitantly with the differentiation of hESCs in response to activation of BMP signaling (8) or inhibition of TGFβ and/or FGF signaling (9). We asked whether or not JMJD1C expression also declines during neural differentiation. It has been known that dual inhibition of TGFβ and BMP signaling (18) and inhibition of FGF signaling synergistically induce neural initiation in hESCs (11). We mimicked these conditions by culturing hESCs in T1/F0 (which lacked external bFGF) supplemented with 10 μm SB431542 (which inhibits TGFβ signaling) and 0.5 μm dorsomorphin (which inhibits BMP signaling). We found that JMJD1C expression decreased, whereas expression of the neural inducing gene NR2F2 increased in hESCs cultured in these conditions for up to 3 days, as determined by RT-PCR (Fig. 1A). Similar results were observed when neural differentiation of hESCs was induced via formation of embryoid bodies (17) (Fig. 1B) and confirmed by Western blotting (Fig. 1C). The association of reduced JMJD1C expression with hESC differentiation prompted us to ask whether or not JMJD1C expression is sustained by pluripotency factors, such as OCT4.
FIGURE 1.

Expression of JMJD1C declines during neural differentiation of hESCs and relies on the OCT-binding site at its promoter. A and B, RT-PCR analysis for gene expression in undifferentiated (Undiff.) H9 hESCs and H9 cells induced for neural differentiation in a two-dimensional culture in T1/F0 supplemented with 10 μm SB431542 (SB) and 0.5 μm dorsomorphin for up to 3 days (A) or a three-dimensional culture through embryoid body (EB) formation for up to 6 days (B). C, Western blotting for JMJD1C protein level in undifferentiated H9 hESCs and day 6 embryoid body differentiated from H9 cells. Quantitation of the signals showed that the ratio of the JMJD1C signal for the embryoid body to that for the hESCs was 0.6, after normalization by the β-ACTIN signals. D, H9 hESCs were transfected with a luciferase reporter plasmid driven by wild-type (WT) JMJD1C promoter or by the promoter with the OCT-binding site mutated (Mut) or deleted (Del). The reporter activity was assayed on day 3 post-transfection, normalized by the Renilla luciferase control, and displayed as mean ± S.D. (error bars) from three independent experiments. *, p < 0.05 compared with WT.
As stated above, the OCT-binding site in the JMJD1C promoter indicates that JMJD1C expression may be directly regulated by OCT4 (10). To address this possibility, we used the luciferase reporter driven by the JMJD1C promoter (WT) or the promoter with the OCT-binding site mutated (Mut) or deleted (Del). Indeed, luciferase activity was reduced to almost half in hESCs transfected with the reporter driven by the Mut or Del promoter, compared with that with the WT promoter (Fig. 1D). Thus, JMJD1C expression may be sustained by OCT4 via direct binding to the JMJD1C promoter.
JMJD1C Knockdown Promotes Neural Differentiation
The declined expression of JMJD1C during hESC differentiation prompted us to ask whether or not this protein is involved in maintenance of pluripotency. We generated stable JMJD1C KD clones from two hESC lines H9 and CT2 using five lentiviral shRNA vectors coded KD1 to -5, respectively (Table 1), and also MM control clones from both hESC lines. Of the five KD shRNA vectors, KD4 caused the most effective KD on JMJD1C in both hESC lines, and the resultant cell lines are named H9-KD and CT2-KD, respectively. JMJD1C expression decreased about 50% in both KD cell lines at mRNA (Fig. 2A) and protein (Fig. 2B) levels, in comparison with that in the MM cells. KD3 also caused remarkable knockdown of JMJD1C (data not shown) and resulted in another stable line H9-KD-v2.
TABLE 1.
Lentiviral human JMJD1C shRNAs (Thermo Scientific) tested in hESCs
| Catalog no. | Sequence | Code |
|---|---|---|
| TRCN0000107563 | ATGATCCGGCTAGTAAATGGG | KD1 |
| TRCN0000107564 | AAATCCTGGCAAATGAGAGGC | KD2 |
| TRCN0000107560 | ATAACATTGAATCACAGGAGC | KD3 |
| TRCN0000107561 | TTTGTACTAGAAGATGGACGC | KD4 |
| TRCN0000107562 | ATCAAGACTAATTGATTCCGC | KD5 |
FIGURE 2.

JMJD1C knockdown promotes neural differentiation of hESCs. A and B, CT2 and H9 hESC lines were stably transduced with lentiviral shRNA vector for JMJD1C KD or an MM control. The resultant cell lines were subjected to RT-qPCR (A) or Western blotting (B) for JMJD1C expression. C, phase-contrast photographs of JMJD1C KD and MM H9 cells cultured in T1 or T1/F0 medium for 6 days. Both KD cell lines (KD and KD-v2 derived using KD4 and KD3 shRNAs, respectively) formed typical neural rosettes in T1/F0. Scale bar, 100 μm. D, RT-PCR analysis for expression of pluripotency (OCT4) and neural (PAX6 and MAP2) marker genes in JMJD1C KD and MM cells cultured in T1 or T1/F0. Day 7 embryoid body (EB) was tested as a control. E and F, JMJD1C KD and MM H9 cells were cultured in T1 or T1/F0 for 5 days followed by flow cytometry analysis for PAX6+ cell ratio (E) or immunostaining for PAX6 (F). Scale bar, 200 μm. Error bars, S.D.
Interestingly, both KD and MM cells expressed the pluripotency markers OCT4 and NANOG at similar levels (Fig. 3A) and formed teratomas including tissues from the three germ layers in immunocompromised mice (Fig. 3B). In addition, both KD and MM cells could be expanded for prolonged culture without differentiation as wild-type hESCs. We reasoned that, although the partial KD of JMJD1C did not have a remarkable impact on pluripotency of hESCs, the KD might be sufficient to affect their lineage specification and differentiation.
FIGURE 3.

hESCs with JMJD1C knockdown remain pluripotent. A, immunostaining for OCT4 or NANOG on JMJD1C KD or MM H9 cells cultured in either CM or T1 medium. Cell nuclei were counterstained with DAPI. B, JMJD1C KD and MM H9 cells were injected into SCID-beige mice. Teratomas exhibiting complex differentiation formed in the mice 5–6 weeks postinoculation and were processed for histological analysis with hematoxylin-eosin staining. Representative images for the three germ layers are shown. C, PluriTest. Two JMJD1C KD, two MM, and one wild-type hESC sample (for both H9 and CT2) were subjected to PluriTest via analysis of their gene expression microarray data, and all of the samples (shown as small circles in the graphs) passed the test.
It has been shown that discrete local changes of H3K9me2 particularly at genic regions are detected and associated with altered gene expression upon neural differentiation of mouse ESCs (21). Furthermore, the neural fate is generally considered to be the intrinsic direction of ESC differentiation. Thus, we asked whether or not JMJD1C KD would affect neural differentiation of hESCs. For this purpose, we used the T1/F0 medium, reasoning that this bFGF-depleted, defined medium would promote neural differentiation because it lacked bFGF, an inhibitor of neural initiation in hESCs (11).
Indeed, the T1/F0-cultured KD cells from either H9 or CT2 started neural differentiation rapidly, reflected by the appearance of neural rosettes as early as on day 3, whereas no neural rosettes were formed from the MM cells within the same time period (Fig. 2C) or longer (data not shown). Neural rosettes also developed from the H9-KD-v2 cells cultured in T1/F0 for 3 days (Fig. 2C). Expression of the pluripotency marker OCT4 decreased, and expression of the neural markers PAX6 and MAP2 increased in the KD cells cultured in T1/F0 within 5 days, compared with the MM cells in T1/F0 or the KD and MM cells in T1 (Fig. 2D). PAX6+ cell ratio was increased to 42.9% in the KD cells, compared with 0.3% in the MM cells (Fig. 2E), which was confirmed by positive immunostaining for PAX6 in the KD, but not MM, cells (Fig, 2F). These data suggest that a sufficient level of JMJD1C is required for preventing hESCs from neural differentiation.
JMJD1C Knockdown Reduces miR-302 Expression, Enhances TGFβ Signaling, and Inhibits BMP Signaling
To gain mechanistic insight into neural differentiation of the JMJD1C KD cells, we analyzed the transcriptomic profiles of the KD and MM cells derived from both H9 and CT2 in duplicates, using the whole genome gene expression microarray. First, consistent with the pluripotency assays above, both KD and MM cell clones passed the PluriTest (Fig. 3C) based on the microarray data using the reported method (19). In general, the pluripotency genes were highly expressed and developmental genes for the three germ layers were expressed at a low level in both the KD and MM cells, based on raw data from the microarray deposited at GEO. After normalization by a corresponding value for each of the genes in wild-type H9 and CT2 cells, the relative RNA levels of these genes in the KD cells were similar to those in the MM cells (supplemental Table S1).
Concomitantly, JMJD1C KD affected the transcriptional levels of a small group of genes in hESCs. Among them, a particularly interesting gene was miR-302c, one of the four isoforms (miR-302a, -b, -c, and -d) transcribed from the miR-302/367 cluster that is exclusively expressed in ESCs and plays a diverse role in their fate decision by supporting pluripotency and repressing neural differentiation (12, 22, 23). Here, we found that miR-302c expression was reduced to about half in both H9-KD and CT2-KD cells compared with that in the MM cells (supplemental Table S1). Interestingly, except for miR-302c, the isoforms (a, b, and d) were not detectable in the data set, probably due to low sensitivity for their probes in the microarray.
To validate the array data, we first performed RT-PCR analysis to determine the expression level of the entire primary miR-302/367 (pri-miR-302) transcript. As shown in Fig. 4A, the KD resulted in about 60 and 80% reductions of this transcript in CT2 and H9 cells, respectively, based on densitometric analysis of the data. Furthermore, Northern blot analysis indicated attenuated levels of all of the four miR-302 isoforms in the KD cells in comparison with the MM cells for both H9 and CT2. It is interesting to note that there were cell line-specific differences in the expression of miR-302, as demonstrated by both the levels of the pri-miR-302 transcript and the miR302 isoforms (Fig. 4, A and B). Another independent Northern blot assay validated the attenuated expression of the miR-302 isoforms in the KD cells (data not shown). These data also suggest that the regulation of miR-302 expression is at the transcriptional level rather than at the post-transcriptional or miRNA-processing level.
FIGURE 4.
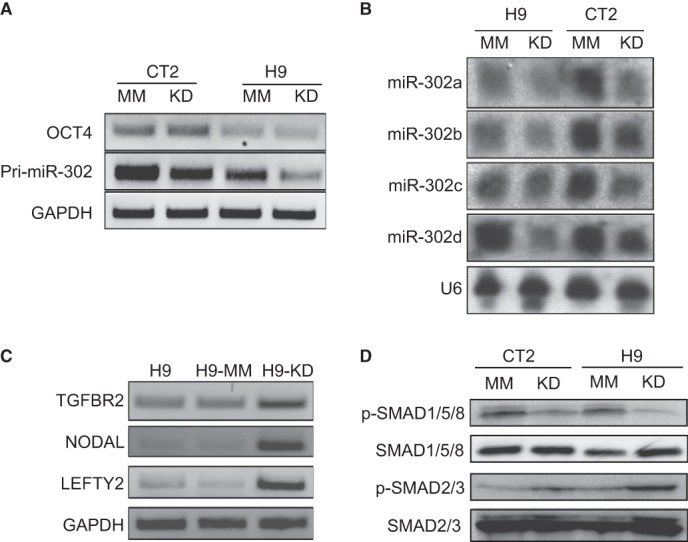
JMJD1C knockdown reduces miR-302 expression, enhances TGFβ signaling, and inhibits BMP signaling. A, RT-PCR analysis for expression of pri-miR-302 in JMJD1C KD and MM cells from both H9 and CT2 hESCs. B, Northern blotting for miR-302 isoforms in the KD and MM cells. The small nuclear RNA U6 was determined as a loading control. C, RT-PCR analysis for expression of some TGFβ ligands and targets in wild-type, JMJD1C KD, and MM H9 cells. D, Western blot analysis for phosphorylated SMAD1/5/8 (p-SMAD1/5/8) and phosphorylated SMAD2/3 (p-SMAD2/3) to determine BMP and TGFβ signaling levels, respectively, in the JMJD1C KD and MM (CT2 and H9) cells. Total SMAD1/5/8 and SMAD2/3 were detected as controls.
Furthermore, we noticed enhanced expression of four genes involved in TGFβ signaling in the KD cells compared with the MM cells (supplemental Table S1). They include NODAL, a ligand to self-activate TGFβ/NODAL signaling (24); TGFBR2, a receptor for TGFβ signaling (25); LEFTY2, an antagonist of NODAL; and CER1 that encodes CERBERUS, an inhibitor of both BMPs and NODAL (26). We confirmed the higher expression of NODAL, TGFBR2, and LEFTY2 in the KD cells than in the MM cells via RT-PCR (Fig. 4C). It has been known that NODAL, LEFTY2 (27), and CER1 (26) are also targets stimulated by TGFβ signaling, whereas both TGFBR2 (28) and LEFTY1/2 (29) are targets inhibited by miR-302. Therefore, the level of TGFβ signaling may be a result of competition between the two groups of antagonizing molecules NODAL and TGFBR2 versus LEFTY2 and CERBURUS. Consistent with a previous study on miR-302 (30), we observed an elevated TGFβ signaling reflected by increase of phosphorylated SMAD2/3 in the KD cells compared with the MM cells (Fig. 4D), which may result from dominance of proteins encoded by the TGFβ signaling-enhancing genes over those encoded by the repressive genes in the KD cells.
It has been reported that three internal inhibitors of BMP signaling, including DAZAP2, SLAIN1, and TOB2, are also targets of miR-302 (28). Although transcriptional levels of these inhibitors in the KD cells were similar to those in the MM cells (supplemental Table S1), down-regulation of miR-302 is supposed to enhance the translation of these inhibitors, thus reducing BMP signaling. We indeed observed lower BMP signaling, reflected by decreased phosphorylated SMAD1/5/8 level, in the KD cells than in the MM cells (Fig. 4D). Due to the neural inhibitory nature of BMP signaling, reduction of this signaling in the KD cells may also promote their neural differentiation.
JMJD1C Promotes Expression of miR-302 by Demethylating H3K9 at Its Promoter
Because methylated H3K9 often acts as a histone repressive mark, demethylation of H3K9 on the neural suppressor miR-302 may help sustain its expression, thus preventing hESCs from precocious neural differentiation. Comparative sequence analysis shows that human and mouse miR-302 promoters are highly conserved (supplemental Fig. S1). Interestingly, mouse H3K36 demethylase Jhdm1b also binds to the region immediately downstream of the dual Oct-binding sites at mouse miR-302 promoter and coordinates with Oct4 to initiate miR-302 expression during fibroblast reprogramming (6). This raises a possibility that human JMJD1C may act similarly as mouse Jhdm1b to promote miR-302 expression.
To address this hypothesis, we first tested JMJD1C binding to the miR-302 promoter in the JMJD1C KD and MM cells, using ChIP-qPCR. Following ChIP with an anti-JMJD1C antibody, five regions downstream of the dual OCT-binding sites were amplified from the precipitated DNA using corresponding primer sets (Table 2), with regions 1–3 being upstream of the transcription start site (TSS), and regions 4 and 5 downstream of the transcription start site (Fig. 5A and supplemental Fig. S1). We found that anti-JMJD1C antibody pulled down these regions about 5–30-fold of those pulled down by an IgG control, and the pull-down peaks for the antibody resided in region 2 and then region 3 (Fig. 5B), suggesting that human JMJD1C binds the most to the proximate promoter of human miR-302.
FIGURE 5.

JMJD1C binds to miR-302 promoter in hESCs and demethylates H3K9 at the promoter. A, a scheme of the miR-302 promoter and primer sets used to determine JMJD1C binding to the promoter by amplifying five regions downstream of the dual OCT-binding sites. TSS, transcription start site. B, qPCR analysis to determine enrichment of the five regions at the miR-302 promoter in H9 hESCs following ChIP with anti-JMJD1C antibody versus IgG control. Results are displayed as mean ± S.D. (error bars) from three independent experiments. C, PCR analysis to determine enrichment of regions 2 and 3 at the miR-302 promoter in JMJD1C KD and MM H9 cells, following ChIP with IgG or antibodies against H3K9me1 and H3K9me2. The PCR amplicons were separated on gel (left). For each tested region, the ratio of signal of the H3K9me2 band over that of the H3K9me1 band in the KD or MM cells is displayed as mean ± S.D. in a bar chart (right). *, p < 0.05 compared with MM. D, JMJD1C KD H9 cells cultured in T1 or T1/F0 in the presence of 4 μm BIX01294 (BIX; an inhibitor of H3K9 methyltransferase G9a) or the vehicle DMSO for 5 days were subjected to flow cytometry analysis for PAX6+ cell ratio (left). Results from three independent experiments are displayed as mean ± S.D. in a bar chart (right). **, p < 0.01 compared with DMSO.
We next determined the ratio of H3K9me2 over H3K9me1 at regions 2 and 3 using ChIP-PCR and found that the ratio in the KD cells was about 3-fold the ratio in the MM cells (Fig. 5C). This suggests that JMJD1C promotes miR-302 expression by demethylating H3K9 at the miR-302 promoter, and the increased H3K9 methylation level at the promoter in the KD cells is responsible for the reduced miR-302 expression.
We further confirmed that the role of JMJD1C in neural differentiation of hESCs relies on its H3K9 demethylase activity by using BIX01294, an inhibitor of H3K9 methyltransferase G9a that methylates H3K9me1 and H3K9me2 de novo. Pretreatment of H9 KD cells with 4 μm BIX01294 for 2 days reduced the PAX6+ cell ratio in T1/F0 from 36 to 12% (Fig. 5D), indicating that JMJD1C indeed prevents neural differentiation via its H3K9 demethylase activity.
miR-302 Repression Contributes to Neural Differentiation of hESCs with JMJD1C KD
Although miR-302 down-regulation is accompanied by H3K9 hypermethylation at the miR-302 promoter in the JMJD1C KD cells, it remains to be elucidated if miR-302 reduction is causative for or a consequence of neural differentiation of the KD cells. We first analyzed whether rescue of miR-302 repression would inhibit neural differentiation of the JMJD1C KD cells. We transfected the KD cells with a mimic for miR-302c and induced neural differentiation of the cells by culturing them in T1/F0 for 5 days. The miR-302c mimic, but not the negative control, inhibited neural rosette formation by the KD cells (Fig. 6A) with 12.7% of the PAX6+ cell ratio, compared with 33.3% for the mock-transfected KD cells (Fig. 6B). Inhibition of the PAX6+ cell ratio (from 22.5 to 12.8%) was also observed with a miR-302b mimic (Fig. 6C).
FIGURE 6.

miR-302 reduction contributes to neural differentiation in hESCs with JMJD1C knockdown. A, phase-contrast photographs of the JMJD1C KD H9 cells transfected with a miR-302c mimic or a negative control (Neg. Ctrl.) and cultured in T1 or T1/F0 for 5 days. Note that neural rosettes were only present in the KD cells transfected with the negative control mimic but not the miR-302c mimic. B–D, flow cytometry analysis for the ratio of PAX6+ cells among H9-KD cells transfected with the miR-302c mimic (B) or a miR-302b mimic (C), wild-type H9 cells transfected with a miR-302c inhibitor (Inh.), or a miR-302b inhibitor (D). E, a model for JMJD1C-miR-302-mediated inhibition on neural differentiation of hESCs. Arrows, induction; hammer-ended line, inhibition.
We also tested whether inhibition of specific miR-302 isoforms would cause heterogeneous differentiation of hESCs into neural and other cell lineages, as reported via other approaches (12, 22, 23). Transfection of wild-type H9 hESCs with a miR-302c inhibitor followed by culture in T1/F0 caused differentiation of the cells into a mixed population, including 13.7% of PAX6+ cells, which was higher than the PAX6+ cell ratio (4.15%) among H9 cells transfected with a negative control (Fig. 6D). However, no remarkable differentiation was observed with H9 cells transfected with a miR-302b inhibitor, as reflected by low PAX6+ cell ratios in both the inhibitor and control groups (Fig. 6D), which is probably due to insufficient inhibition of miR-302b and/or compensation by the other miR-302 isoforms.
As controls, wild-type H9 cells transfected with the mimics for either miR-302c or -b remained undifferentiated with sustained OCT4 expression (data not shown), consistent with a report on hESCs with overexpression of pri-miR-302 (31). Thus, all of the data above suggest that decreased miR-302 in the JMJD1C KD cells indeed contributes to their neural differentiation.
DISCUSSION
During neural ectoderm formation in mammals, various epigenetic factors regulate the activities of external and intracellular signaling pathways and sequential activation/inactivation of fate-determining transcription factors in an orchestrated manner, leading to the transition from the inner cell mass to the epiblast and then to the neural ectoderm (32). Similar events occur during ESC differentiation into neural progenitors (33). We have found here that the decline of the H3K9 demethylase JMJD1C during hESC differentiation is not just a consequence of the differentiation but an epigenetic event required for initiation of the neural program. In other words, the presence of JMJD1C in undifferentiated hESCs represses the program.
It appears that miR-302 expression promoted by JMJD1C plays a key role in the neural repression. JMJD1C directly bound to the miR-302 promoter and demethylated H3K9 near the dual OCT-binding sites. JMJD1C KD caused down-regulation of miR-302, including the entire pri-miR-302 cluster and the four miR-302 isoforms in hESCs. A mimic for miR-302b or -c inhibited neural differentiation of the KD hESCs, whereas an inhibitor for miR-302c enhanced neural differentiation of wild-type hESCs. These data suggest that all of the miR-302 isoforms may sustain pluripotency and repress neural differentiation, and a threshold of the pool of the isoforms is critical to prevent hESCs from differentiation. In addition, neural differentiation only occurred in the KD cells cultured in T1/F0, but not T1, which suggests that JMJD1C KD (with partial reduction of its mRNA and protein) is not sufficient to trigger neural differentiation of hESCs unless external bFGF is removed.
FGF signaling promotes pluripotency by antagonizing BMP signaling (34) and preventing NANOG degradation (35). Sustained NANOG maintains the integrity and activity of the pluripotency circuitry formed by NANOG, OCT4, and SOX2 (36). Inhibition of FGF/ERK signaling leads to elevated expression of both HAND1 (which induces extraembryonic and mesodermal differentiation) and PAX6 (which induces neural differentiation) in hESCs; however, inhibition of both FGF and BMP signaling leads to repressed HAND1 expression and enhanced PAX6 expression (11). This latter fact indicates why neural differentiation rapidly occurs in the KD cells cultured in T1/F0.
It has also been known that dual inhibition of BMP and TGFβ signaling (the so-called 2i method) can induce neural differentiation of hESCs (18). We did observe reduced BMP signaling in the JMJD1C KD cells compared with the MM cells. However, TGFβ signaling level was even higher in the KD cells than the MM cells, which might result from the following possibilities: (i) because the TGFβ receptor TGFBR2 is targeted by miR-302 (30), miR-302 reduction derepresses TGFBR2, thus enhancing TGFβ signaling; (ii) NODAL is self-stimulated by activated TGFβ signaling, which might form a positive feedback; and (iii) JMJD1C might repress TGFβ signaling in a miR-302-independent manner. Nevertheless, down-regulation of miR-302 in the JMJD1C KD cells may trigger NR2F2 expression, thus inducing neural differentiation without the need for inhibition of TGFβ signaling. Elucidation of these possibilities awaits further investigations.
In summary, we have demonstrated here that JMJD1C promoted the expression of miR-302, a key pluripotency-supportive miRNA, in hESCs via direct binding to its promoter and demethylating the repressive mark H3K9. To our knowledge, this is the first evidence for epigenetic modification of the miR-302 locus in hESCs. In the absence of external bFGF, hESCs with JMJD1C KD, but not control hESCs, differentiated into neural progenitors faster than ever reported to our knowledge. This suggests that JMJD1C plays a central role in control of neural differentiation, and only inhibition of JMJD1C and removal of external bFGF are sufficient to rapidly induce neural progenitors from hESCs. In addition, BMP signaling is reduced, whereas TGFβ signaling is elevated in hESCs with JMJD1C KD, suggesting that, when JMJD1C is knocked down, inhibition of both signalings, as proposed for the 2i model (18), is not necessary for neural differentiation. However, these events might happen in parallel or sequentially.
A schematic model is proposed (Fig. 6E) to accommodate all of the results in this study in the context of previous findings. At present, we are not sure whether individual miR302 isoforms, such as miR-302c and -b, or all of the four isoforms together mediate the inhibitory effect of JMJD1C on neural differentiation and whether JMJD1C regulates the isoforms indirectly at the post-transcriptional level. It is also worth noting that JMJD1C should have a broad spectrum of targets in the genome, perhaps including other neurally related and unrelated genes. It may be involved in other functions of hESCs and differentiation into other cell lineages to play various physiological roles during development. Although it has not yet been reported about Jmjd1c functions during development in animal models, a study on patients using positional cloning has identified JMJD1C as a candidate gene for autism (37). With miR-302 targets being extensively explored (38), the global search for JMJD1C targets in hESCs and their progeny should provide in-depth insight into the biological functions of JMJD1C during human development.
CONCLUSION
JMJD1C helps sustain the expression of miR-302, a key pluripotency-supportive miRNA, in hESCs via direct binding to its promoter and demethylation of H3K9 at the promoter. JMJD1C knockdown is sufficient to rapidly induce neural differentiation from hESCs in a defined medium without bFGF. When JMJD1C is silenced, inhibition of TGFβ signaling is no longer necessary for neural differentiation.
Acknowledgments
We thank Leann Crandall and Kristen Martins-Taylor for technical assistance and Adam Lazorchak for critical reading of the manuscript.
This work was supported by Connecticut Stem Cell Research Grants 06SCB14 and 09SCD01 (to R. X.), 10SCA21 (to X. W.), and 11SCB08 (to H. D.).

This article contains supplemental Table S1 and Fig. S1.
The microarray data sets have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE46485.
- ESC
- embryonic stem cell
- hESC
- human ESC
- H3K9
- histone 3 lysine 9
- CM
- mouse embryonic fibroblast-conditioned medium
- T1
- defined medium mTeSR1
- bFGF
- basic fibroblast growth factor
- KD
- knockdown
- MM
- mismatch
- qPCR
- quantitative PCR.
REFERENCES
- 1. Greer E. L., Shi Y. (2012) Histone methylation. A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 13, 343–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Black J. C., Van Rechem C., Whetstine J. R. (2012) Histone lysine methylation dynamics. Establishment, regulation, and biological impact. Mol. Cell 48, 491–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kooistra S. M., Helin K. (2012) Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell Biol. 13, 297–311 [DOI] [PubMed] [Google Scholar]
- 4. Loh Y. H., Zhang W., Chen X., George J., Ng H. H. (2007) Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev. 21, 2545–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang J., Zhang M., Zhang Y., Kou Z., Han Z., Chen D. Y., Sun Q. Y., Gao S. (2010) The histone demethylase JMJD2C is stage-specifically expressed in preimplantation mouse embryos and is required for embryonic development. Biol. Reprod. 82, 105–111 [DOI] [PubMed] [Google Scholar]
- 6. Wang T., Chen K., Zeng X., Yang J., Wu Y., Shi X., Qin B., Zeng L., Esteban M. A., Pan G., Pei D. (2011) The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C-dependent manner. Cell Stem Cell 9, 575–587 [DOI] [PubMed] [Google Scholar]
- 7. Kim S. M., Kim J. Y., Choe N. W., Cho I. H., Kim J. R., Kim D. W., Seol J. E., Lee S. E., Kook H., Nam K. I., Kook H., Bhak Y. Y., Seo S. B. (2010) Regulation of mouse steroidogenesis by WHISTLE and JMJD1C through histone methylation balance. Nucleic Acids Res. 38, 6389–6403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu R. H., Chen X., Li D. S., Li R., Addicks G. C., Glennon C., Zwaka T. P., Thomson J. A. (2002) BMP4 initiates human embryonic stem cell differentiation to trophoblast. Nat. Biotechnol. 20, 1261–1264 [DOI] [PubMed] [Google Scholar]
- 9. Xu R. H., Sampsell-Barron T. L., Gu F., Root S., Peck R. M., Pan G., Yu J., Antosiewicz-Bourget J., Tian S., Stewart R., Thomson J. A. (2008) NANOG is a direct target of TGFβ/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell 3, 196–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Katoh M., Katoh M. (2007) Comparative integromics on JMJD1C gene encoding histone demethylase. Conserved POU5F1 binding site elucidating mechanism of JMJD1C expression in undifferentiated ES cells and diffuse-type gastric cancer. Int. J. Oncol. 31, 219–223 [PubMed] [Google Scholar]
- 11. Greber B., Coulon P., Zhang M., Moritz S., Frank S., Müller-Molina A. J., Araüzo-Bravo M. J., Han D. W., Pape H. C., Schöler H. R. (2011) FGF signalling inhibits neural induction in human embryonic stem cells. EMBO J. 30, 4874–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosa A., Brivanlou A. H. (2011) A regulatory circuitry comprised of miR-302 and the transcription factors OCT4 and NR2F2 regulates human embryonic stem cell differentiation. EMBO J. 30, 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thomson J. A., Itskovitz-Eldor J., Shapiro S. S., Waknitz M. A., Swiergiel J. J., Marshall V. S., Jones J. M. (1998) Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 [DOI] [PubMed] [Google Scholar]
- 14. Lin G., Martins-Taylor K., Xu R. H. (2010) Human embryonic stem cell derivation, maintenance, and differentiation to trophoblast. Methods Mol. Biol. 636, 1–24 [DOI] [PubMed] [Google Scholar]
- 15. Xu C., Inokuma M. S., Denham J., Golds K., Kundu P., Gold J. D., Carpenter M. K. (2001) Feeder-free growth of undifferentiated human embryonic stem cells. Nat. Biotechnol. 19, 971–974 [DOI] [PubMed] [Google Scholar]
- 16. Ludwig T. E., Bergendahl V., Levenstein M. E., Yu J., Probasco M. D., Thomson J. A. (2006) Feeder-independent culture of human embryonic stem cells. Nat. Methods 3, 637–646 [DOI] [PubMed] [Google Scholar]
- 17. Zhang S. C., Wernig M., Duncan I. D., Brüstle O., Thomson J. A. (2001) In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 19, 1129–1133 [DOI] [PubMed] [Google Scholar]
- 18. Chambers S. M., Fasano C. A., Papapetrou E. P., Tomishima M., Sadelain M., Studer L. (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Müller F. J., Schuldt B. M., Williams R., Mason D., Altun G., Papapetrou E. P., Danner S., Goldmann J. E., Herbst A., Schmidt N. O., Aldenhoff J. B., Laurent L. C., Loring J. F. (2011) A bioinformatic assay for pluripotency in human cells. Nat. Methods 8, 315–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim S. W., Li Z., Moore P. S., Monaghan A. P., Chang Y., Nichols M., John B. (2010) A sensitive non-radioactive Northern blot method to detect small RNAs. Nucleic Acids Res. 38, e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lienert F., Mohn F., Tiwari V. K., Baubec T., Roloff T. C., Gaidatzis D., Stadler M. B., Schübeler D. (2011) Genomic prevalence of heterochromatic H3K9me2 and transcription do not discriminate pluripotent from terminally differentiated cells. PLoS Genet. 7, e1002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barroso-delJesus A., Romero-López C., Lucena-Aguilar G., Melen G. J., Sanchez L., Ligero G., Berzal-Herranz A., Menendez P. (2008) Embryonic stem cell-specific miR302–367 cluster. Human gene structure and functional characterization of its core promoter. Mol. Cell Biol. 28, 6609–6619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Card D. A., Hebbar P. B., Li L., Trotter K. W., Komatsu Y., Mishina Y., Archer T. K. (2008) Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell Biol. 28, 6426–6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miyazono K. (2000) Positive and negative regulation of TGF-β signaling. J. Cell Sci. 113, 1101–1109 [DOI] [PubMed] [Google Scholar]
- 25. Kawabata M., Chytil A., Moses H. L. (1995) Cloning of a novel type II serine/threonine kinase receptor through interaction with the type I transforming growth factor-β receptor. J. Biol. Chem. 270, 5625–5630 [DOI] [PubMed] [Google Scholar]
- 26. Katoh M. (2006) CER1 is a common target of WNT and NODAL signaling pathways in human embryonic stem cells. Int. J. Mol. Med. 17, 795–799 [PubMed] [Google Scholar]
- 27. Besser D. (2004) Expression of nodal, lefty-a, and lefty-B in undifferentiated human embryonic stem cells requires activation of Smad2/3. J. Biol. Chem. 279, 45076–45084 [DOI] [PubMed] [Google Scholar]
- 28. Lipchina I., Elkabetz Y., Hafner M., Sheridan R., Mihailovic A., Tuschl T., Sander C., Studer L., Betel D. (2011) Genome-wide identification of microRNA targets in human ES cells reveals a role for miR-302 in modulating BMP response. Genes Dev. 25, 2173–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anokye-Danso F., Trivedi C. M., Juhr D., Gupta M., Cui Z., Tian Y., Zhang Y., Yang W., Gruber P. J., Epstein J. A., Morrisey E. E. (2011) Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell 8, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Subramanyam D., Lamouille S., Judson R. L., Liu J. Y., Bucay N., Derynck R., Blelloch R. (2011) Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nat. Biotechnol. 29, 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barroso-delJesus A., Lucena-Aguilar G., Sanchez L., Ligero G., Gutierrez-Aranda I., Menendez P. (2011) The Nodal inhibitor Lefty is negatively modulated by the microRNA miR-302 in human embryonic stem cells. FASEB J. 25, 1497–1508 [DOI] [PubMed] [Google Scholar]
- 32. Wen S., Li H., Liu J. (2009) Epigenetic background of neuronal fate determination. Prog. Neurobiol. 87, 98–117 [DOI] [PubMed] [Google Scholar]
- 33. Coskun V., Tsoa R., Sun Y. E. (2012) Epigenetic regulation of stem cells differentiating along the neural lineage. Curr. Opin. Neurobiol. 22, 762–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu C., Rosler E., Jiang J., Lebkowski J. S., Gold J. D., O'Sullivan C., Delavan-Boorsma K., Mok M., Bronstein A., Carpenter M. K. (2005) Basic fibroblast growth factor supports undifferentiated human embryonic stem cell growth without conditioned medium. Stem Cells 23, 315–323 [DOI] [PubMed] [Google Scholar]
- 35. Wang X., Lin G., Martins-Taylor K., Zeng H., Xu R. H. (2009) Inhibition of caspase-mediated anoikis is critical for basic fibroblast growth factor-sustained culture of human pluripotent stem cells. J. Biol. Chem. 284, 34054–34064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jaenisch R., Young R. (2008) Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 132, 567–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Castermans D., Vermeesch J. R., Fryns J. P., Steyaert J. G., Van de Ven W. J., Creemers J. W., Devriendt K. (2007) Identification and characterization of the TRIP8 and REEP3 genes on chromosome 10q21.3 as novel candidate genes for autism. Eur. J. Hum. Genet. 15, 422–431 [DOI] [PubMed] [Google Scholar]
- 38. Lipchina I., Studer L., Betel D. (2012) The expanding role of miR-302–367 in pluripotency and reprogramming. Cell Cycle 11, 1517–1523 [DOI] [PubMed] [Google Scholar]