

Background: RGS4 and RGS6 are regulator of G protein signaling (RGS) proteins, and both have been proposed to modulate parasympathetic regulation of heart rate (HR).
Results: RGS6 ablation enhances parasympathetic influence on the heart; RGS4 ablation does not.
Conclusion: RGS6 is the primary RGS modulator of parasympathetic influence on the heart.
Significance: Understanding the parasympathetic regulation of HR will improve treatment of arrhythmias.
Keywords: G Proteins, Gene Knockout, Heart, Patch Clamp Electrophysiology, Potassium Channels, RGS Proteins, GIRK/Kir3, Muscarinic, Sinoatrial Node
Abstract
Parasympathetic activity decreases heart rate (HR) by inhibiting pacemaker cells in the sinoatrial node (SAN). Dysregulation of parasympathetic influence has been linked to sinus node dysfunction and arrhythmia. RGS (regulator of G protein signaling) proteins are negative modulators of the parasympathetic regulation of HR and the prototypical M2 muscarinic receptor (M2R)-dependent signaling pathway in the SAN that involves the muscarinic-gated atrial K+ channel IKACh. Both RGS4 and RGS6-Gβ5 have been implicated in these processes. Here, we used Rgs4−/−, Rgs6−/−, and Rgs4−/−:Rgs6−/− mice to compare the relative influence of RGS4 and RGS6 on parasympathetic regulation of HR and M2R-IKACh-dependent signaling in the SAN. In retrogradely perfused hearts, ablation of RGS6, but not RGS4, correlated with decreased resting HR, increased heart rate variability, and enhanced sensitivity to the negative chronotropic effects of the muscarinic agonist carbachol. Similarly, loss of RGS6, but not RGS4, correlated with enhanced sensitivity of the M2R-IKACh signaling pathway in SAN cells to carbachol and a significant slowing of M2R-IKACh deactivation rate. Surprisingly, concurrent genetic ablation of RGS4 partially rescued some deficits observed in Rgs6−/− mice. These findings, together with those from an acute pharmacologic approach in SAN cells from Rgs6−/− and Gβ5−/− mice, suggest that the partial rescue of phenotypes in Rgs4−/−:Rgs6−/− mice is attributable to another R7 RGS protein whose influence on M2R-IKACh signaling is masked by RGS4. Thus, RGS6-Gβ5, but not RGS4, is the primary RGS modulator of parasympathetic HR regulation and SAN M2R-IKACh signaling in mice.
Introduction
Cardiac output reflects a balance between input from the parasympathetic and sympathetic branches of the autonomic nervous system. Parasympathetic tone is dominant under resting conditions, slowing heart rate (HR)3 by decreasing the spontaneous pacemaker activity of the sinoatrial node (SAN) (1, 2). Excessive parasympathetic influence can lead to atrioventricular block, and dysregulation of parasympathetic activity has been linked to sinus node dysfunction and arrhythmia (3–5). The importance of parasympathetic regulation to cardiac physiology and pathophysiology has prompted much interest in characterizing the molecular basis of parasympathetic actions in the heart.
The negative chronotropic effect of acetylcholine (ACh) is largely mediated by activation of M2 muscarinic receptors (M2R) and inhibitory (Gi/o-dependent) G protein signaling in SAN cells (1, 2). Activated Gi/o G proteins inhibit adenylyl cyclase, leading to suppression of hyperpolarization-activated cyclic nucleotide-gated cation channels and L-type Ca2+ channels while activating the muscarinic-gated atrial K+ channel IKACh (1, 2). IKACh is a tetramer formed by GIRK1 (G protein-gated inwardly-rectifying potassium (K) 1)/Kir3.1 and GIRK4/Kir3.4 subunits (6) and is found in SAN cells, atrial myocytes, and atrioventricular node cells (1). IKACh activation accounts for a substantial fraction of the negative chronotropic influence of parasympathetic stimulation on HR in mice (7). Moreover, loss of this signaling pathway correlates with elevated resting HR, a decrease in heart rate variability (HRV), resistance to pacing-induced arrhythmia, and protracted recovery times from stress, physical activity, and direct sympathetic stimulation (7–10).
The parasympathetic regulation of HR and M2R-IKACh signaling in cardiac myocytes are negatively modulated by RGS (regulator of G protein signaling) proteins (4, 11–13). RGS6 in complex with the atypical Gβ subunit Gβ5 (RGS6-Gβ5) has been implicated in the parasympathetic regulation of HR (14, 15). Indeed, Rgs6−/− mice exhibit slower resting HR, enhanced sensitivity to M2R-dependent bradycardia, elevated HRV, and enhanced M2R-IKACh signaling characterized primarily by a prominent slowing of current deactivation (14–16). Moreover, a loss-of-function mutation in the human RGS6 gene correlates with elevated HRV (16), and an RGS6 polymorphism correlate with altered HR recovery after exercise (17).
Interestingly, RGS4 was the first RGS protein implicated in the parasympathetic regulation of HR and modulation of M2R-IKACh signaling in SAN cells (18, 19). Indeed, the isolated heart and SAN cell phenotypes reported in Rgs4−/− and Rgs6−/− mice related to parasympathetic HR regulation and M2R-IKACh signaling are virtually identical (14, 15), suggesting the possibility that SAN cells employ parallel RGS4- and RGS6-Gβ5-dependent mechanisms to modulate the parasympathetic regulation of HR. To test this hypothesis, we generated mice lacking both RGS4 and RGS6 and evaluated the impact of single or dual RGS ablation on parasympathetic HR regulation and M2R-IKACh signaling in SAN cells. Our findings argue that RGS6-Gβ5 provides the dominant RGS influence on parasympathetic regulation of HR in the mouse.
EXPERIMENTAL PROCEDURES
Animals
Rgs4−/− mice (B6;129P2-Rgs4tm1Dgen/J) were purchased from The Jackson Laboratory (Bar Harbor, ME). The generation of Rgs6−/− mice was described previously (14). Mice were housed in groups on a 12-h light/dark cycle with food and water available ad libitum. All procedures were approved by the Institutional Animal Care and Use Committees of the University of Minnesota and The Scripps Research Institute.
Quantitative RT-PCR
RGS4 expression in the SAN and hippocampal tissue from wild-type and Rgs4−/− mice was compared using quantitative RT-PCR. Intact hippocampi were extracted from freshly isolated brain tissue, and total RNA was isolated using TRIzol (Invitrogen) according to the manufacturer's recommendations and treated with recombinant DNase I (Roche Applied Science). Total RNA from mouse heart (SAN and left atria) was isolated using an RNeasy fibrous tissue mini kit (Qiagen). Reverse transcription was performed using an iScriptTM cDNA synthesis kit (Bio-Rad). Quantitative PCR was performed on a LightCycler® 480 II system in a final volume of 20 μl with a LightCycler® 480 SYBR Green I Master kit (Roche Applied Science). After preincubation at 95 °C for 5 min, amplification consisted of 45 cycles of denaturation for 10 s at 95 °C, followed by annealing for 30 s at 60 °C and extension for 10 s at 72 °C. Intron-spanning RGS4 primer sets were as follows: set A (A+, 5′-gtcggaatacagcgaggagaac-3′; and A−, 5′-ggaaggattggtcaggtcaagatag-3′) and set B (B+, 5′-ttcaccatgaatgtggactggca-3′; and B−, 5′-gtccaggttcacctcttttgttgc-3′). All samples were tested in triplicate; the average of replicates was used in the data analysis. GAPDH (Mm_Gapd_2_SG QuantiTect primers, Qiagen) was used as the internal control in each test.
Quantitative Immunoblotting
Preparation of hippocampal neuron cultures was described previously (20). After 10 days, cultures were treated with vehicle (dimethyl sulfoxide) or 50 μm MG132 (American Peptide Co., Sunnyvale, CA) for 6 h. Neurons were lysed in radioimmune precipitation assay buffer containing protease inhibitors (10 mg/ml pepstatin A, 10 mg/ml aprotinin, 10 mg/ml PMSF, 1 mg/ml leupeptin, and 50 μm MG132) and incubated on ice for 20 min. Lysates were centrifuged at 16,000 × g for 15 min at 4 °C. The supernatant was mixed with 4× SDS sample buffer, heated to 75 °C, and then stored at 4 °C. DTT was added to each sample to a final concentration of 0.2 m. Samples were heated at 75 °C for 10 min, loaded onto 12% BisTris gels, and run in Tris glycine buffer. Samples were transferred to nitrocellulose membranes and blocked in 5% milk for 1 h. Primary antibodies against RGS4 (EMD Millipore) and β-actin (Abcam, Cambridge, MA) were diluted in 5% milk to 1:200 and 1:10,000, respectively, and then incubated overnight at 4 °C with shaking. Blots were washed three times with PBS and 0.1% Tween 20 and incubated with IRDye 680CW-labeled donkey anti-rabbit and IRDye 800CW-labeled donkey anti-mouse secondary antibodies (LI-COR Biosciences, Lincoln, NE) diluted 1:2000 and 1:5000 in 5% milk, respectively, for 1 h at room temperature with shaking. After three more washes with PBS and 0.1% Tween 20, blots were developed using an Odyssey infrared imaging system (LI-COR Biosciences); integral band intensities were measured using LI-COR Odyssey software.
Ex Vivo Cardiac Physiology
Mice (8–12 weeks) were heparinized (100 IU) and anesthetized using isoflurane (Halocarbon, River Edge, NJ). Hearts were rapidly excised and immediately cannulated for retrograde aortic perfusion in a constant pressure mode (60 mm Hg) with modified Krebs-Henseleit buffer containing 118.5 mm NaCl, 25 mm NaHCO3, 4.7 mm KCl, 1.2 mm KH2PO4, 11 mm d-glucose, 1.2 mm MgSO4, 1.8 mm CaCl2, and 2 mm sodium pyruvate. The buffer solution was filtered (0.22 μm) and saturated with 95% O2 and 5% CO2 at 38 °C. Hearts were allowed to stabilize for 30 min and were excluded from pharmacologic experiments and HRV analysis if any of the following was present: (i) persistent arrhythmia for >5 min, (ii) HR below 250 beats/minute, and (iii) stable steady-state HR not attained within the first 20 min. Hearts with signs of ischemia upon dismounting from the apparatus were also excluded. Ex vivo data were acquired using a PowerLab data acquisition system (ADInstruments, Colorado Springs, CO) and digitized at a sampling rate of 2 kHz. LabChart Pro v7 software with HRV and dose-response plug-ins (ADInstruments) was used for all data analysis. Basal HRs were quantified within a 10-min window using the HRV plug-in of LabChart Pro v7 as described below. The HR response to various doses of carbachol (CCh) was measured as the 7-min average following the beginning of drug application. The effect of CCh in isoproterenol-treated hearts was evaluated in a similar manner. After stabilizing the hearts by Krebs-Henseleit buffer perfusion, they were treated with 50 nm isoproterenol alone (to establish the appropriate baseline) or in combination with increasing doses of CCh. All drugs were added to the perfusate immediately prior to the start of the treatment to minimize effects on stability and concentration. The HR response was measured as the 7-min average following the beginning of drug application (CCh + isoproterenol) or the beginning of the response plateau (isoproterenol alone).
HRV Analysis
A “maximum after threshold” algorithm was used for R peak detection. Noisy data segments and ectopic beats were manually excluded from analysis. Signal pre-processing, threshold, and retrigger delay values were altered when necessary to ensure that all of the peaks within the selected window were labeled correctly. All HRV parameters were analyzed in the 5-min interval preceding drug treatment for isolated hearts, over a 5-min total of appended consecutive intervals of telemetry recording (baseline), or within the last 5-min window of drug application for isolated hearts. For time domain analysis, the following parameters were calculated: mean normal-to-normal interval (NN, ms), S.D. of all normal-to-normal intervals (SDNN, ms), and square root of the mean square of successive differences between adjacent normal-to-normal intervals (RMSSD, ms). Frequency domain analysis was done with a fast Fourier transformation size of 1024 and a Welch window with half-overlap. Frequency bands were defined as follows: 0.4–1.5 Hz, low frequency; and 1.5–5 Hz, high frequency. Power in each band and total power (TP, 0.0–10 Hz; ms2) were calculated. Low (LF) and high (HF) frequencies were also expressed in normalized units ((LF or HF) × 100/(TP − VLF)) (where VLF is very low frequency), and the low frequency/high frequency ratio was determined.
SAN Preparation and Electrophysiology
SAN cells were isolated from adult mice (2–3 months) as described (18) and used within 8 h of isolation. In brief, hearts were excised into Tyrode's solution (140 mm NaCl, 5.4 mm KCl, 1.2 mm KH2PO4, 1.0 mm MgCl2, 1.8 mm CaCl2, 5.5 mm glucose, and 5 mm HEPES (pH 7.4 with NaOH)). The SAN was identified as the narrow band of tissue located on the inner wall of the right atrium, medial to the crista terminalis and between the superior and inferior vena cavae. Two incisions were made to the superficial side of the superior and inferior vena cavae, followed by a longer cut along the outer atrial wall, to expose the SAN region. SAN-containing tissue was excised into modified Tyrode's solution containing 140 mm NaCl, 5.4 mm KCl, 1.2 mm KH2PO4, 0.2 mm CaCl2, 50 mm taurine, 18.5 mm glucose, 5 mm HEPES, and 0.1% BSA (pH 6.9 with NaOH) with elastase (0.3 mg/ml; Worthington) and collagenase II (0.21 mg/ml; Worthington). SAN tissue was digested at 37 °C for 30 min and then washed three times with solution containing 100 mm l-glutamic acid/potassium salt, 10 mm l-aspartic acid/potassium salt, 25 mm KCl, 10 mm KH2PO4, 2 mm MgSO4, 20 mm taurine, 5 mm creatine, 0.5 mm EGTA, 20 mm glucose, 5 mm HEPES, and 0.1% BSA (pH 7.2 with KOH). SAN tissue was triturated in wash solution and plated onto poly-l-lysine-coated coverslips. Coverslips containing SAN cells were transferred to a perfusion chamber, and electrophysiological recordings were conducted as described (14, 16). SAN cells targeted for electrophysiological characterization were spindle-shaped and striated and, in most cases, exhibited spontaneous beating. Most experiments used a fast-step perfusion system to allow for fast application and removal of CCh (within 100 ms) and have been described previously (14, 16). A small subset of experiments was done with gravity-flow perfusion of CCh (within 10–20 s).
Analysis
Statistical analyses were performed using Prism 5 (GraphPad Software, La Jolla, CA) and SigmaPlot 11 (Systat Software, San Jose, CA). Nonlinear fitting of dose-response data, Hill coefficient analysis, and EC50 analysis were all done with Prism 5 software using the least-squares fitting method. The impact of genotype on CCh-induced current responses (steady-state current density and kinetics) was evaluated using one-way (single-saturating concentration study) and two-way (concentration-response study) analysis of variance (ANOVA). The impact of genotype on CCh-induced HR response was evaluated using two-way (time-response study) ANOVA. Activation and deactivation kinetics were determined by fitting regions of current traces with a one-term Boltzmann equation as described previously (14). Newman-Keuls multiple comparison (one-way ANOVA) and Bonferroni (two-way ANOVA) post hoc tests were used as appropriate. The level of significance was set at p < 0.05.
RESULTS
Characterization of Rgs4−/− Mice
We acquired the Rgs4 mutant strain B6;129P2-Rgs4tm1Dgen/J used previously to implicate RGS4 in the parasympathetic regulation of HR (18). This strain harbors a lacZ-containing insert (LacO-SA-IRES-lacZ-Neo555G/Kan) that spans part of exons 1 and 2, resulting in the loss of 58 bp of coding sequence (and intervening intronic sequence), predicted to result in a frameshift and early truncation of RGS4 (Fig. 1, A and B). Sequencing of the amplicon generated in mutant mice using the supplier's genotyping conditions confirmed the presence of a foreign DNA element positioned between exons 1 and 2 in the Rgs4 gene (data not shown).
FIGURE 1.

Characterization of Rgs4−/− mice. A, schematic depiction of the Rgs4 gene, which consists of five exons (boxes 1–5). The translation initiation (ATG) and termination (STOP) codons are shown in green and red, respectively. The gray regions in exons 1 and 2 (along with intervening intronic sequence) were replaced with the lacZ-containing cassette (LacO-SA-IRES-lacZ-Neo555G/Kan) to generate B6;129P2-Rgs4tm1Dgen/J mice. B, schematic depiction of RGS4 mRNA, with exon boundaries denoted by vertical lines and numbers. The gray domain corresponds to the 58-bp fragment of coding sequence missing in the RGS4 mutant mice. Yellow highlighting shows the location of coding sequence for the catalytic (RGS) domain of RGS4. Arrows denote the positions and identities of the primer sets (A+/− and B+/−) used for quantitative RT-PCR. C, quantitative RT-PCR analysis of RGS4 expression in cardiac tissues (SAN and left atria) and hippocampi from wild-type and B6;129P2-Rgs4tm1Dgen/J mice using the primer sets depicted in C. Expression levels were normalized within samples to GAPDH (the levels of which were comparable in all tissues examined) and to wild-type samples for each primer set. ***, p < 0.001 versus wild-type mice (within tissue and primer set by t test). D, immunoblotting for RGS4 in primary hippocampal cultures (10 days in vitro) from wild-type and B6;129P2-Rgs4tm1Dgen/J mice. Cultures were pretreated with MG132 (50 μm) for 6 h prior to protein isolation. E, quantification of RGS4 immunoblotting data (n = three separate experiments). A significant impact of group was observed (F3,11 = 29.8; p < 0.001). ***, p < 0.001 versus wild-type mice (untreated).
We probed the levels of residual RGS4-like mRNAs in tissue samples from the heart (SAN and left atria) and brain (hippocampus) from RGS4 mutant mice. We used two distinct intron-spanning primer pairs targeting sequence in exons 3–5, which encode the catalytic domain of RGS4, including one pair (“A”) used previously to characterize RGS4 expression in B6;129P2-Rgs4tm1Dgen/J mice (18). Residual RGS4 expression levels were 10–100-fold lower in tissue samples from B6;129P2-Rgs4tm1Dgen/J mice than in corresponding wild-type samples (Fig. 1C).
RGS4 protein is difficult to detect in cells, as it is efficiently degraded via the ubiquitin-dependent N-end rule pathway (21, 22). Indeed, we were unable to detect recombinant RGS4 by immunoblotting in HEK cells transfected with RGS4 unless cells were pretreated with the proteasome inhibitor MG132 (data not shown). Thus, we used MG132 pretreatment to probe for RGS4 protein in hippocampal cultures from wild-type and B6;129P2-Rgs4tm1Dgen/J mice. Although RGS4 was observed in MG132-treated wild-type cultures, no immunoreactive band was seen in cultures from RGS4 mutant mice (Fig. 1, D and E). Collectively, these data indicate that the Rgs4 gene is targeted as described in B6;129P2-Rgs4tm1Dgen/J mice (hereafter referred to as Rgs4−/− mice), yielding a dramatic reduction in mRNA levels and no detectable residual RGS4 protein. Rgs4−/− mice were bred with Rgs6−/− mice to generate mice lacking both RGS4 and RGS6 (Rgs4−/−:Rgs6−/−); Rgs4−/−:Rgs6−/− mice were viable and did not display obvious phenotypic abnormalities.
Impact of RGS Ablation
Isolated Hearts
We first compared the contributions of RGS4 and RGS6 to automaticity and HRV using a retrogradely perfused Langendorff heart preparation. Basal HR in hearts from Rgs6−/− mice was significantly lower than measured in wild-type counterparts, consistent with published observations (14, 16), whereas HR in hearts from Rgs4−/− mice was normal (Fig. 2A). Interestingly, HR measured in hearts from Rgs4−/−:Rgs6−/− mice was also normal. Using time- and frequency-based analyses to compare HRV across genotypes (16), we observed that HRV was significantly elevated in hearts from Rgs6−/− mice (Table 1), whereas all HRV measures in Rgs4−/− mice were normal. Similar to the effect on HR, concurrent RGS4 ablation rescued in part the HRV abnormalities seen in hearts from Rgs6−/− mice.
FIGURE 2.
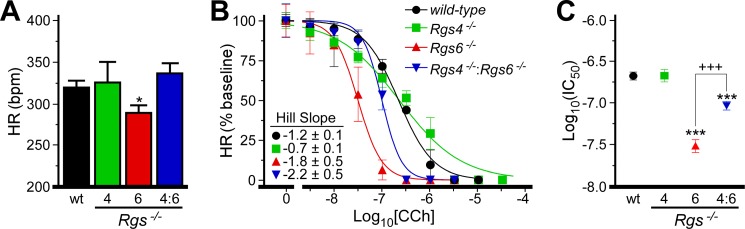
Impact of RGS ablation on HR and CCh-induced bradycardia. A, HR in isolated hearts from wild-type (wt) and Rgs−/− mice. A significant effect of genotype on HR was observed (F3,94 = 2.7, p < 0.05; n = 4–12/genotype). B, impact of CCh on HR in wild-type and Rgs−/− hearts (n = 4–8/genotype). Data are normalized to baseline HR. A significant impact of genotype was observed for normalized HR (F3,101 = 21.0, p < 0.001). Hill coefficients for each curve are listed. A significant impact of genotype was observed for Hill coefficients (F3,135 = 3.7, p = 0.013). Specifically, Rgs4−/− was significantly different from Rgs4−/−:Rgs6−/− (*, p > 0.05). C, IC50 values calculated from dose-response curves in B (F3,135 = 38.1, p < 0.001). * and ***, p < 0.05 and 0.001, respectively, versus wild-type mice; +++, p < 0.001.
TABLE 1.
HRV analysis of wild-type and Rgs−/− hearts
Shown are the results from baseline HRV analysis of 5-min recordings from 4–12 mice/genotype (8–16 weeks). NN range, range of normal-to-normal intervals between successive heart beats; SDNN, standard deviation of normal-to-normal intervals; RMSSD, square root of the mean squared difference of successive normal-to-normal intervals; TP, total power (0–10 Hz); VLF, very low frequency (0–0.4 Hz); LF, low frequency (0.4–1.5 Hz); LF norm, 100 × (LF/(TP − VLF)); HF, high frequency (1.5–5 Hz); HF norm, 100 × (HF/(TP − VLF)); LF/HF, ratio of low to high frequency. Results of corresponding ANOVAs are indicated; pairwise comparisons were made using Tukey's honest significant difference test when appropriate.
| HRV parameter | Genotype |
ANOVA | |||
|---|---|---|---|---|---|
| Wild-type | Rgs4−/− | Rgs6−/− | Rgs4−/−:Rgs6−/− | ||
| NN range (ms) | 16.2 ± 1.6 | 18.2 ± 3.7 | 30.2 ± 4.8a | 22.6 ± 7.2 | F3,30 = 3.0, p < 0.05 |
| SDNN (ms) | 2.70 ± 0.20 | 3.20 ± 0.43 | 5.22 ± 0.87a | 4.06 ± 1.28 | F3,30 = 3.4, p < 0.05 |
| RMSSD (ms) | 1.35 ± 0.28 | 1.20 ± 0.29 | 3.90 ± 1.13 | 2.31 ± 1.30 | F3,30 = 2.7, p = 0.06 |
| TP (ms2) | 5.2 ± 0.9 | 8.7 ± 2.2 | 29.0 ± 8.5a | 17.5 ± 10.0 | F3,30 = 3.8, p < 0.05 |
| VLF (ms2) | 4.2 ± 0.8 | 7.7 ± 1.9 | 18.2 ± 5.8 | 11.5 ± 5.5 | F3,30 = 2.7, p = 0.06 |
| LF (ms2) | 0.48 ± 0.17 | 0.68 ± 0.39 | 4.50 ± 1.94 | 3.53 ± 3.25 | F3,30 = 2.1, p = 0.12 |
| LF norm | 49.9 ± 3.8 | 51.7 ± 8.0 | 48.6 ± 5.4 | 46.5 ± 5.1 | F3,30 = 0.1, p = 0.96 |
| HF (ms2) | 0.54 ± 0.17 | 0.32 ± 0.11 | 6.26 ± 3.49 | 2.54 ± 2.25 | F3,30 = 1.7, p = 0.19 |
| HF norm | 50.1 ± 3.8 | 48.2 ± 8.0 | 51.4 ± 5.4 | 51.2 ± 4.5 | F3,30 = 0.1, p = 0.98 |
| LF/HF | 1.13 ± 0.17 | 1.82 ± 0.86 | 1.44 ± 0.54 | 1.00 ± 0.17 | F3,30 = 0.4, p = 0.75 |
a p < 0.05 versus wild-type mice.
We next evaluated the impact of RGS ablation on the bradycardic effects of the cholinergic agonist CCh (Fig. 2B). CCh suppressed HR in all genotypes, although differences in sensitivity were evident (Fig. 2C). The IC50 for CCh-induced bradycardia was nearly 10-fold lower in Rgs6−/− hearts (31 ± 6 nm) compared with wild-type hearts (219 ± 24 nm). In contrast, CCh sensitivity was normal in hearts from Rgs4−/− mice. The dose-response relationship in Rgs4−/− mice was shallower, however, than in other genotypes. Finally, the CCh sensitivity of hearts from Rgs4−/−:Rgs6−/− mice was significantly lower compared with wild-type mice and higher compared with Rgs6−/− mice, indicating that RGS4 ablation partially rescued the enhanced CCh sensitivity seen in Rgs6−/− mice.
M2R-IKACh Signaling in SAN Cells
We next examined the impact of RGS ablation on M2R-IKACh signaling in SAN cells (Fig. 3). Previous studies have shown that Girk4 gene ablation completely eliminates the whole-cell inward currents induced by CCh in SAN cells, confirming the IKACh dependence of the response (10, 16). No significant genotype-dependent differences in peak or steady-state CCh-induced IKACh currents were observed in response to a saturating CCh concentration (10 μm) (Fig. 3, A and B), although responses seen in cells from Rgs4−/− mice tended to be larger. Similarly, no genotype-dependent difference in acute desensitization was found (Fig. 3E). Consistent with previous reports (14, 15), the M2R-IKACh current deactivation rate was profoundly slower in Rgs6−/− SAN cells, and a modest but significant slowing of current activation was also seen (Fig. 3, C and D). In agreement with results from the isolated heart study, CCh-induced IKACh currents in Rgs4−/− cells exhibited normal activation and deactivation kinetics, and concurrent RGS4 ablation partially rescued M2R-IKACh kinetic deficits seen in Rgs6−/− cells.
FIGURE 3.

Impact of RGS ablation on M2R-IKACh signaling in SAN cells. A, IKACh currents evoked by CCh (10 μm) in SAN cells from wild-type and Rgs−/− mice. Peaks were normalized to allow for comparison of deactivation kinetics. Scale bars = 10 s/400 pA. B, no impact of genotype on peak (F3,36 = 2.0, p = 0.13) or steady-state (F3,36 = 2.2, p = 0.10) current densities was observed (n = 8–16/genotype). pF, picofarads. C and D, a significant impact of genotype was observed for activation (F3,35 = 5.0, p < 0.01) and deactivation (F3,36 = 29.1, p < 0.001) kinetics of the CCh-induced current in wild-type and Rgs−/− SAN cells. E, there was no impact of genotype on the acute desensitization of the CCh-induced IKACh current (F3,36 = 1.3, p = 0.29). F, concentration-response curves for CCh-induced IKACh activation (steady-state current amplitudes normalized to response measured with 10 μm CCh) in wild-type and Rgs−/− mice (n = 6–8/group). Hill coefficients for each curve are listed. No significant impact of genotype was observed for Hill coefficients (F3,151 = 0.4, p = 0.8). G, EC50 values calculated from concentration-response curves shown in F (F3,151 = 24.8, p < 0.0001). H and I, activation and deactivation kinetics of the CCh-induced currents in wild-type and Rgs−/− SAN cells. An interaction of genotype and dose was observed for activation kinetics (F6,95 = 7.4, p < 0.001) but not deactivation kinetics (F6,95 = 1.5, p = 0.18). However, a significant impact of group on deactivation kinetics was observed for both genotype (F3,95 = 66.2, p < 0.001) and concentration (F2,95 = 15.3, p < 0.001), so within-concentration comparisons were performed by one-way ANOVA. * and ***, p < 0.05 and 0.001, respectively, versus wild-type mice; + and +++, p < 0.05 and 0.001 respectively.
We also evaluated the sensitivity of M2R-IKACh signaling in SAN cells from wild-type and Rgs−/− mice. The EC50 values for IKACh activation by CCh in wild-type and Rgs4−/− SAN cells were indistinguishable (Fig. 3, F and G). In contrast, the M2R-IKACh signaling pathway in SAN cells from Rgs6−/− mice was ∼5-fold more sensitive to CCh. The EC50 measured in Rgs4−/−:Rgs6−/− SAN cells was slightly larger than that in Rgs6−/− cells, but the difference was not significant. Genotype- and dose-dependent differences in M2R-IKACh activation and deactivation kinetics were also observed (Fig. 3, H and I), and these differences were consistent with outcomes from the saturating CCh experiments (Fig. 3, C and D).
Influence of Another R7 RGS Protein
The partial rescue of RGS6-dependent phenotypes seen with concurrent ablation of RGS4 suggested that another cardiac RGS protein(s) is present that can modulate parasympathetic signaling when RGS4 is absent. To test this hypothesis and to explore the possibility that adaptations secondary to constitutive RGS4 ablation play a role in these phenomena, we employed a pharmacologic strategy involving CCG-63802, a compound that inhibits RGS4 (and perhaps other RGS and non-RGS proteins) by forming a covalent adduct to cysteine residues (23, 24). With CCG-63802 (50 μm) included in the pipette solution, the M2R-IKACh deactivation rate was significantly accelerated in SAN cells from Rgs6−/− mice, similar to the effect of concurrent genetic ablation of RGS4 and RGS6 (Fig. 4, A and C). There was no effect of CCG-63802 on activation kinetics, and whereas peak and steady-state current responses tended to be larger than those in control cells, this difference was not significant (Fig. 4, B, D, and E).
FIGURE 4.

Influence of another R7 RGS protein revealed by RGS4 ablation. A, IKACh currents evoked by CCh (10 μm) in Rgs6−/− and Gβ5−/− SAN cells treated with vehicle or CCG-63802 (50 μm, delivered via the pipette). Peak currents were normalized for comparison. B, activation kinetics of the CCh-induced current in Rgs6−/− (t14 = 0.1, p = 0.9) or Gβ5−/− (t8 = 0.2, p = 1.0) SAN cells. C, deactivation kinetics of the CCh-induced current in Rgs6−/− (t18 = 3.0, p < 0.01) or Gβ5−/− (t6 = 0.3, p = 0.8) SAN cells. D, peak IKACh current densities in Rgs6−/− (t15 = 1.7, p = 0.1) or Gβ5−/− (t9 = 0.4, p = 0.7) SAN cells. pF, picofarads. E, steady-state IKACh current densities in Rgs6−/− (t16 = 1.1, p = 0.3) or Gβ5−/− (t8 = 0.2, p = 0.8) SAN cells. Note that only within-genotype comparisons were made (n = 4–12/genotype). ***, p < 0.001 versus vehicle for each genotype.
As RGS6 belongs to the R7 subfamily of RGS proteins (which consists of RGS6, RGS7, RGS9, and RGS11 (25)), we next asked whether the unusual influence of genetic ablation of RGS4 or acute CCG-63802 application on M2R-IKACh signaling seen in SAN cells from Rgs6−/− mice was attributable to another R7 RGS family member. We exploited the observation that all four R7 RGS proteins are undetectable in Gβ5−/− mice (26). Consistent with the involvement of multiple R7 RGS proteins, CCh-induced IKACh current deactivation was significantly longer in Gβ5−/− SAN cells than in Rgs6−/− SAN cells (t13 = 2.30, p < 0.05). Moreover, although CCG-63802 accelerated the M2R-IKACh deactivation rate in Rgs6−/− SAN cells, it had no such effect in Gβ5−/− SAN cells (Fig. 4, A and C). The differential impact of RGS4 inhibition (genetic or pharmacologic) on M2R-IKACh signaling in SAN cells from Rgs6−/− and Gβ5−/− mice argues that RGS4 suppresses the influence of another R7 RGS family member when RGS6 is absent.
DISCUSSION
Our findings extend the previously reported roles for RGS6 in the parasympathetic regulation of HR and M2R-IKACh signaling in SAN cells (14–16). RGS6 ablation correlates with decreased HR (in vivo and ex vivo), increased HRV, enhanced sensitivity to the negative chronotropic effects of CCh, and multiple effects on M2R-IKACh signaling in SAN cells that should collectively enhance the influence of this signaling pathway on cardiac output. Under equivalent conditions, no similar impact of RGS4 ablation was observed.
To the contrary, we did observe that the ablation of RGS4 seemed to enhance slightly M2R-IKACh signaling in SAN cells rather than disrupt it. Although these effects were not statistically significant, the loss of RGS4 (by either genetic or pharmacologic manipulation) tended to correlate with larger peak CCh-induced responses and higher EC50 values for CCh-induced IKACh activation and exhibited faster activation and deactivation kinetics. In addition, isolated heart experiments with Rgs4−/− mice revealed a shallower CCh dose-response curve compared with wild-type mice even though the IC50 was the same for both genotypes. Collectively, these observations suggest that RGS4 can impact the parasympathetic regulation of HR and underlying signaling pathways, albeit in manner distinct from that proposed previously. Furthermore, the mild effect observed with RGS4 ablation appears to be exacerbated with the concurrent ablation of RGS6, leading to the partial rescue of the prominent phenotypes linked to RGS6 ablation in both single-cell and isolated heart assays.
The lack of negative influence of RGS4 on parasympathetic signaling was unexpected in light of a previous report involving the same mice (18). Although some of the cardiac phenotypes in Rgs4−/− mice reported previously could be due to an influence of RGS4 in the central nervous system (e.g. the enhanced negative chronotropic effect of CCh seen in conscious Rgs4−/− mice), there are few differences between studies with respect to the design of the isolated heart experiments. In this context, it is worth noting that we also examined the influence of RGS4 ablation on CCh-induced bradycardia under conditions of sympathetic (isoproterenol) stimulation. Although the previous study reported that CCh-induced bradycardia was more pronounced in isoproterenol-treated hearts from Rgs4−/− mice compared with wild-type controls (18), we observed no genotype-dependent difference in this assay (Fig. 5, A–C). Interestingly, the baseline HR of retrogradely perfused hearts was slower (300–350 beats/minute) in our hands than in the previous study (>400 beats/minute). This may be due to the difference in the perfusion protocol used in the ex vivo heart studies, which involved constant pressure (our study) or constant flow (previous study). Moreover, although the CCh sensitivity of hearts from Rgs4−/− mice was apparently similar in both studies, hearts from wild-type mice appeared to be slightly more sensitive to CCh-induced bradycardia in our study.
FIGURE 5.
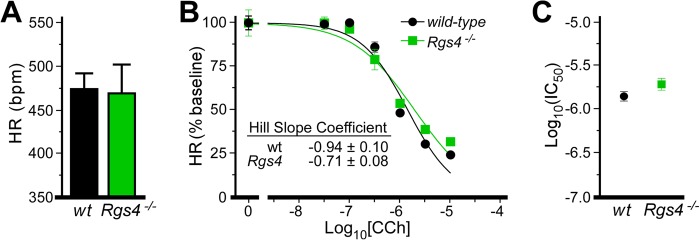
RGS4 ablation in isoproterenol-stimulated hearts. A, HR in isolated hearts from wild-type (wt) and Rgs4−/− mice stimulated with isoproterenol. HRs in isoproterenol-treated wild-type and Rgs4−/− hearts were not significantly different (t4 = 0.14, p = 0.9). bpm, beats/min. B, impact of CCh on HR in wild-type and Rgs4−/− hearts (n = 3/genotype). Data are normalized to baseline HR. There was no significant impact of genotype observed for normalized HR (F2,37 = 2.4, p = 0.1). Hill coefficients for each curve are listed and were not significantly different (t37 = 1.8, p = 0.09). C, IC50 values calculated from dose-response curves in B were not significant (t37 = 1.6, p = 0.12).
With respect to the measurement of M2R-IKACh signaling in SAN cells, there were also few study design-related differences that might explain the divergent outcomes. We used a rapid solution-exchange system to apply and remove CCh to/from SAN cells. Accordingly, the rates of current activation and deactivation were ∼100-fold faster in our study. Rapid solution exchange (complete solution exchange in <100 ms) ensures that the time course of IKACh activation and deactivation is controlled by G protein cycling rather than gradually changing levels of agonist and/or the potentially confounding influence of signaling pathway desensitization. This difference in experimental design is, however, unlikely to explain the discrepancy between studies. Indeed, using a slower gravity-flow perfusion approach to deliver and remove CCh, we found no difference in CCh-induced IKACh current densities, desensitization, or kinetics in SAN cells from wild-type and Rgs4−/− mice (Fig. 6, A–D).
FIGURE 6.
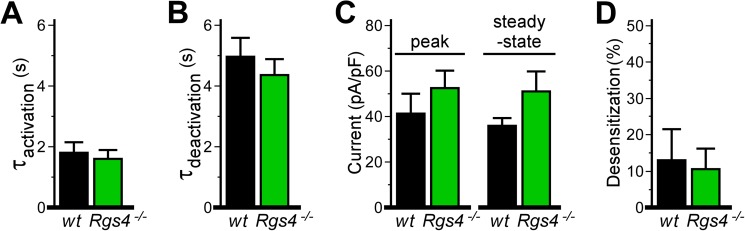
M2R-IKACh signaling evaluated by slow perfusion of CCh. Shown are summary data of IKACh currents evoked by CCh (10 μm) in wild-type and Rgs4−/− SAN cells, taken from experiments in which CCh was allowed to slowly fill the recording chamber by gravity flow and was washed out of the chamber by gravity flow of CCh-free bath solution. Although responses in SAN cells from Rgs4−/− mice showed slightly faster kinetics and larger current densities, there were no significant genotype-dependent differences in any of the following parameters: activation rate (t8 = 0.4, p = 0.7) (A), deactivation rate (t7 = 0.7, p = 0.5) (B), peak (t8 = 0.9, p = 0.4) or steady-state (t7 = 1.5, p = 0.2) CCh-induced current densities (C), or acute desensitization of the CCh-induced IKACh current (t8 = 0.2, p = 0.8; n = 4–6/genotype) (D). pF, picofarads.
Although we did not observe a marked influence of RGS4 on the parasympathetic regulation of HR or M2R-IKACh signaling in SAN cells, our data suggest that RGS4 could influence such signaling under certain circumstances. Indeed, although the sensitivity of the isolated heart to CCh-induced bradycardia was normal in Rgs4−/− mice, the slope of curve was shallower. Moreover, M2R-IKACh current densities tended to be higher with inhibition of RGS4. Although the mechanisms underlying these observations are unclear, RGS4 can serve as a GTPase-activating protein for Gαq (27–29), which should enhance the activity of phospholipase Cβ and increase the levels of phosphoinositol 4,5-bisphosphate, a required cofactor for GIRK channel gating (30, 31). As RGS4 is up-regulated in the failing human heart (32), a more robust influence of RGS4 on parasympathetic HR regulation and related signaling might be expected in this setting.
Studies with embryonic stem cell-derived cardiomyocytes expressing RGS-insensitive G proteins implicated Gαi2 and Gαo in M2R-dependent signaling (11). RGS6-Gβ5 strictly acts on members of the Gαi/o family (33), whereas RGS4 can serve as a GTPase-activating protein for Gαo, Gαi1, Gαi2, Gαi3, and Gαq (34–36). Because RGS4 can regulate M2R-GIRK signaling mediated by Gαi2 and Gαo in expression systems (37, 38), some compartmentalization mechanism in SAN cells must facilitate the interaction between RGS6-Gβ5 and M2R-IKACh and/or preclude the interaction between RGS4 and M2R-GIRK. Macromolecular complex formation may play a role in this process, as RGS-Gβ5 can interact directly with cardiac and neuronal GIRK channels (14, 39). In contrast, although RGS4 can interact with receptor-GIRK complexes (38, 40), the association appears to be driven by an interaction between RGS4 and the receptor.
Our data suggest that another member of the R7 RGS protein family, whose influence on M2R-IKACh signaling is masked by RGS4 in the absence of RGS6, can partially compensate for the loss of RGS6. RGS7 and/or RGS9 may be the compensatory factor(s), as expression of both RGS proteins has been detected in the heart (32, 41). The presence of another R7 RGS protein would explain the comparably larger impact of Gβ5 ablation on IKACh deactivation kinetics in SAN cells than is seen in SAN cells from Rgs6−/− mice. It remains unclear how RGS4 masks its influence on M2R-IKACh activity. It is possible that RGS4 is expressed at levels sufficiently higher than the compensatory R7 RGS protein(s) in SAN cells, which allows it to out-compete residual RGS-Gβ5 complexes for binding to the receptor and/or channel.
Enhanced parasympathetic input to the heart facilitates the induction of atrial fibrillation, whereas decreased parasympathetic influence (and decreased IKACh) confers resistance to atrial fibrillation (12). A mutation in the human GIRK4/KCNJ5 gene that yields reduced surface expression of IKACh has been linked to long QT syndrome (42, 43). Furthermore, loss-of-function mutations in RGS6 that increase IKACh function result in elevated HRV and increased susceptibility to atrial fibrillation induction (16). These observations suggest directly that certain arrhythmias may be effectively treated or prevented by decreasing (atrial fibrillation) or enhancing (long QT syndrome) M2R-IKACh signaling. Although direct-acting IKACh agonists and antagonists may eventually prove useful in these settings, the data presented herein argue that RGS6-Gβ5 should be considered as a novel target for the treatment or prevention of arrhythmias.
Acknowledgments
We thank Daniele Young, Jennifer Kutzke, and Natalia Martemyanova for maintaining the mouse colonies.
This work was supported, in whole or in part, by National Institutes of Health Grants HL105550 (to K. W. and K. A. M.), MH061933 (to K. W.), and DA007234 (to N. W.).
- HR
- heart rate
- SAN
- sinoatrial node
- ACh
- acetylcholine
- M2R
- M2 muscarinic receptor
- HRV
- heart rate variability
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- CCh
- carbachol
- ANOVA
- analysis of variance.
REFERENCES
- 1. Mangoni M. E., Nargeot J. (2008) Genesis and regulation of the heart automaticity. Physiol. Rev. 88, 919–982 [DOI] [PubMed] [Google Scholar]
- 2. DiFrancesco D. (2010) The role of the funny current in pacemaker activity. Circ. Res. 106, 434–446 [DOI] [PubMed] [Google Scholar]
- 3. Dobrzynski H., Boyett M. R., Anderson R. H. (2007) New insights into pacemaker activity: promoting understanding of sick sinus syndrome. Circulation 115, 1921–1932 [DOI] [PubMed] [Google Scholar]
- 4. Fu Y., Huang X., Piao L., Lopatin A. N., Neubig R. R. (2007) Endogenous RGS proteins modulate SA and AV nodal functions in isolated heart: implications for sick sinus syndrome and AV block. Am. J. Physiol. Heart Circ. Physiol. 292, H2532–H2539 [DOI] [PubMed] [Google Scholar]
- 5. Vaseghi M., Shivkumar K. (2008) The role of the autonomic nervous system in sudden cardiac death. Prog. Cardiovasc. Dis. 50, 404–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Krapivinsky G., Gordon E. A., Wickman K., Velimirović B., Krapivinsky L., Clapham D. E. (1995) The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+-channel proteins. Nature 374, 135–141 [DOI] [PubMed] [Google Scholar]
- 7. Wickman K., Nemec J., Gendler S. J., Clapham D. E. (1998) Abnormal heart rate regulation in GIRK4 knockout mice. Neuron 20, 103–114 [DOI] [PubMed] [Google Scholar]
- 8. Kovoor P., Wickman K., Maguire C. T., Pu W., Gehrmann J., Berul C. I., Clapham D. E. (2001) Evaluation of the role of IKACh in atrial fibrillation using a mouse knockout model. J. Am. Coll. Cardiol. 37, 2136–2143 [DOI] [PubMed] [Google Scholar]
- 9. Bettahi I., Marker C. L., Roman M. I., Wickman K. (2002) Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J. Biol. Chem. 277, 48282–48288 [DOI] [PubMed] [Google Scholar]
- 10. Mesirca P., Marger L., Toyoda F., Rizzetto R., Audoubert M., Dubel S., Torrente A. G., Difrancesco M. L., Muller J. C., Leoni A. L., Couette B., Nargeot J., Clapham D. E., Wickman K., Mangoni M. E. (2013) The G-protein-gated K+ channel, IKACh, is required for regulation of pacemaker activity and recovery of resting heart rate after sympathetic stimulation. J. Gen. Physiol. 142, 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fu Y., Huang X., Zhong H., Mortensen R. M., D'Alecy L. G., Neubig R. R. (2006) Endogenous RGS proteins and Gα subtypes differentially control muscarinic and adenosine-mediated chronotropic effects. Circ. Res. 98, 659–666 [DOI] [PubMed] [Google Scholar]
- 12. Mighiu A. S., Heximer S. P. (2012) Controlling parasympathetic regulation of heart rate: a gatekeeper role for RGS proteins in the sinoatrial node. Front. Physiol. 3, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stewart A., Huang J., Fisher R. A. (2012) RGS proteins in heart: brakes on the vagus. Front. Physiol. 3, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Posokhova E., Wydeven N., Allen K. L., Wickman K., Martemyanov K. A. (2010) RGS6/Gβ5 complex accelerates IKACh gating kinetics in atrial myocytes and modulates parasympathetic regulation of heart rate. Circ. Res. 107, 1350–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang J., Huang J., Maity B., Gao Z., Lorca R. A., Gudmundsson H., Li J., Stewart A., Swaminathan P. D., Ibeawuchi S. R., Shepherd A., Chen C. K., Kutschke W., Mohler P. J., Mohapatra D. P., Anderson M. E., Fisher R. A. (2010) RGS6, a modulator of parasympathetic activation in heart. Circ. Res. 107, 1345–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Posokhova E., Ng D., Opel A., Masuho I., Tinker A., Biesecker L. G., Wickman K., Martemyanov K. A. (2013) Essential role of the M2R-RGS6-IKACh pathway in controlling intrinsic heart rate variability. PLoS ONE 8, e76973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vasan R. S., Larson M. G., Aragam J., Wang T. J., Mitchell G. F., Kathiresan S., Newton-Cheh C., Vita J. A., Keyes M. J., O'Donnell C. J., Levy D., Benjamin E. J. (2007) Genome-wide association of echocardiographic dimensions, brachial artery endothelial function and treadmill exercise responses in the Framingham Heart Study. BMC Med. Genet. 8, S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cifelli C., Rose R. A., Zhang H., Voigtlaender-Bolz J., Bolz S. S., Backx P. H., Heximer S. P. (2008) RGS4 regulates parasympathetic signaling and heart rate control in the sinoatrial node. Circ. Res. 103, 527–535 [DOI] [PubMed] [Google Scholar]
- 19. Neubig R. R. (2008) And the winner is … RGS4! Circ. Res. 103, 444–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wydeven N., Young D., Mirkovic K., Wickman K. (2012) Structural elements in the Girk1 subunit that potentiate G protein-gated potassium channel activity. Proc. Natl. Acad. Sci. U.S.A. 109, 21492–21497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davydov I. V., Varshavsky A. (2000) RGS4 is arginylated and degraded by the N-end rule pathway in vitro. J. Biol. Chem. 275, 22931–22941 [DOI] [PubMed] [Google Scholar]
- 22. Lee M. J., Tasaki T., Moroi K., An J. Y., Kimura S., Davydov I. V., Kwon Y. T. (2005) RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 102, 15030–15035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blazer L. L., Roman D. L., Chung A., Larsen M. J., Greedy B. M., Husbands S. M., Neubig R. R. (2010) Reversible, allosteric small-molecule inhibitors of regulator of G protein signaling proteins. Mol. Pharmacol. 78, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vashisth H., Storaska A. J., Neubig R. R., Brooks C. L., 3rd (2013) Conformational dynamics of a regulator of G-protein signaling protein reveals a mechanism of allosteric inhibition by a small molecule. ACS Chem. Biol. 10.1021/cb400568g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anderson G. R., Posokhova E., Martemyanov K. A. (2009) The R7 RGS protein family: multi-subunit regulators of neuronal G protein signaling. Cell Biochem. Biophys. 54, 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen C. K., Eversole-Cire P., Zhang H., Mancino V., Chen Y. J., He W., Wensel T. G., Simon M. I. (2003) Instability of GGL domain-containing RGS proteins in mice lacking the G protein β-subunit Gβ5. Proc. Natl. Acad. Sci. U.S.A. 100, 6604–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang C., Hepler J. R., Gilman A. G., Mumby S. M. (1997) Attenuation of Gi- and Gq-mediated signaling by expression of RGS4 or GAIP in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 94, 6159–6163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yan Y., Chi P. P., Bourne H. R. (1997) RGS4 inhibits Gq-mediated activation of mitogen-activated protein kinase and phosphoinositide synthesis. J. Biol. Chem. 272, 11924–11927 [DOI] [PubMed] [Google Scholar]
- 29. Hepler J. R., Berman D. M., Gilman A. G., Kozasa T. (1997) RGS4 and GAIP are GTPase-activating proteins for Gqα and block activation of phospholipase Cβ by γ-thio-GTP-Gqα. Proc. Natl. Acad. Sci. U.S.A. 94, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lüscher C., Slesinger P. A. (2010) Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hibino H., Inanobe A., Furutani K., Murakami S., Findlay I., Kurachi Y. (2010) Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol. Rev. 90, 291–366 [DOI] [PubMed] [Google Scholar]
- 32. Mittmann C., Chung C. H., Höppner G., Michalek C., Nose M., Schüler C., Schuh A., Eschenhagen T., Weil J., Pieske B., Hirt S., Wieland T. (2002) Expression of ten RGS proteins in human myocardium: functional characterization of an upregulation of RGS4 in heart failure. Cardiovasc. Res. 55, 778–786 [DOI] [PubMed] [Google Scholar]
- 33. Hooks S. B., Waldo G. L., Corbitt J., Bodor E. T., Krumins A. M., Harden T. K. (2003) RGS6, RGS7, RGS9, and RGS11 stimulate GTPase activity of Gi family G-proteins with differential selectivity and maximal activity. J. Biol. Chem. 278, 10087–10093 [DOI] [PubMed] [Google Scholar]
- 34. Watson N., Linder M. E., Druey K. M., Kehrl J. H., Blumer K. J. (1996) RGS family members: GTPase-activating proteins for heterotrimeric G-protein α-subunits. Nature 383, 172–175 [DOI] [PubMed] [Google Scholar]
- 35. Lan K. L., Sarvazyan N. A., Taussig R., Mackenzie R. G., DiBello P. R., Dohlman H. G., Neubig R. R. (1998) A point mutation in Gαo and Gαi1 blocks interaction with regulator of G protein signaling proteins. J. Biol. Chem. 273, 12794–12797 [DOI] [PubMed] [Google Scholar]
- 36. Heximer S. P., Watson N., Linder M. E., Blumer K. J., Hepler J. R. (1997) RGS2/G0S8 is a selective inhibitor of Gqα function. Proc. Natl. Acad. Sci. U.S.A. 94, 14389–14393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang Q., Pacheco M. A., Doupnik C. A. (2002) Gating properties of GIRK channels activated by Gαo- and Gαi-coupled muscarinic m2 receptors in Xenopus oocytes: the role of receptor precoupling in RGS modulation. J. Physiol. 545, 355–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jaén C., Doupnik C. A. (2006) RGS3 and RGS4 differentially associate with G protein-coupled receptor-Kir3 channel signaling complexes revealing two modes of RGS modulation. Precoupling and collision coupling. J. Biol. Chem. 281, 34549–34560 [DOI] [PubMed] [Google Scholar]
- 39. Xie K., Allen K. L., Kourrich S., Colón-Saez J., Thomas M. J., Wickman K., Martemyanov K. A. (2010) Gβ5 recruits R7 RGS proteins to GIRK channels to regulate the timing of neuronal inhibitory signaling. Nat. Neurosci. 13, 661–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fowler C. E., Aryal P., Suen K. F., Slesinger P. A. (2007) Evidence for association of GABAB receptors with Kir3 channels and regulators of G protein signalling (RGS4) proteins. J. Physiol. 580, 51–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kardestuncer T., Wu H., Lim A. L., Neer E. J. (1998) Cardiac myocytes express mRNA for ten RGS proteins: changes in RGS mRNA expression in ventricular myocytes and cultured atria. FEBS Lett. 438, 285–288 [DOI] [PubMed] [Google Scholar]
- 42. Yang Y., Yang Y., Liang B., Liu J., Li J., Grunnet M., Olesen S. P., Rasmussen H. B., Ellinor P. T., Gao L., Lin X., Li L., Wang L., Xiao J., Liu Y., Liu Y., Zhang S., Liang D., Peng L., Jespersen T., Chen Y. H. (2010) Identification of a Kir3.4 mutation in congenital long QT syndrome. Am. J. Hum. Genet. 86, 872–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang F., Liu J., Hong L., Liang B., Graff C., Yang Y., Christiansen M., Olesen S. P., Zhang L., Kanters J. K. (2013) The phenotype characteristics of type-13 long QT syndrome with mutation in KCNJ5 (Kir3.4-G387R). Heart Rhythm 10, 1500–1506 [DOI] [PubMed] [Google Scholar]