

Background: The role of apoA-IV in hepatic gluconeogenesis is unknown.
Results: ApoA-IV interacts with NR1D1and stimulates its expression and decreases hepatic Glc-6-Pase and PEPCK expression and glucose production.
Conclusion: ApoA-IV inhibits hepatic gluconeogenesis by suppressing Glc-6-Pase and PEPCK expression through the NR1D1 pathway.
Significance: This finding links lipid and carbohydrate metabolism and is crucial for understanding apoA-IV physiologic processes on glucose homeostasis.
Keywords: Apolipoproteins, Diabetes, Gluconeogenesis, Nuclear Receptors, Phosphoenolpyruvate Carboxykinase, Apolipoprotein A-IV, NR1D1
Abstract
We showed recently that apoA-IV improves glucose homeostasis by enhancing pancreatic insulin secretion in the presence of elevated levels of glucose. Therefore, examined whether apolipoprotein A-IV (apoA-IV) also regulates glucose metabolism through the suppression of hepatic gluconeogenesis. The ability of apoA-IV to lower gluconeogenic gene expression and glucose production was measured in apoA-IV−/− and wild-type mice and primary mouse hepatocytes. The transcriptional regulation of Glc-6-Pase and phosphoenolpyruvate carboxykinase (PEPCK) by apoA-IV was determined by luciferase activity assay. Using bacterial two-hybrid library screening, NR1D1 was identified as a putative apoA-IV-binding protein. The colocalization and interaction between apoA-IV and NR1D1 were confirmed by immunofluorescence, in situ proximity ligation assay, and coimmunoprecipitation. Enhanced recruitment of NR1D1 and activity by apoA-IV to Glc-6-Pase promoter was verified with ChIP and a luciferase assay. Down-regulation of apoA-IV on gluconeogenic genes is mediated through NR1D1, as illustrated in cells with NR1D1 knockdown by siRNA. We found that apoA-IV suppresses the expression of PEPCK and Glc-6-Pase in hepatocytes; decreases hepatic glucose production; binds and activates nuclear receptor NR1D1 and stimulates NR1D1 expression; in cells lacking NR1D1, fails to inhibit PEPCK and Glc-6-Pase gene expression; and stimulates higher hepatic glucose production and higher gluconeogenic gene expression in apoA-IV−/− mice. We conclude that apoA-IV inhibits hepatic gluconeogenesis by decreasing Glc-6-Pase and PEPCK gene expression through NR1D1. This novel regulatory pathway connects an influx of energy as fat from the gut (and subsequent apoA-IV secretion) with inhibition of hepatic glucose production.
Introduction
Human apolipoprotein A-IV (apoA-IV) is a 46-kDa protein produced by enterocytes in response to fat absorption (1). It is secreted in association with chylomicrons. Following the metabolism of chylomicron triacylglycerol by lipoprotein lipase, apoA-IV rapidly dissociates from chylomicrons and is transferred to high density lipoproteins or to a lipoprotein-free fraction of the plasma in the postprandial state (2). Unlike in humans, its synthesis and secretion in rodents are not restricted to enterocytes of the small intestine because the liver and the hypothalamus have also been demonstrated to produce apoA-IV (3–5). ApoA-IV is involved in numerous aspects of lipid metabolism, including the intestinal absorption of lipids (6), inhibition of the oxidation of plasma lipoproteins (7, 8), promotion of free cholesterol efflux via the ATP-binding cassette transporter A1 (ABCA1) pathway (9), and the activation of plasma lecithin:cholesterol acyltransferase (LCAT), and cholesteryl ester transfer protein (CETP) (10, 11). However, neither the apoA-IV receptor nor its pathway of action in target cells has been identified.
Hepatic gluconeogenesis is required for survival during prolonged fasting or starvation but is inappropriately activated in diabetes mellitus. Glucocorticoids and glucagon have strong gluconeogenic actions on the liver. In contrast, insulin suppresses hepatic gluconeogenesis (12, 13). We reported recently that apoA-IV improves glucose homeostasis by enhancing insulin secretion in the presence of elevated glucose (14). Furthermore, this action of apoA-IV on glucose-induced insulin secretion was not compromised in diet-induced obese or diabetic KKAy mice, suggesting that the action of apoA-IV is probably not limited to insulin secretion. In this experiment, we determined that apoA-IV additionally acts to reduce hepatic gluconeogenesis, mediated through the nuclear receptor subfamily 1, group D, member 1 (NR1D1), also known as REV-ERBα. It is an important transcription factor regulating glucose metabolism by suppressing the expression of gluconeogenic genes (15–17). Here we provide evidence that apoA-IV suppresses the expression of the gluconeogenic genes PEPCK2 and Glc-6-Pase and, therefore, hepatic glucose production through the NR1D1 pathway. This is a novel and important finding, not only regarding this important role of apoA-IV in glucose metabolism but also linking lipid and glucose metabolism.
EXPERIMENTAL PROCEDURES
Reagents, Antibodies, and Plasmids
For primary mouse hepatocyte isolation, collagenase II was obtained from Worthington (Lakewood, NJ). Percoll was purchased from Sigma. 8-(4-chlorophenylthio)-cAMP was obtained from Enzo (Plymouth Meeting, PA). All other chemicals and materials were obtained from Thermo Fisher Scientific (Carlsbad, CA) or Sigma. For recombinant protein purification, full-length human apoA-IV (NM_000482, nucleotides 165–1293), full-length human apoA-IV fused with GFP, and full-length mouse apoA-IV (NM_007468, 371–1498) were subcloned into the pET21 Escherichia coli expression plasmid (Novagen, Merck KGaA, Darmstadt, Germany) containing an N-terminal His6 tag. Recombinant mouse apoA-IV (r-m-apoA-IV) protein and recombinant human apoA-IV (r-h-apoA-IV) protein from the above two plasmids were purified as described previously (14). The pcDNA3.1-Nr1d1 expression plasmid and Glc-6-Pase promoter plasmid pGL3-Glc-6-PaseP-luciferase were provided by Mitchell A. Lazar (17). The sequence of the inserted DNA in all above plasmids was confirmed by sequencing.
Animal Experiments
Four- to five-month-old male apoA-IV−/− and C57BL/6J WT mice were the same as described previously (14). The mice were individually housed in a temperature-controlled vivarium on a 12-h light, 12-h dark cycle (lights on at 05:00 h). Laboratory chow (Teklad 7912, Madison, WI) and water were provided ad libitum until the time of sacrifice, except where noted. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Cincinnati. For blood glucose and insulin measurements (as described previously) (14), tail blood was collected at the indicated times after r-m-apoA-IV was injected intraperitoneally in mice fed ad libitum and fasted for 5 h (07:00–12:00, apoA-IV injection at 12:00). For hepatic gene expression measurements, r-m-apoA-IV was injected 2 h before sacrifice (on the basis of previous studies), and livers were isolated for RT-PCR analyses.
Euglycemic-Hyperinsulinemic Clamps
The experiment was performed as described previously (18, 19) in 4- to 5-month-old male apoA-IV−/− and WT mice. Briefly, the chow-fed mice underwent surgical placement of a chronic indwelling catheter into the right jugular vein and were allowed to recover for about 4 days. Following a 5-h fast, the following infusions were initiated. Tracer glucose was administered as a primed dose, followed by continuous infusion of [3-3H]d-glucose (bolus of 2 μCi followed by 0.1 μCi/min, PerkinElmer Life Sciences, Waltham, MA), and a primed dose was administered, followed by continuous infusion of insulin (bolus of 62 milliunit/kg followed by 3.5 milliunits/min/kg, Novolin Regular, Novo Nordisk, Clayton, NC). Euglycemia (130–150 mg/dl) was maintained by an infusion of 20% glucose at a variable rate. After the initiation of infusions, blood samples were taken from the tail at t = 5 min as the 0 time point and then at 20, 40, 60, 70, 80, 90, and 100 min. The glucose infusion rate (GIR) and glucose utilization (GU) were calculated as the means of the values obtained at 10-min intervals during 70–100 min of the clamp. Endogenous glucose production was calculated as the difference between GU and GIR.
Cell Culture and Treatment with ApoA-IV
Primary mouse hepatocytes were isolated from 12-week-old male C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) and cultured in a similar way as the HEK-293 and HepG2 cells (ATCC) in high glucose DMEM with 10% FBS as described previously (17). The primary mouse hepatocytes were incubated in DMEM with 10% FBS medium with 1 nm dexamethasone overnight and then switched to glucose-free medium with or without r-m-apoA-IV (20 μg/ml), along with sodium pyruvate (2 mm), sodium lactate (20 mm), dexamethasone (1 nm), and 8-(4-chlorophenylthio)-cAMP (500 μm) for 6 h (PEPCK and Glc-6-Pase mRNA measurement) or overnight ( glucose measurement) (17). The culture medium was collected for a glucose assay (Diagnostic Chemicals Limited, Charlottetown, PE, Canada) and normalized to the cellular protein concentrations. For the measurement of NR1D1 expression, the primary hepatocytes, HepG2, or HEK-293 cells were starved in DMEM with 1% BSA overnight and then treated with r-m-apoA-IV (20 μg/ml) or r-h-apoA-IV (50 μg/ml) for the indicated times. mRNA levels of genes from the cells were determined by real-time RT-PCR. Protein levels of genes were detected by Western blotting.
Real-time RT-PCR
Total RNA from tissues or cells was isolated with an RNeasy mini kit (Qiagen, Germantown, MD). First strand cDNA was synthesized with a ScriptsTM cDNA synthesis kit and then used for real-time quantitative PCR by using iQ SYBR Green Supermix (Bio-Rad). The sequence of the primers (Integrated DNA Technologies, Coralville, IA) were as follows: mouse NR1D1, 5′-ACGACCCTGGACTCCAATAA-3′ (forward) and 5′-CCATTGGAGCTGTCACTGTAGA-3′ (reverse); mouse PEPCK, 5′-TTTTTGGTTTT-TCTTTGTTTTGC-3′ (forward) and 5′-TGTG-TCTCCCAACGCTCA-3′ (reverse); mouse Glc-6-Pase, 5′-TCTGTCCCGGATCTACCTTG-3′ (forward) and 5′-GAAAGTTTCAGCCA-CAGCAA-3′ (reverse); and cyclophilin, 5′-ATTCATGTGCCAGGGTGGTGACT-3′ (forward) and 5′-TCAGTCTTGGCAGTGCAGAT-3′ (reverse).
Bacterial Two-hybrid Library Screening
Bacterial two-hybrid library screening was carried out using a BacterioMatch II two-hybrid system and rat liver BacterioMatch II cDNA library (Stratagene, La Jolla, CA). Rat apoA-IV (NM_012737, nucleotides 152–1356) was subcloned into the pBT E. coli plasmid. Several control transformations were performed, as suggested by the manufacturer. Verification of true positive clones was carried out by retransformation of each purified target plasmid paired with the recombinant pBT-rat-apoA-IV. The protein encoded by the target DNA plasmid was determined by comparison to the nucleotide sequence database.
Immunofluorescence
1 × 106 HepG2 cells were transfected with 1.5 μg of human pcDNA-Nr1d1 plasmid DNA by electroporation with a Nucleofector kit (Amaxa, Gaithersburg, MD). Cells were seeded on 8-well chamber slides, maintained for 48 h with growth medium, serum-starved overnight in growth medium with 1% BSA, and then incubated with recombinant human apoA-IV protein fused with GFP (r-h-apoA-IV-GFP) or GFP only as a negative control for 2 h. After fixation, permeabilization, and blocking, the cells were incubated with anti-human NR1D1 (1:200) and anti-GFP (1:200) antibodies (Cell Signaling Technology, Beverly, MA) and then with Alexa Fluor 594-conjugated goat anti-rabbit and FITC-conjugated goat anti-mouse secondary antibodies (Invitrogen). The slides were viewed with a Zeiss LSM-510 confocal fluorescence microscope.
In Situ PLA
An in situ proximity ligation assay (PLA) was performed using Duolink II detection reagents (Olink Bioscience, Uppsala, Sweden) with anti-NR1D1 and anti-GFP antibodies by following the instructions of the manufacturer. Briefly, as the above process for immunofluorescence, after permeabilization and blocking, this was followed by PLA probe incubation and, ultimately, ligation and polymerization steps, generating a concatameric oligonucleotide product linked to the fluorescent antibody complex. The interaction between apoA-IV and NR1D1 resulted in a red PLA signal, visualized by fluorescence microscopy (Zeiss Axiovert 200). As negative controls, cells were transfected with the empty plasmid or Nr1d1 plasmid but with the omission of anti-NR1D1 primary antibody.
Western Immunoblotting
Western blotting was performed as described in the Western immunoblotting protocol from Cell Signaling Technology. Briefly, tissues or cells were lysed in radioimmune precipitation assay buffer with proteinase inhibitors (Santa Cruz Biotechnology, Dallas, Texas). The lysates (10 μg of protein) were separated by SDS-PAGE gels (Bio-Rad) and transferred to PVDF membranes. Blots were probed with anti-NR1D1 and anti-β-actin (Millipore, Billerica, MA) as indicated. The bands were detected using chemiluminescent HRP substrate (Millipore) and quantitated by TotalLab TL100 (Nonlinear USA Inc., Durham, NC).
Coimmunoprecipitation
To immunoprecipitate apoA-IV and its associated proteins, HepG2 cells were switched to DMEM with 1% FBS serum overnight and then treated with r-h-apoA-IV-GFP or GFP as a control for 6 h. The nuclear proteins were extracted from the cells according to the instruction manual of NE-PER nuclear extraction reagents (Thermo Fisher Scientific). 100 μg of nuclear protein was incubated at 4 °C with 25 μl of protein G magnetic beads and 3 μg of anti-GFP antibody. The immunoprecipitated complexes were further cleared by washing the beads. Precipitates were eluted with 2× SDS loading buffer and boiled at 95 °C for 10 min to separate protein complexes. The presence of apoA-IV and NR1D1 in the immunoprecipitated complexes was analyzed by SDS-PAGE gel and immunoblotting with specific antibodies.
ChIP Assay
A ChIP assay was performed, according to the instruction manual of Active Motif, Inc. (Carlsbad, CA), with a ChIP-IT Express enzymatic kit. Immunoprecipitation was performed at 4 °C overnight with anti-NR1D1 (Sigma) and anti-apoA-IV (Santa Cruz Biotechnology). An anti-GFP immunoprecipitation sample was used as a negative control. As described previously (17), the primers were used to amplify the potential RORE region of human Glc-6-Pase promoter area, and primers encompassing a portion of the GAPDH promoter were used as additional negative control.
Luciferase Activity Assay
As described previously (17), HEK-293 cells were seeded in 24-well plates for 24 h and then transfected by using Lipofectamine 2000 (Invitrogen), with 0.3 μg of Glc-6-Pase promoter reporter plasmids and 5 ng of Renilla luciferase control reporter vector with or without 0.3 μg of pcDNA- Nr1d1 expression vector to each well. The total amount of DNA was kept constant by adding empty vector pcDNA. 24 h after transfection, the cells were treated with or without 50 μg/ml r-h-apoA-IV for 24 h. Relative luciferase activities were determined using a Dual-Luciferase reporter assay system kit (Promega, Madison, WI). The relative luciferase activity was determined by dividing the light units generated by the firefly luciferase by those generated by the Renilla luciferase in the same reaction.
RNA Interference
siRNA targeting mouse Nr1d1 (siNr1d1, SASI_Mm01_00116940) was purchased from Sigma, and a negative control siRNA (catalog no. R0017) was from Abnova (Taipei, Taiwan). To deliver siRNA into mouse primary hepatocyte cells, an Amaxa-based electroporation method (Amaxa) was used according to the instructions of the manufacturer. Briefly, 30 pmol of RNAi was used to transfect 1 × 106 cells in each electroporation. Cells were then seeded in 12-well plates, incubated with RNAi for 48 h, and then treated with or without r-m-apoA-IV (20 μg/ml) for the indicated time.
Statistics
All data represent the mean ± S.E. For cell culture experiments, results are from three independent experiments with at least three samples each time. Significant differences were determined by unpaired two-tailed Student's t test. p < 0.05 was considered significant.
RESULTS
ApoA-IV has been found recently to decrease blood glucose by enhancing glucose-stimulated insulin release (14). To determine whether apoA-IV per se also impacts hepatic glucose homeostasis without insulin secretion, apoA-IV was injected intraperitoneally without glucose administration in mice fed ad libitum or fasted for 5 h. A bolus injection with 1 μg/g body weight of apoA-IV (a physiologic dose on the basis of calculations explained under “Discussion”) lowered blood glucose levels in both ad libitum-fed and fasted mice (Fig. 1). The plasma insulin level was unaffected (data not shown). This reduction of glucose by apoA-IV lasted from 2–4 h in fed animals and from 4–6 h in 5-h fasted mice. To determine whether this effect of apoA-IV on blood glucose is a result of alterations in hepatic gluconeogenesis, 2 h after treatment with exogenous apoA-IV, hepatic gluconeogenic gene expression was determined. Both Glc-6-Pase and PEPCK gene expressions were significantly decreased in both the fed and fasted group (Fig. 1, B and D), indicating decreased gluconeogenesis. However, the decrease in PEPCK mRNA was less than Glc-6-Pase in both fed and 5-h fasted animals.
FIGURE 1.
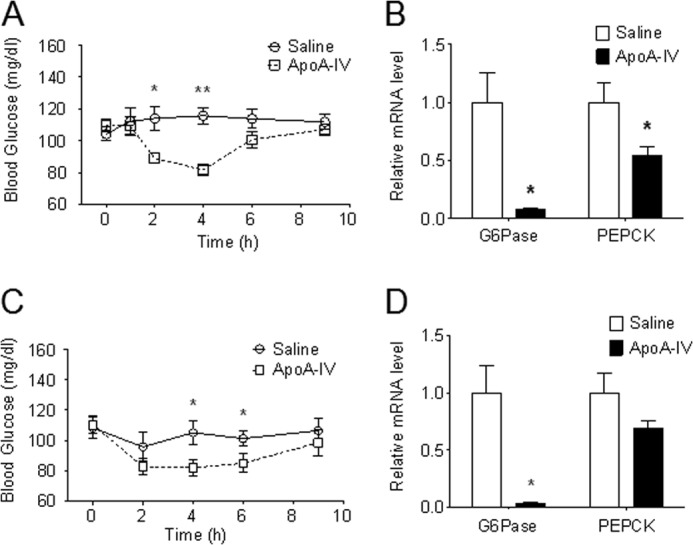
ApoA-IV lowers blood glucose and suppresses the expression of hepatic gluconeogenic genes in fed and fasted WT mice. A bolus of r-m-apoA-IV (1 μg/g body weight) was injected intraperitoneally into mice fed ad libitum and mice fasted for 5 h. Tail blood was collected at the indicated times for glucose determination. n = 5–6 mice/group. Data are presented as mean ± S.E. *, p < 0.05; **, p < 0.01 versus saline controls. For gene expression measurements, liver RNA was extracted from animals that had the same treatments as described above. Glc-6-Pase and PEPCK mRNA levels were quantitated by real-time RT-PCR and normalized to cyclophilin. n = 5–6 mice/group. Data are presented as mean ± S.E. *, p < 0.05 versus saline controls. A and B, blood glucose and gene expression in ad libitum-fed mice. C and D, blood glucose and gene expression in 5-h fasted mice.
ApoA-IV−/− mice have impaired glucose tolerance relative to the WT controls (14). To test whether apoA-IV−/− mice have impaired regulation of gluconeogenesis, we determined hepatic gluconeogenic gene expression in 5-h fasted apoA-IV−/− mice and also performed euglycemic-hyperinsulinemic clamp studies. The mRNA levels of hepatic Glc-6-Pase and PEPCK were significantly higher in apoA-IV−/− mice compared with the WT animals (Fig. 2A). During the glucose clamp period (Fig. 2B), the GIR required to maintain euglycemia was significantly lower in apoA-IV−/− mice compared with the WT controls (C). Although the rate of GU did not change significantly, endogenous glucose production was significantly higher in apoA-IV−/− compared with WT mice, indicating that insulin-dependent suppression of hepatic glucose production is impaired in apoA-IV deficient mice. This result is consistent with higher hepatic Glc-6-Pase and PEPCK gene expression in apoA-IV−/− mice. These data further support our above observations that apoA-IV regulates hepatic gluconeogenesis by suppressing the expression of these gluconeogenic genes.
FIGURE 2.
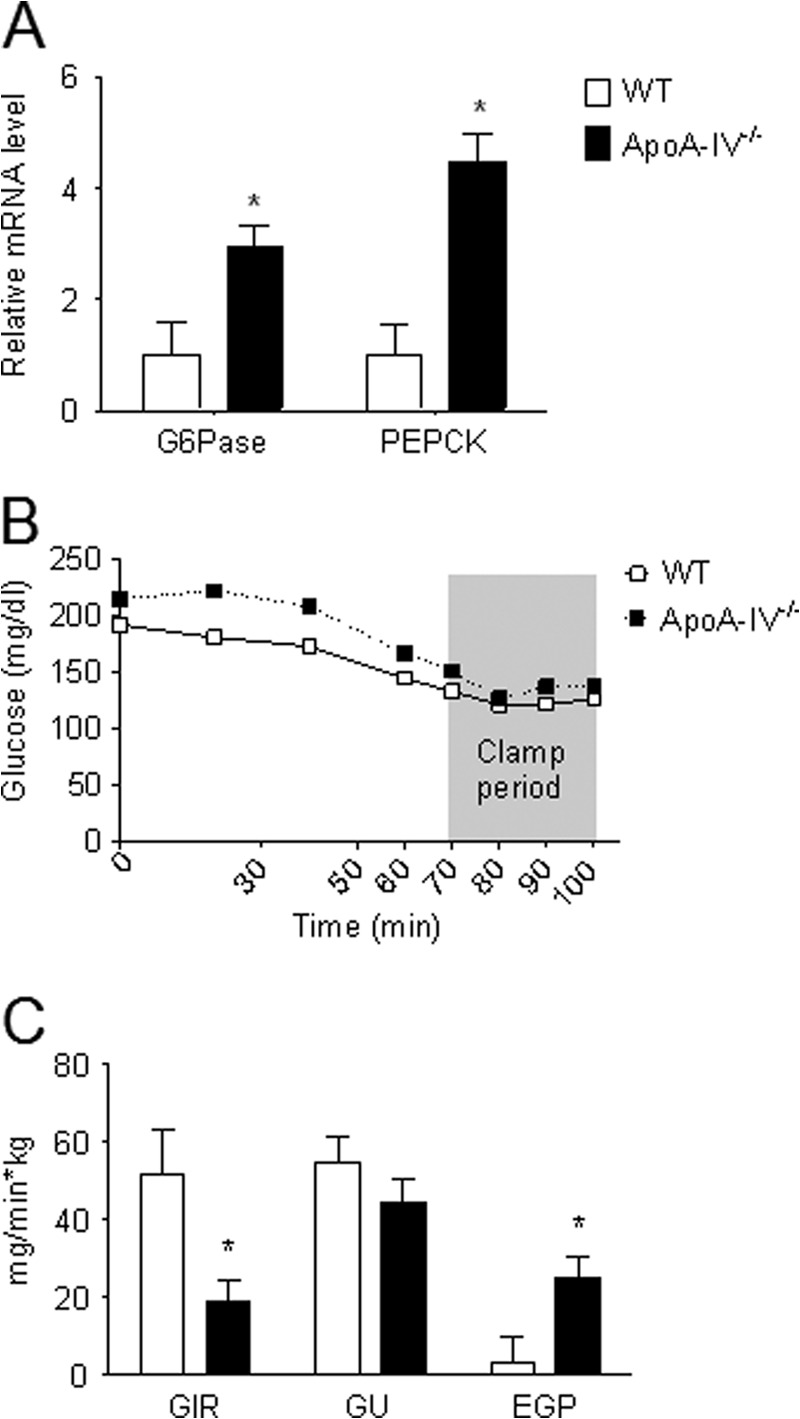
Hepatic gluconeogenic gene expression and euglycemic-hyperinsulinemic clamps in apoA-IV−/− mice. A, hepatic gluconeogenic gene expression in apoA-IV−/− and WT mice following a 5-h fast. Total RNAs were isolated from the livers in 5-h fasted apoA-IV−/− and WT mice, and Glc-6-Pase and PEPCK mRNA levels were quantitated by real-time RT-PCR and normalized to cyclophilin. n = 6–7 mice/group. Data are presented as mean ± S.E. *, p < 0.05 versus WT mice. B and C, euglycemic-hyperinsulinemic clamps in apoA-IV−/− mice. B, blood glucose levels during the clamp period. C, GIR, GU, and endogenous glucose production (EGP) at 70–100 min during the euglycemic clamp. n = 6–7 mice/group. Data are presented as mean ± S.E. *, p < 0.05 versus WT mice.
To determine whether this is a direct effect of apoA-IV on hepatic function, primary mouse hepatocytes were isolated and treated with r-m-apoA-IV. Consistent with the effects observed in vivo, apoA-IV significantly decreased PEPCK and Glc-6-Pase mRNA levels as well as glucose output into the medium by 71.4, 57.4, and 19.3%, respectively, relative to the vehicle control (Fig. 3). These data suggest that apoA-IV acts directly on hepatocytes to suppress glucose production, in part by reducing gluconeogenic gene expression.
FIGURE 3.
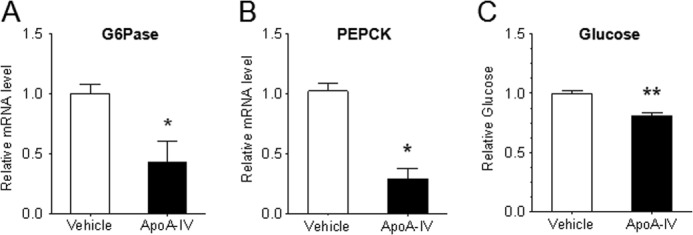
ApoA-IV decreases gluconeogenic gene expression and glucose output in primary mouse hepatocytes. Primary mouse hepatocytes were isolated from WT mice and cultured in DMEM with 10% FBS medium with 1 nm dexamethasone overnight. The cells were then switched to glucose-free medium with or without r-m-apoA-IV (20 μg/ml) along with sodium pyruvate (2 mm), sodium lactate (20 mm), dexamethasone (1 nm), and 8-(4-chlorophenylthio)-cAMP (500 μm) for 6 h (mRNA measurement) or overnight (glucose measurement). Glc-6-Pase (A) and PEPCK (B) mRNA levels were then determined by real-time RT-PCR normalized to cyclophilin, and the glucose in the medium (C) was measured. The results are from three independent experiments with at least three samples for each experiment. Data are presented as mean ± S.E. *, p < 0.05; **, p < 0.01 versus vehicle.
To determine the intrahepatic mechanism of how apoA-IV regulates gluconeogenesis, we used a bacterial two-hybrid screen, with rat apoA-IV plasmid as the bait, to screen a rat liver cDNA library. A positive clone was identified as the fragment of nuclear receptor NR1D1, a transcription factor that has been demonstrated to suppress Glc-6-Pase and PEPCK gene expression (15, 17). To corroborate this finding, immunofluorescence microscopy was used to determine whether apoA-IV and NR1D1 were colocalized in mammalian cells. Using HepG2 cells overexpressing human NR1D1, apoA-IV and NR1D1 were colocalized in the cytoplasm as well as in the nucleus (Fig. 4A). To further validate the interaction between apoA-IV and NR1D1 in mammalian cells, we employed the sensitive in situ PLA. We observed that apoA-IV and NR1D1 interacted with one another both in the cytoplasm and the nucleus (Fig. 4B). Collectively, these data suggest that apoA-IV can be taken up by HepG2 cells and interact with NR1D1 intracellularly.
FIGURE 4.
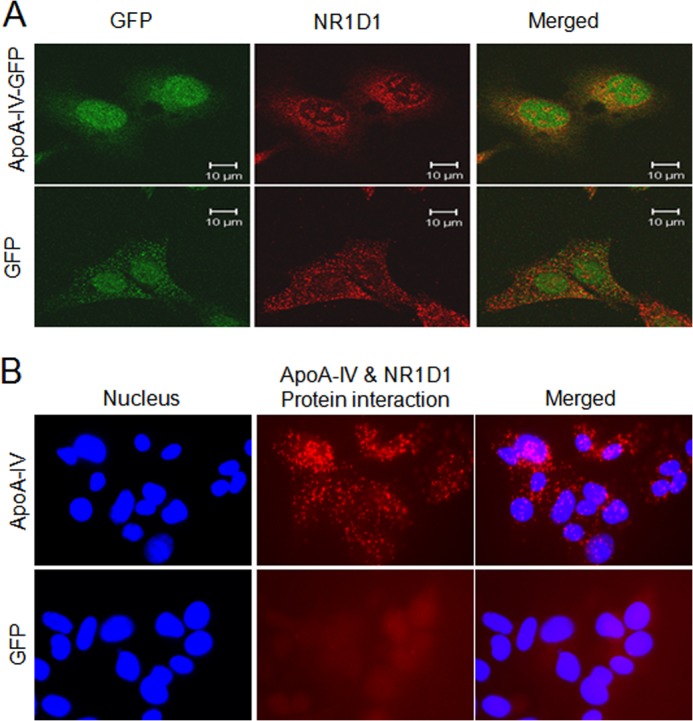
Subcellular colocalization and interaction between apoA-IV and NR1D1. A, the uptake of apoA-IV protein and subcellular colocalization with NR1D1 shown by immunofluorescence. Red represents NR1D1, and green represents r-h-apoA-IV-GFP or GFP as a negative control. B. the interaction between apoA-IV and NR1D1 detected by in situ proximity ligation assay. The interaction was visualized as red fluorescent spots. The cells were counterstained with DAPI (blue) to visualize the nuclei. This figure is a representative slide from three independent experiments.
To further confirm the binding of apoA-IV with NR1D1, we performed coimmunoprecipitation with nuclear proteins from HepG2 cells treated with r-h-apoA-IV-GFP. We observed the presence of apoA-IV and NR1D1 in the immunoprecipitated complex isolated by anti-GFP antibodies (Fig. 5A). These results demonstrate that apoA-IV binds to NR1D1 located in the nucleus. Moreover, with ChIP, we observed that apoA-IV, along with NR1D1, has the ability to directly bind to the DNA RORE region of the human Glc-6-Pase promoter area (Fig. 5B). To determine the transcriptional repression activity of apoA-IV on gluconeogenic genes, luciferase activity driven by the human Glc-6-Pase promoter was analyzed in HEK-293 cells treated with r-h-apoA-IV. ApoA-IV not only suppressed luciferase activity but also enhanced the NR1D1 suppression of Glc-6-Pase transcription by 30% (Fig. 5C). Taken together, these results suggest that apoA-IV binds with NR1D1 to directly reduce the expression of gluconeogenic genes.
FIGURE 5.
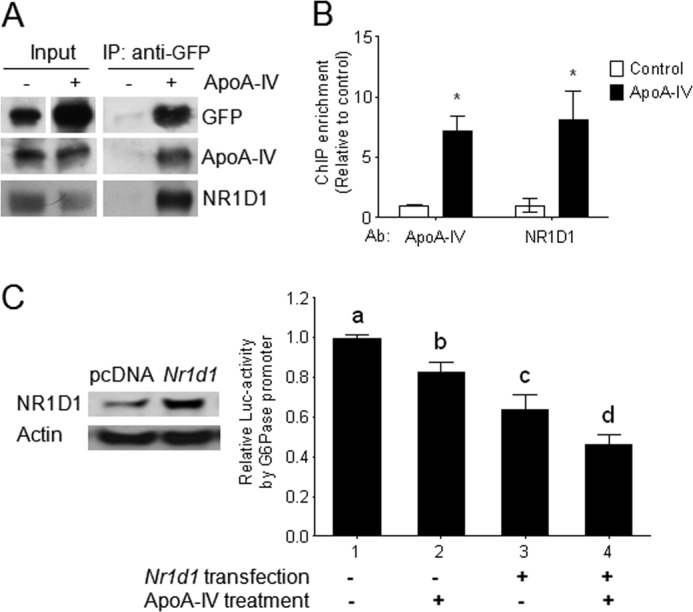
Transcriptional regulation of the Glc-6-Pase promoter by apoA-IV acting through NR1D1. A, protein-protein interaction between exogenous apoA-IV-GFP and endogenous NR1D1 in nuclei of HepG2 cells. HepG2 cells were treated with r-h-apoA-IV-GFP (50 μg/ml) for 6 h. Nuclear proteins from the cells were then isolated and immunoprecipitated (IP) with anti-GFP and assayed for apoA-IV-GFP and NR1D1by immunoblotting. GFP not conjugated to apoA-IV was used as the negative control. B, ChIP assays to detect association of exogenous apoA-IV and endogenous NR1D1 with the proximal region of the human Glc-6-Pase promoter as described under “Experimental Procedures.” The mean ± S.E. of four samples is shown. *, p < 0.05 versus vehicle control. Ab, antibody. C, activity of the luciferase reporter gene driven by the human Glc-6-Pase promoter after treatment with r-h-apoA-IV in HEK-293 cells. Inset, protein expression of Nr1d1 plasmids or empty vector pcDNA as a control in HEK-293 cells was determined by Western immunoblotting. Data are presented as mean ± S.E. from three independent experiments with at least three samples each time. Where noted by a different letter (a, b, c, or d), we found significant differences from all other groups (p < 0.05).
To confirm that the effect of apoA-IV on PEPCK and Glc-6-Pase requires NR1D1, we knocked down NR1D1 in primary mouse hepatocytes using siRNA (Fig. 6A), and the increased glucose output was observed (B). As expected, the expression of PEPCK and Glc-6-Pase and glucose production were no longer affected by r-m-apoA-IV treatment when NR1D1 gene expression was diminished (Fig. 6, C–E), clearly supporting the NR1D1 dependence of apoA-IV effects on gluconeogenic genes.
FIGURE 6.
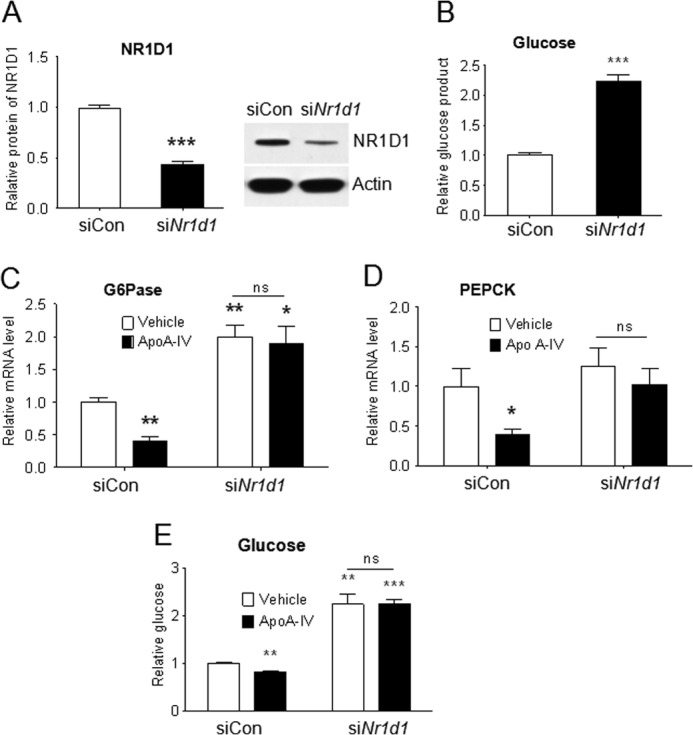
ApoA-IV represses Glc-6-Pase and glucose production through NR1D1. Primary mouse hepatocytes were transfected with siRNA against mouse NR1D1 (siNr1d1) or control siRNA (siCon) for 48 h and then treated with or without 20 μg/ml r-m-apoA-IV for 6 h or overnight as described in Fig. 3. A, NR1D1 protein detected by Western immunoblotting to determine the magnitude of inhibition of protein expression. B, glucose in the medium after NR1D1 inhibition. ***, p < 0.001 versus control siRNA. C and D, mRNA levels of Glc-6-Pase (C) and PEPCK (D) were quantitated by real-time RT-PCR and normalized to cyclophilin. ns, not significant. E, glucose in the medium. Data are presented as mean ± S.E. from three independent experiments with at least three samples each time. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus vehicle of control siRNA.
NR1D1 expression is regulated by a complex interaction of factors, including glucocorticoids and clock proteins, and also undergoes autoregulation (15–17). Regulation also differs in different tissues. We performed an initial set of experiments to investigate whether apoA-IV regulates hepatic gluconeogenesis, in part, by regulating expression of NR1D1. As shown in Fig. 7, A and B, NR1D1 gene expression and protein levels are stimulated in mouse primary hepatocytes by r-m-apoA-IV. This effect of apoA-IV on NR1D1 expression was also reproduced in the human hepatic carcinoma cell line HepG2 and human kidney cell line HEK-293 (Fig. 7, C and D).
FIGURE 7.
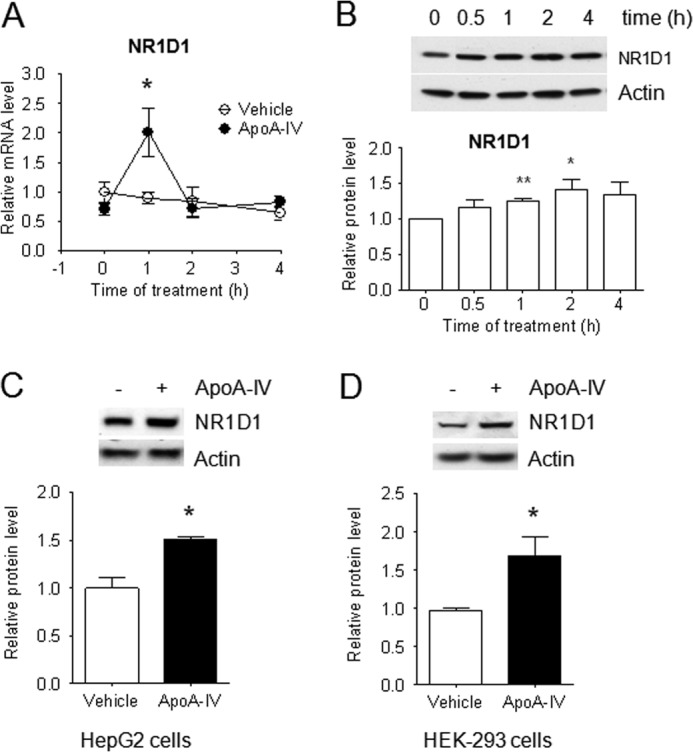
ApoA-IV induces NR1D1 expression in primary mouse hepatocytes and HepG2 and HEK-293 cells. The cells were starved in DMEM with 1% BSA overnight and then treated with or without 20 μg/ml r-m-apoA-IV in mouse hepatocytes for the indicated times or 50 μg/ml r-h-apoA-IV in HepG2 and HEK-293 for 2 h. mRNA levels in primary mouse hepatocytes were quantitated by real-time RT-PCR and normalized to cyclophilin. NR1D1 protein levels were detected by Western immunoblotting with anti-NR1D1 antibody and then standardized against actin. The results are from three independent experiments with at least three samples each time. Data are presented as mean ± S.E. *, p < 0.05; **, p < 0.01 versus vehicle. A and B, NR1D1 mRNA level (A) and NR1D1 protein level (B) in primary mouse hepatocytes. C, NR1D1 protein level in HepG2 cells. D, NR1D1 protein level in HEK-293 cells.
DISCUSSION
During active fat absorption, the production and secretion of apoA-IV by enterocytes increase dramatically (3, 20). It is estimated that the intestine contributes ∼60% of the plasma apoA-IV pool in the fasting or fat-fed rat (21). From a report of a 40% increase of plasma apoA-IV in rats after fat infusion (22), one can calculate that the increase in plasma apoA-IV is about 24 μg/ml (a calculation on the basis of the fasting plasma apoA-IV level). Lipid feeding in normal healthy fasting male subjects resulted in a 31.3% rise in plasma apoA-IV from 374 μg/ml into 491 μg/ml (1), implying that the increase in plasma apoA-IV in humans after fat absorption is about 117 μg/ml. Our previous study has shown that 1 μg of recombinant apoA-IV/g body weight of mice increases plasma apoA-IV about 10 μg/ml, as detected by Western blotting (14). Therefore, the dose of 1 μg of recombinant apoA-IV/g body weight for mice, 20 μg/ml for mouse cells, and 50 μg/ml for human cells used in our experiments are all within the physiologic range of plasma apoA-IV.
We reported recently that apoA-IV is important in blood glucose homeostasis by stimulating the secretion of insulin in the presence of elevated levels of glucose (14). Importantly, this action of apoA-IV on blood glucose modulation was not restricted to apoA-IV−/− and WT mice but was also observed in KKAy mice, which exhibit hyperglycemia and insulin resistance and are widely used to model obesity and non-insulin-dependent diabetes mellitus (14, 23). In addition to insulin secretion by pancreatic β cells, we wondered whether apoA-IV affects other aspects of glucose metabolism, such as gluconeogenesis. In this study, we demonstrate that apoA-IV also regulates blood glucose homeostasis by suppressing hepatic gluconeogenesis. It has been reported that insulin suppresses hepatic gluconeogenesis by suppressing the expression of PEPCK and Glc-6-Pase (24, 25). Because apoA-IV itself did not stimulate insulin secretion unless glucose was administered (14), we tested the effect of apoA-IV in fed and fasted mice without glucose administration and in isolated primary mouse hepatocytes to avoid the influence of insulin changes caused by apoA-IV in vivo. We observed that apoA-IV acts to lower blood glucose and suppress PEPCK and Glc-6-Pase gene expression in the absence of its effect on insulin secretion and in isolated hepatocytes in vitro. This direct and important physiologic role of apoA-IV is further supported by two additional observations: 1) higher endogenous glucose production in the apoA-IV knockout animals relative to the WT animals during the steady-state hyperinsulinemic-euglycemic clamp study and 2) higher basal hepatic gluconeogenic gene expression in apoA-IV−/− mice relative to WT animals.
The effect of apoA-IV on hepatic Glc-6-Pase is more marked than on PEPCK in fed and 5-h fasted mice, reflecting the differential regulation of PEPCK and Glc-6-Pase gene expression. The differential regulation of Glc-6-Pase and PEPCK expression has been demonstrated in other studies (26). Blood glucose represents the net balance between glucose uptake by peripheral tissues and glucose secretion by the liver. This may explain why there was a more profound decrease in blood glucose in vivo (33%) than glucose output data observed in isolated hepatocytes (19%) by apoA-IV.
A critical finding of our study is that apoA-IV inhibits gluconeogenesis through an NR1D1 pathway. We identified NR1D1 as a potential binding protein of apoA-IV using bacterial two-hybrid library screening with a rat liver cDNA library. We then used immunofluorescent microscopy to further confirm that the two proteins colocalize both in the cytoplasm and in the nucleus. In addition, we used the highly sensitive approach of the in situ PLA to validate the interaction of these proteins in HepG2 cells. Furthermore, with coimmunoprecipitation, our data confirmed that the binding of apoA-IV with NR1D1 is located in the nucleus. In addition, the ability of NR1D1 stimulated by apoA-IV to directly bind to the RORE region of the Glc-6-Pase promoter and suppress luciferase activity demonstrates that apoA-IV modulates gluconeogenesis by interacting with NR1D1 and enhancing its suppression of gluconeogenic gene transcription.
PLA is a method that is suitable for visualizing protein interactions in both tissue sections and in vitro cultured cells (27, 28). First, this method is highly sensitive and detects minute and transient protein-protein interactions. Second, it has the advantage of not only detecting the presence of interaction complexes but also their localization. Third, visualization of apoA-IV protein inside the cell indicates the uptake of exogenous apoA-IV by HepG2 cells. Using radiolabeled apoA-IV, Gotto and colleagues (29) proposed an effective uptake of plasma apoA-IV by the liver. However, the mechanism of how apoA-IV is taken up by the hepatocytes is a question that warrants further investigation. It has been proposed that the passive membrane diffusion of lipid-soluble molecules, such as steroid hormones, followed by random association with cytosolic or nuclear receptors according to intracellular concentration gradients is a universal mechanism for the cellular uptake of nuclear receptor ligands (30–32), e.g. vitamin D, sex hormones, corticosteroids, retinoids, and thyroid hormone. However, recent reports have proposed a more active role of serum-binding proteins in the delivery of the ligands for the nuclear receptors through endocytic pathways (33–35). Whether apoA-IV utilizes any of the above mentioned mechanism for delivery to the hepatocytes remains to be discovered.
Mechanistically, nuclear receptors (NRs) such as NR1D1, which belong to a superfamily of structurally related, ligand-dependent transcription factors, have been shown to be involved in the regulation of numerous aspects of energy homeostasis in an organ-specific manner (14). The endogenous/native ligands have not yet been identified. Thus, these NRs were designated as orphan NRs (36). Several agonists have since been discovered that may interact with these receptors (37, 38). It has also been reported that heme is a ligand of NR1D1 to coordinate hepatic glucose metabolism (17).
Our data show that apoA-IV not only binds to and increases activity of NR1D1 but also stimulates NR1D1 gene expression in primary mouse hepatocytes, the human hepatic cell line HepG2, and the human kidney cell line HEK-293. Actions of NR1D1 result not only from the ligand-dependent enhanced recruitment of NR1D1 to the promoter of target genes but also from the gene expression of NR1D1 itself (16, 39). For example, NR1D1 is highly induced during adipogenesis. Overexpression of NR1D1 in 3T3-L1 pre-adipocytes results in increased expression of markers of adipogenesis (40). Overexpression of NR1D1 in HepG2 cells results in decreased expression of the target genes Bmal1, Glc-6-Pase, and PEPCK (17). Thus, we propose that both interaction of NR1D1 with apoA-IV and the up-regulation of NR1D1 by apoA-IV play important roles in mediating the impact of apoA-IV on NR1D1 transactivation on gluconeogenesis.
Our results showing that NR1D1 was required for apoA-IV suppression of Glc-6-Pase and PEPCK suggest that apoA-IV is a novel endogenous protein stimulus for NR1D1, thus linking this important nuclear receptor to a protein made by the enterocytes whose synthesis and secretion is stimulated exclusively by fat absorption. NR1D1 is a downstream mediator of apoA-IV for its action on target cells. NR1D1 is not the only transcription factor regulated by apoA-IV. A most recent publication reported that apoA-IV stimulated intestinal lipid absorption by increasing transcriptional factors FOXO1 and FOXA2 gene expression (41). Their observations, along with our data, raise the possibility that apoA-IV acts to exert its function directly through multiple transcription factors in different tissues to execute different important physiological actions. ApoA-IV and NR1D1 display overlapping functions such as lipid metabolism, inflammation, and glucose homeostasis. Expression of NR1D1 is extremely high in a number of tissues involved in energy metabolism and storage, such as the liver, adipose tissue, skeletal muscle, and brain, suggestive of its important functional role in energy metabolism (24, 37, 42). The finding that apoA-IV acts through NR1D1 lends further support for a new role of apoA-IV in energy metabolism.
Our findings also highlight an important intestine-liver communication between a gut protein stimulated by fat absorption and hepatic gluconeogenesis, thus linking lipid and carbohydrate metabolism. From a clinical perspective, low-carbohydrate, high-fat diets (such as the ketogenic diet) benefit individuals with type II diabetes by improving glucose homeostasis and reducing the need for glucose-lowering drugs (43). Our finding of a direct effect of apoA-IV on the suppression of hepatic gluconeogenesis provides a potential mechanistic link between the ketogenic diet and glycemic control. Fig. 8 shows the proposed mechanism for the effect of apoA-IV on glucose homeostasis through NR1D1. ApoA-IV, thus, has utility in both type I and type II diabetes, especially under pathophysiological conditions such as non-insulin-dependent diabetes.
FIGURE 8.
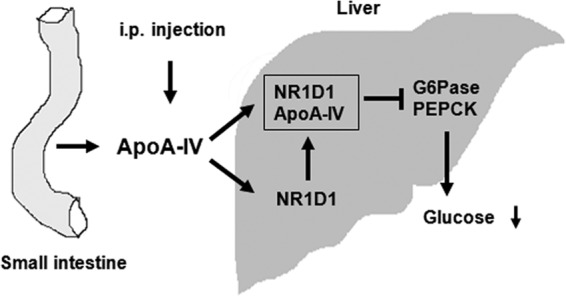
Proposed mechanism for the effect of apoA-IV on glucose homeostasis through NR1D1. ApoA-IV synthesis and secretion by the small intestine is markedly stimulated by meal feeding. Any exogenously administered apoA-IV, as well as those released from the intestine, enter the circulation and are taken up by the hepatocytes. In hepatocytes, apoA-IV binds to NR1D1, activates NR1D1 to suppress the expression of gluconeogenic genes, and increases NR1D1 expression to further suppress gluconeogenesis. These actions of apoA-IV result in the decrease of glucose output from liver and the decrease of blood glucose.
Acknowledgments
We thank Dr. Mitchell A. Lazar (University of Pennsylvania, School of Medicine, Philadelphia, PA) for the Nr1d1 and luciferase expression plasmids. We also thank James Dressman, Li Yang, Sarah Huesman, Jamie Morris, Chia-Yang Liu, and Yujin Zhang (University of Cincinnati, Cincinnati, OH) for technical assistance and Dr. Stephen C. Woods, Philip Howles, and Yong Ji (University of Cincinnati, Cincinnati, OH) for advice and manuscript preparation.
This work was supported, in whole or in part, by National Institutes of Health Grants DK 59630, DK 92138, and DK 76928 (to P. T.) and F32-091173-01 (to A. B. K.).
- PEPCK
- phosphoenolpyruvate carboxykinase
- r-m-apoA-IV
- recombinant mouse apolipoprotein A-IV
- r-h-apoA-IV
- recombinant human apolipoprotein A-IV
- GIR
- glucose infusion rate
- GU
- glucose utilization
- PLA
- proximity ligation assay
- NR
- nuclear receptor.
REFERENCES
- 1. Bisgaier C. L., Sachdev O. P., Megna L., Glickman R. M. (1985) Distribution of apolipoprotein-A-IV in human-plasma. J. Lipid Res. 26, 11–25 [PubMed] [Google Scholar]
- 2. Weinberg R. B. (2002) Apolipoprotein A-IV polymorphisms and diet-gene interactions. Curr. Opin. Lipidol. 13, 125–134 [DOI] [PubMed] [Google Scholar]
- 3. Green P. H., Glickman R. M., Riley J. W., Quinet E. (1980) Human apolipoprotein-A-IV. Intestinal origin and distribution in plasma. J. Clin. Invest. 65, 911–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu M., Doi T., Shen L., Woods S. C., Seeley R. J., Zheng S., Jackman A., Tso P. (2001) Intestinal satiety protein apolipoprotein AIV is synthesized and regulated in rat hypothalamus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R1382-R1387 [DOI] [PubMed] [Google Scholar]
- 5. Wu A. L., Windmueller H. G. (1979) Relative Contributions by liver and intestine to individual plasma apolipoproteins in the rat. J. Biol. Chem. 254, 7316–7322 [PubMed] [Google Scholar]
- 6. Lu S., Yao Y., Cheng X., Mitchell S., Leng S., Meng S., Gallagher J. W., Shelness G. S., Morris G. S., Mahan J., Frase S., Mansbach C. M., Weinberg R. B., Black D. D. (2006) Overexpression of apolipoprotein A-IV enhances lipid secretion in IPEC-1 cells by increasing chylomicron size. J. Biol. Chem. 281, 3473–3483 [DOI] [PubMed] [Google Scholar]
- 7. Ferretti G., Bacchetti T., Bicchiega V., Curatola G. (2002) Effect of human Apo AIV against lipid peroxidation of very low density lipoproteins. Chem. Phys. Lipids. 114, 45–54 [DOI] [PubMed] [Google Scholar]
- 8. Qin X. F., Swertfeger D. K., Zheng S. Q., Hui D. Y., Tso P. (1998) Apolipoprotein AIV. A potent endogenous inhibitor of lipid oxidation. Am. J. Physiol. Heart Circ. Physiol. 43, H1836-H1840 [DOI] [PubMed] [Google Scholar]
- 9. Thorngate F. E., Yancey P. G., Kellner-Weibel G., Rudel L. L., Rothblat G. H., Williams D. L. (2003) Testing the role of apoA-I, HDL, and cholesterol efflux in the atheroprotective action of low-level apoE expression. J. Lipid Res. 44, 2331–2338 [DOI] [PubMed] [Google Scholar]
- 10. Emmanuel F., Steinmetz A., Rosseneu M., Brasseur R., Gosselet N., Attenot F., Cuiné S., Séguret S., Latta M., Fruchart J. C. (1994) Identification of specific amphipathic α-helical sequence of human apolipoprotein A-IV involved in lecithin-cholesterol acyltransferase activation. J. Biol. Chem. 269, 29883–29890 [PubMed] [Google Scholar]
- 11. Main L.A., Ohnishi T., Yokoyama S. (1996) Activation of human plasma cholesteryl ester transfer protein by human apolipoprotein A-IV. Biochim. Biophys. Acta Lipids and Lipid Metabol. 1300, 17–24 [DOI] [PubMed] [Google Scholar]
- 12. Hall R. K., Granner D. K. (1999) Insulin regulates expression of metabolic genes through divergent signaling pathways. J. Basic. Clin. Physiol. Pharmacol. 10, 119–133 [DOI] [PubMed] [Google Scholar]
- 13. Unger R. H. (1985) Glucagon physiology and patho-physiology in the light of new advances. Diabetologia 28, 574–578 [DOI] [PubMed] [Google Scholar]
- 14. Wang F., Kohan A. B., Kindel T. L., Corbin K. L., Nunemaker C. S., Obici S., Woods S. C., Davidson W. S., Tso P. (2012) Apolipoprotein A-IV improves glucose homeostasis by enhancing insulin secretion. Proc. Natl. Acad. Sci. 109, 9641–9646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Adelmant G., Bègue A., Stéhelin D., Laudet V. (1996) A functional Rev-erb α responsive element located in the human Rev-erb α promoter mediates a repressing activity. Proc. Natl. Acad. Sci. 93, 3553–3558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duez H., Staels B. (2008) The nuclear receptors Rev-erbs and RORs integrate circadian rhythms and metabolism. Diab. Vasc. Dis. Res. 5, 82–88 [DOI] [PubMed] [Google Scholar]
- 17. Yin L., Wu N., Curtin J. C., Qatanani M., Szwergold N. R., Reid R. A., Waitt G. M., Parks D. J., Pearce K. H., Wisely G. B., Lazar M. A. (2007) Rev-erb α, a heme sensor that coordinates metabolic and circadian pathways. Science 318, 1786–1789 [DOI] [PubMed] [Google Scholar]
- 18. Bajzer M., Olivieri M., Haas M. K., Pfluger P. T., Magrisso I. J., Foster M. T., Tschöp M. H., Krawczewski-Carhuatanta K. A., Cota D., Obici S. (2011) Cannabinoid receptor 1 (CB1) antagonism enhances glucose utilisation and activates brown adipose tissue in diet-induced obese mice. Diabetologia 54, 3121–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lo C. M., Obici S., Dong H. H., Haas M., Lou D., Kim D. H., Liu M., D'Alessio D., Woods S. C., Tso P. (2011) Impaired insulin secretion and enhanced insulin sensitivity in cholecystokinin-deficient mice. Diabetes 60, 2000–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalogeris T. J., Fukagawa K., Tso P. (1994) Synthesis and Lymphatic transport of intestinal apolipoprotein A-IV in response to graded doses of triglyceride. J. Lipid Res. 35, 1141–1151 [PubMed] [Google Scholar]
- 21. Windmueller H. G., Wu A. L. (1981) Biosynthesis of plasma apolipoproteins by rat small-intestine without dietary or biliary fat. J. Biol. Chem. 256, 3012–3016 [PubMed] [Google Scholar]
- 22. Kalogeris T. J., Painter R. G. (2001) Adaptation of intestinal production of apolipoprotein A-IV during chronic feeding of lipid. Am. J. Physiol. Regul. Integr. Comp. Physiol. 280, R1155-R1161 [DOI] [PubMed] [Google Scholar]
- 23. Iwatsuka H., Shino A., Suzuoki Z. (1970) General survey of diabetic features of yellow Kk mice. Endocrinol. Jpn. 17, 23. [DOI] [PubMed] [Google Scholar]
- 24. Puigserver P., Rhee J., Donovan J., Walkey C. J., Yoon J. C., Oriente F., Kitamura Y., Altomonte J., Dong H., Accili D., Spiegelman B. M. (2003) Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1 α interaction. Nature 423, 550–555 [DOI] [PubMed] [Google Scholar]
- 25. Yoon J. C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C. R., Granner D. K., Newgard C. B., Spiegelman B. M. (2001) Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature 413, 131–138 [DOI] [PubMed] [Google Scholar]
- 26. Pei L., Waki H., Vaitheesvaran B., Wilpitz D. C., Kurland I. J., Tontonoz P. (2006) NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat. Med. 12, 1048–1055 [DOI] [PubMed] [Google Scholar]
- 27. Bottermann K., Reinartz M., Barsoum M., Kötter S., Gödecke A. (2013) Systematic analysis reveals elongation factor 2 and a-enolase as novel interaction partners of AKT2. PLOS ONE 8, e66045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Söderberg O., Leuchowius K. J., Gullberg M., Jarvius M., Weibrecht I., Larsson L. G., Landegren U. (2008) Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 45, 227–232 [DOI] [PubMed] [Google Scholar]
- 29. Ghiselli G., Crump W. L., 3rd, Musanti R., Sherrill B. C., Gotto A. M., Jr. (1989) Metabolism of apolipoprotein A-IV in rat. Biochim. Biophys. Acta 1006, 26–34 [DOI] [PubMed] [Google Scholar]
- 30. Mendel C. M. (1989) The free hormone hypothesis. A physiologically based mathematical-model. Endocr. Rev. 10, 232–274 [DOI] [PubMed] [Google Scholar]
- 31. Mendel C. M., Kuhn R. W., Weisiger R. A., Cavalieri R. R., Siiteri P. K., Cunha G. R., Murai J. T. (1989) Uptake of cortisol by the perfused rat-liver. Validity of the free hormone hypothesis applied to cortisol. Endocrinology 124, 468–476 [DOI] [PubMed] [Google Scholar]
- 32. Hammond G. L. (2002) Access of reproductive steroids to target tissues. Obstet. Gynecol. Clin. North Am. 29, 411–423 [DOI] [PubMed] [Google Scholar]
- 33. de Wolf F. A., Brett G. M. (2000) Ligand-binding proteins. Their potential for application in systems for controlled delivery and uptake of ligands. Pharmacol. Rev. 52, 207–236 [PubMed] [Google Scholar]
- 34. Hammes A., Andreassen T. K., Spoelgen R., Raila J., Hubner N., Schulz H., Metzger J., Schweigert F. J., Luppa P. B., Nykjaer A., Willnow T. E. (2005) Role of endocytosis in cellular uptake of sex steroids. Cell 122, 751–762 [DOI] [PubMed] [Google Scholar]
- 35. Watson C. S., Gametchu B. (2003) Proteins of multiple classes may participate in nongenomic steroid actions. Exp. Biol. Med. 228, 1272–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Andreassen T. K. (2006) The role of plasma-binding proteins in the cellular uptake of lipophilic vitamins and steroids. Horm. Metab. Res. 38, 279–290 [DOI] [PubMed] [Google Scholar]
- 37. Pearen M. A., Muscat G. E. (2010) Minireview. Nuclear hormone receptor 4A signaling. Implications for metabolic disease. Mol. Endocrinol. 24, 1891–1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hazel T. G., Nathans D., Lau L. F. (1988) A gene inducible by serum growth-factors encodes a member of the steroid and thyroid-hormone receptor superfamily. Proc. Natl. Acad. Sci. 85, 8444–8448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meng Q. J., McMaster A., Beesley S., Lu W. Q., Gibbs J., Parks D., Collins J., Farrow S., Donn R., Ray D., Loudon A. (2008) Ligand modulation of REV-ERB α function resets the peripheral circadian clock in a phasic manner. J. Cell Sci. 121, 3629–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Burris T. P. (2008) Nuclear hormone receptors for heme: REV-ERB α and REV-ERB β are ligand-regulated components of the mammalian clock. Mol. Endocrinol. 22, 1509–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pan X., Munshi M. K., Iqbal J., Queiroz J., Sirwi A. A., Shah S., Younus A., Hussain M. M. (2013) Circadian regulation of intestinal lipid absorption by apolipoprotein AIV involves forkhead transcription factors A2/O1 and microsomal triglyceride transfer protein. J. Biol. Chem. 288, 20464–20476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Laitinen S., Fontaine C., Fruchart J. C., Staels B. (2005) The role of the orphan nuclear receptor Rev-erb α in adipocyte differentiation and function. Biochimie 87, 21–25 [DOI] [PubMed] [Google Scholar]
- 43. Badman M. K., Pissios P., Kennedy A. R., Koukos G., Flier J. S., (2007) Hepatic fibroblast growth factor 21 is regulated by PPAR α and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 5, 426–437 [DOI] [PubMed] [Google Scholar]