

Abstract
The objective of the present study was to identify a camptothecin (CPT) prodrug with optimal release and cytotoxicity properties for immobilization on a passively targeted microparticle delivery system. A series of α-amino acid ester prodrugs of CPT were synthesized, characterized and evaluated. Four CPT prodrugs were synthesized with increasing aliphatic chain length (glycine (Gly) (2a), alanine (Ala) (2b), aminobutyric acid (Abu) (2c) and norvaline (Nva) (2d)). Prodrug reconversion was studied at pH 6.6, 7.0 and 7.4 corresponding to tumor, lung and extracellular/physiological pH, respectively. Cytotoxicity was evaluated in A549 human lung carcinoma cells using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The hydrolytic reconversion rate to parent CPT increased with decreasing side chain length as well as increasing pH. The Hill slope of 2d was significantly less than CPT and the other prodrugs tested, indicating a higher cell death rate at lower concentrations. These results suggest that 2d is the best candidate for a passively targeted sustained release lung delivery system.
Keywords: Amino acid prodrug, camptothecin, bioconjugate, lung cancer
Introduction
Camptothecin (CPT), a cytotoxic quinoline alkaloid, is a potent anticancer agent that inhibits both DNA and RNA synthesis.1-3 The lactone form of CPT (i.e., the active form) is responsible for its anticancer activity and the stabilization of the DNA-topoisomerase complex.4, 5 DNA enzyme topoisomerase I is a nuclear enzyme that is needed for DNA replication. The lactone form of CPT binds to the topoisomerase I-DNA complex to form a ternary complex that is very stable.6-8 CPT primarily exhibits cytotoxicity during the period of DNA synthesis.9 S-phase cells are 100-1000 times more sensitive to CPT than those in G1 or G2.10 Since typically less than 26% of cells are in the S-phase at any given point in time,11 maintaining continuous low dose CPT exposure would appear to be an optimal clinical strategy for treating cancer.
There are two major drawbacks to CPT and similar chemotherapeutic agents that have made them less attractive for clinical use. First, CPT has unfavorable physical-chemical properties and second, it has severe clinical toxicities. CPT has very poor aqueous solubility (2.5 μg/mL) making it difficult to formulate.12, 13 Under physiological conditions, CPT is rapidly hydrolyzed to the carboxylate form.14 In addition, the carboxylate form is known to induce severe cumulative hematological toxicity, diarrhea and chemical or hemorrhagic cystitis, which are often formidable and unpredictable.15 Therefore, approaches that stabilize the lactone ring prior to entry into the site of action are a critical feature in order to maximize therapeutic activity and minimize toxicity. Several CPT analogues possessing stabilized lactone structures have been developed to overcome this challenge.16-18 For example, exatecan mesylate (DX-8951f), a novel hexacyclic topoisomerase I inhibitor analogue, in Phase I studies demonstrated activity in solid tumors and less toxicity than CPT even though myelosuppression remained a dose limiting toxicity.19-21 Phase II studies demonstrated moderate activity in patients with anthracycline-refractory and taxane-refractory, metastatic breast carcinoma. In addition, the toxicity profile was considered to be acceptable.22
Esterification of the 20-position hydroxyl group of CPT to a molecule with a carboxylic acid group is an often employed strategy because the linkage can be broken under physiological conditions releasing the lactone form of CPT, which is essential for activity.4, 5 A variety of water-soluble prodrug conjugates that link CPT via the hydroxyl group at the 20-position have been reported.23-31 For example, polyacetal poly(1-hydroxymethylethylenehydroxymethylformal)succinyl-Gly-CPT (XMT-1001) is a novel polymeric CPT prodrug conjugate that utilizes a glycine linker, that releases the lactone form of CPT via a two-step mechanism over an extended period of time. It is initially degraded by esterases to form two lipophilic intermediates (camptothecin-20-O-(N-succinimidoglycinate) and camptothecin-20-O-(N-succinamidoyl-glycinate)). These intermediates readily enter the cell and are converted to active CPT (lactone form) intracellularly.5 Other CPT prodrug conjugates using Gly linkers have also been reported with improved physiochemical and biological properties including poly (N-(2-hydroxypropyl) methacrylamide) copolymer,32 poly-(l-glutamic acid)-Gly-CPT conjugates,33, 34 PEG-β-camptothecin,35 PEG-Gly-CPT,35 carboxymethyldextran-(Gly)3-CPT conjugates (T-0128),36 CPT-PEG-biotin bioconjugate,37 and CPT-PEG-folic acid bioconjugates.38 Recently, NMR spectroscopy was used to characterize the kinetics of CPT release from a 2a conjugate and to identify intermediates.39
Previously, our group reported on CPT-prodrug conjugates in which an ester bond with the carboxylic acid group of Gly was cleaved to generate the active form of CPT.37 A novel CPT-Gly-PEG-folate bioconjugate was prepared for a targeted CPT delivery system.37 Gly was used as a linker with the added advantage of ‘locking’ the lactone ring in its active form by virtue of esterification at the 20-OH group in CPT.38 Results indicated significantly higher efficacy of the CPT-Gly-PEG-folate in comparison to unconjugated CPT in which the conjugated CPT-Gly-PEG-folate released the active form of CPT slowly enough for folate receptor mediated endocytosis to occur. Although the CPT-Gly-PEG-folate design suggests the importance of a linker, it is noted that in vivo release is expected to be quite rapid since Gly does not offer a controlled rate of CPT release like other hydrophobic amino acids. The current study was therefore designed to explore the stability of more hydrophobic amino acid linkers in order to achieve the best CPT-prodrug candidate for a passively targeted sustained release drug delivery system.
In the current report, the synthesis and characterization of a series of four α-amino acid ester CPT prodrugs with increasing aliphatic side chain length (Gly (2a), Ala(2b), Abu(2c), and Nva(2d)) are presented (Figure 1). Attachment of these amino acids to CPT offers several advantages including: stabilization of the lactone ring, the ability to control CPT release rate, and the ability to link the prodrugs to a drug delivery scaffold by means of a peptidic bond. Prodrug hydrolysis studies were performed using three sodium phosphate buffers (PB) with pH values 6.6, 7.0 (i.e., representative pH in tumor and lung),40, 41 and 7.4 (i.e., extracellular/physiological pH). Hydrolysis studies were monitored by HPLC, and the cytotoxicity of the prodrugs were evaluated by MTT assay using the A549 cell line. The current results demonstrate that 2c and 2d have increased cytotoxic potential as compared to parent CPT, suggesting that sustained delivery (i.e., slow release, constant exposure) may be an effective method to control lung cancer.
Figure 1.
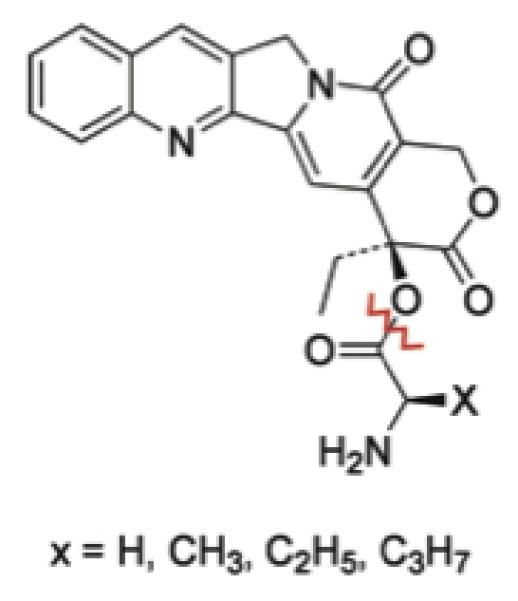
Schematic representation of prodrugs. Hydrolysis site is shown by the red zig-zag line.
Results
Synthesis of 2a-d Prodrugs
The 2a-d prodrugs were synthesized using a two-step procedure with DCM as the solvent. CPT was coupled to four different Boc protected amino acids using the coupling reagent DIPC, resulting in compound 1a-d (Scheme 1). Cleavage of the Boc group from compound 1a-d was performed using 30% TFA in DCM, resulting in 2a-d. The progress of the reaction was monitored by thin layer chromatography (TLC). All reactions proceeded with good yields and high purity. Each intermediate and the final product were confirmed by 1H NMR and mass spectroscopy.
Scheme 1.
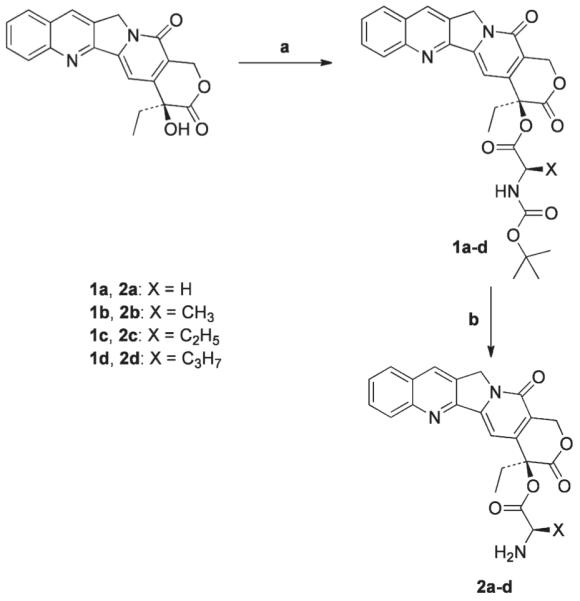
Synthesis of 2a-d: Reagents and conditions: (a) DIPC, DMAP, DCM, RT, 4 h; (b) 30% TFA in DCM, RT, 1 h.
Analysis of CPT, Prodrugs and Intermediates
HPLC with fluorescence detection was used to identify the hydrolysis of prodrug and formation of CPT from the parent prodrug. Triethylamine acetate buffer was used in the mobile phase to serve as the ion-pairing reagent, provide an adequate retention of the carboxylate form of the drug, act as a masking reagent for underivatized silanols to prevent peak tailing, and to serve as the major buffer component.42 The retention time of CPT and 2a-d using this HPLC method are shown in Table 1. The lower limit of detection for CPT was 0.1 ng/mL.
Table 1.
Retention Times of 2a–d (tR,ester) and CPT (tR,CPT)
| CPT ester prodrug |
no.of intermediates |
tR,ester (min) |
tR,CPT (min) |
|---|---|---|---|
| 2a | 2 | 3.5 | 6.8 |
| 2b | 2 | 3.8 | 6.8 |
| 2c | 2 | 5.0 | 6.8 |
| 2d | 2 | 7.0 | 6.8 |
Aliquots (2 mL) were taken from each of the four prodrug reaction mixtures at 6.5 h. Samples were dried and 2a-d and their corresponding intermediates 1 ((lactam (I-1) and hemiorthoester (I′-1)) were analyzed using 1H NMR and I-1 and intermediate 2 (I-2) were analyzed using mass spectroscopy.
The base catalalyzed mechanism proceeds via nucleophilic terminal amine attack at the CPT E-ring carbonyl lactone producing I-1. This intermediate converts to I′-1 and I-2 via intermolecular reactions. The lactone and carboxylate form of CPT are subsequently generated from I-2 through either an intermolecular (pathway e) or intramolecular (pathway f) reaction (Scheme 2). Based on the previously characterized 2a prodrug data,39 the structure of 2b, 2c and 2d prodrug primary intermediates appear to be hemiortho esters.
Scheme 2.
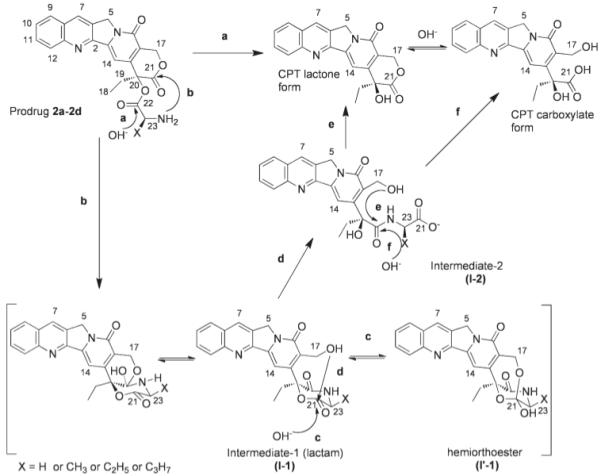
The proposed degradation pathway of 2a-d in three pH values: pH 6.6, 7.0 and 7.4. Prodrug hydrolysis involves steps b and d whereas CPT formation is possible via pathways a, e, or f. However, results in this study demonstrate that only pathway e is responsible for CPT formation.
Notable differences are evident in the chemical shifts in the 1H spectra for all of the prodrugs at each pH value (6.6, 7.0 and 7.4) studied for peaks representing the H-17, H-23, H-5, H-14 and H-7 protons in 2a-d as compared to the corresponding I′-1. The H-17 singlet signal for 2a-d [2a: 5.51 (2H, H-17); 2b: 5.52 (2H, H-17); 2c: 5.53 (2H, H-17); 2d: 5.53 (2H, H-17)] has shifted up field and split to an AB spin system (2a: δA 4.8 ppm, δB 5.0 ppm, JAB = 16.2 Hz; 2b: δA 4.83 ppm, δB 5.02 ppm, JAB = 16.2 Hz; 2c: δA 4.83 ppm, δB 4.81 ppm, JAB = 16.2 Hz; 2d: δA 4.82 ppm, δB 5.02 ppm, JAB = 16.0 Hz) in the hemiortho ester because the two protons are becoming more magnetically nonequivalent after the formation of the bicyclic intermediate.
This intermediate structure I-1 suggests intramolecular nucleophilic attack by the 17-hydroxyl at the ester carbonyl to form a tetrahedral I′-1. The H-17 signal for 2a-d (2a: δ 4.63-4.66 ppm; 2b: δ 4.61-4.69 ppm; 2c: δ 4.69-4.65 ppm; 2d: δ 4.66-4.69 ppm) in the I′-1 are shifted up field in the I-1, becoming an ABX system due to the adjacent hydroxyl group. (Figure 2A)
Figure 2.

1H NMR spectrum of all four prodrugs in DMSO-d6 at 37 °C after 6.5 h of incubation in PB pH7.4: (A) section of 1H NMR spectrum showing changes in the H-5 and H-17 signals of 2a-d, I-1, and I0-1; (B) section of 1H NMR spectrum showing changes in the H-23 signals of 2a-d, and I-1; (C) section of 1H NMR spectrum showing changes in the H-7 and H-14 signals of 2a-d, I-1, and I0-1. Similar spectra were observed at pH 7.0 and 6.6.
The H-23 signal for 2a-d (2a: δA 4.16 ppm, δB 4.22 ppm, JAB =25 Hz, 2b: 4.41-4.24, 2c: 4.13-4.18, 2d: 4.21-4.25) are shifted up field in I-1 (2a: 3.81-3.96; 2b: 3.92-4.09, 2c: 3.86-3.88, 2d: 3.88-3.90), due to the role of the C-22 carbonyl carbon in the formation of I′-1, H-23 proton signal are shifted up field of the I′-1. The H-23 proton signal for I′-1 is not visible for any prodrugs because its resonances have merged with the water signal that is appears at 3.2 ppm (Figure 2C).
The proton signals for H-5, H-7 and H-14 for 2a-d [(2a: δ 5.39 (s, H-5), 8.83 (s, H-7), 7.31 (s, H-14) ppm; 2b: δ 5.40 (s, H-5), 8.99 (s, H-7), 7.35 (s, H-14); 2c: δ 5.26 (s, H-5), 8.93 (s, H-7), 7.24 (s, H-14) ppm; 2d: δ 5.27 (s, H-5), 8.94 (s, H-7), 7.22 (s, H-14) ppm)] are also shifted up field in the I-1 [(2a: δ 5.26 (s, H-5), 8.65 (s, H-7), 7.19 (s, H-14) ppm; 2b: δ 5.27 (s, H-5), 8.67 (s, H-7), 7.24 (s, H-14) ppm; 2c: δ 5.23 (s, H-5), 8.66 (s, H-7), 7.18 (s, H-14) ppm; 2d: δ 5.24 (s, H-5), 8.67 (s, H-7), 7.19 (s, H-14) ppm)]. The tetrahedral I′-1 was formed by the intramolecular nucleophilic attack by the 17-hydroxyl group at the ester carbonyl group. The H-5, H-7 and H-14 signal for 2a-2d [(2a: δ 5.21(s, H-5), 8.62 (s, H-7), 7.0 (s, H-14); 2b: δ 8.67 (s, H-5), 8.63 (s, H-7), 7.22 (s, H-14); 2c: δ 5.21 (s, H-5), 8.63 (s, H-7), 7.07 (s, H-14) ppm; 2d: δ 5.22 (s, H-5), 8.64 (s, H-7), 7.08 (s, H-14) ppm)] in the I′-1 are shifted up field in the I-1, shown in Figure 2A and Figure 2B.
LC-MS results confirm the identity of two intermediate species generated during hydrolysis. LC-MS spectra of the I-1 and I-2 are shown in Figure 3. The mass spectrum of I-1 indicates that it has the same molecular mass as its respective parent prodrug.
Figure 3.
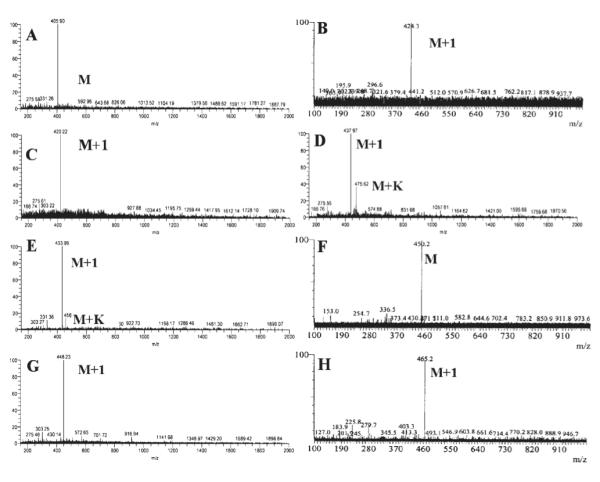
LC-MS spectra of the intermediates obtained during the hydrolysis studies of 2a-d: (A) 2a (I-1); (B) 2a (I-2); (C) 2b (I-1); (D) 2b (I-2); (E) 2c (I-1); (F) 2c (I-2); (G) 2d (I-1); (H) 2d (I-2).
Hydrolysis of 2a-d Prodrugs
The hydrolysis studies of 2a-d were performed in PB at three pH values (pH 6.6, 7.0 and 7.4) at 37°C. The disappearance of 2a-d and the formation of CPT were quantified using HPLC. The percentage of remaining 2a-d and reconverted CPT were plotted as a function of time (Figures 4 and 5, respectively). The results were fit to single exponential decay (eq 1) and association models (eq 2), and the best-fit apparent rate constants were determined. Equation 3 was used to calculate the half-life from the rate constant. The half-lives of 2a-d hydrolysis and CPT formation at three pH values are reported in supporting information.
| (1) |
Figure 4.
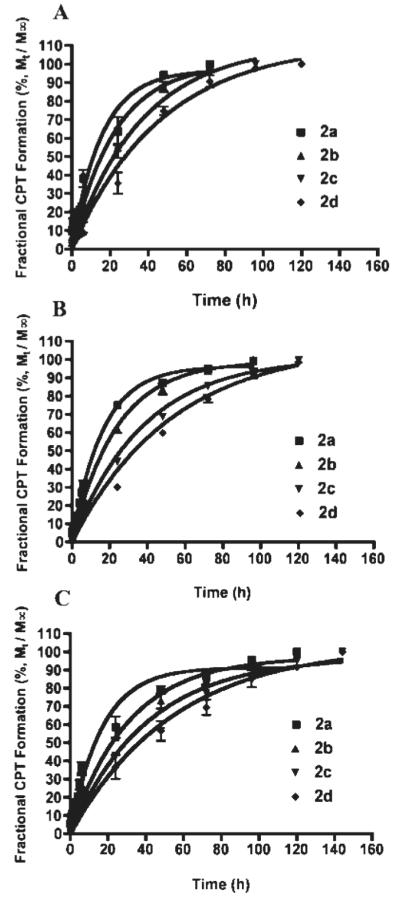
Release of CPT in PB at three pH values at 37 °C: (A) pH 7.4; (B) pH 7.0; (C) pH 6.6.
Figure 5.

Hydrolysis of 2a-d in PB at three pH values at 37°C: (A) pH 7.4; (B) pH 7.0; (C) pH 6.6.
For eq 1, Ct is the concentration of 2a-d at time t, C0 is the initial concentration of 2a-d and k is the first order rate constant.
| (2) |
For eq 2, Ct is CPT concentration at the sampling time indicated, C∞ is the final CPT concentration at time ∞ and k is the first order rate constant.
| (3) |
For eq 3, t1/2 is the half-life and k is the first order rate constant determined from equation 1 or equation 2.
Cytotoxicity of 2a-d Prodrugs
The in vitro biological efficacy of 2a-d was evaluated in A549 human lung carcinoma cells using an MTT assay. The data were fit to a sigmoidal nonlinear regression model and the concentration at which 50% of the cells were viable (IC50) was calculated based on the best-fit model.
| (4) |
Equation 4 is a sigmoidal nonlinear regression model with variable Hill slope (HS), where V is the viability of cells in %, IC50 is the drug concentration at which 50 % of the cells are viable, and DC is the drug concentration.
The IC50 value of CPT administered to A549 cells was determined to be 5.4 nM, which was within its reported range.43, 44 The IC50 values of both 2c (4.1 nM) and 2d (5.2 nM) were in the same range as CPT; however, 2a (12.24 nM) and 2b (120.1 nM) were about 2- and 20- fold less potent than their parent drug, respectively. The order of toxicity of the drugs tested was 2c > 2d ~ CPT > 2a > 2b (Table 2).
Table 2.
Best-Fit Parameters of Cytotoxic Activity of CPT and 2a–d to the Human Lung Carcinoma A549 Cellsa
| drug and prodrugs |
IC50 (95% CI) | Hill slope (95% CI) | R 2 |
|---|---|---|---|
| CPT | 5.379 (4.409/6.562) | −0.692(−0768/−0.617) | 0.9796 |
| 2a | 12.24 (10.04/14.93) | −0.622 (−690/−0.555) | 0.9769 |
| 2b | 120.1 (94.15/153.3) | −0.565 (−0.634/−0.495) | 0.9594 |
| 2c | 4.103 (3.226/5.218) | −0.649 (−0.739/−0.560) | 0.9662 |
| 2d | 5.203 (4.141/6.537) | −0.443 (−0.483/−0.403) | 0.9734 |
IC50 is expressed in units of nM
Discussion
The development of CPT prodrugs is advantageous because of their increased stability and enhanced solubility. CPT prodrugs were found to be more water soluble,34 providing a way to formulate the poorly soluble CPT. CPT prodrugs are expected to display better S-phase efficacy and a possibly synergistic role in cyclotherapy with other anticancer reagents.45 Irinotecan has show antiangiogenic effects in vitro and in vivo in a metronomic regimen, which refers to chemotherapy at low, nontoxic doses on a frequent schedule with no prolonged breaks.46 To stabilize the ester bond, a series of four α-amino acid ester prodrugs of CPT containing an increasing aliphatic side chain length 2a-d were synthesized and characterized. Hydrolysis studies were performed at pH 6.6, 7.0 (i.e. lung pH and representative of a common pH range in tumors)40, 41 and 7.4. In order to understand the degradation pathway of 2a-d, HPLC was used to follow the hydrolysis of the prodrug and the formation of CPT and intermediates generated from the parent prodrug. The hydrolysis of 2a-d is complex, resulting in two degradation intermediates. In addition to the CPT lactone, two additional peaks corresponding to the I-1 and the ring-opened amide I-2 were observed in 2a (Scheme 2), which is consistent with a previously published hydrolysis mechanism.39, 47 Similar results were observed for 2a-d. The structures of the intermediates, which were produced during hydrolysis, were characterized using 1H NMR and ESI-MS (Figures 2 and 3).
Hydrolysis Studies
The hydrolysis studies of the 2a-d were performed in PB at three pH values (pH 6.6, 7.0 and 7.4). 2a was found to have the fastest CPT reconversion rate. The rate of 2a-d hydrolysis was independent of pH. 2d was the most stable among all of the prodrugs tested. The rate of CPT formation from both 2c and 2d was slower than from 2a and 2b. Therefore, the former two esters had longer CPT release profiles than the latter two prodrugs at all three pH values. Furthermore, it was observed that the apparent hydrolysis rates of 2c and 2d were slower than 2a and 2b at all three pH values (Figures 4 and 5).
The ester bond in 2a hydrolyzed within 72 h at pH 7.4 and within 96 h at pH 7.0 and pH 6.6 (Figures 4 and 5). The ester bond in 2b, cleaves within 72 h at pH 7.4 whereas the ester bond in 2c and 2d, cleaves within 96 h and 120 h, respectively at pH 7.4. However, hydrolysis studies at pH 7.0 show that the hydrolysis time for 2a, 2b and 2c increases by 24 h. 2a and 2b take 96 h and 2c takes ~120 h for total ester bond hydrolysis. The ester bond in 2d hydrolyzed by 120 h at pH 7.4 and 7.0. However 2d hydrolysis time increased by 24 h at tumor pH (pH 6.6), whereas complete hydrolysis of the ester bond was observed in 144 h. 2b-d were completely hydrolyzed at pH 6.6 in 96 h, 120 h and 144 h, respectively.
Standard kinetics models were applied to the hydrolysis data. The data shows a good fit to first order kinetics. It was observed that degradation is faster at pH 7.4 than pH 7.0 and 6.6. The half-lives of both prodrug hydrolysis and CPT formation are shown in the supporting information for 2a-d at three pH values (pH 7.4, 7.0 and 6.6). The correlation between the apparent first order pH-dependent half-life of prodrug hydrolysis or CPT formation and hydrophobicity constant are shown in Figures 6 and 7. The hydrophobicity constant appears to be directly proportional to the half-life of both CPT formation and 2a-d hydrolysis. These results suggest that increasing the length of the side chain of the hydrophobic amino acids would sustain (i.e., slow) the release of CPT.
Figure 6.
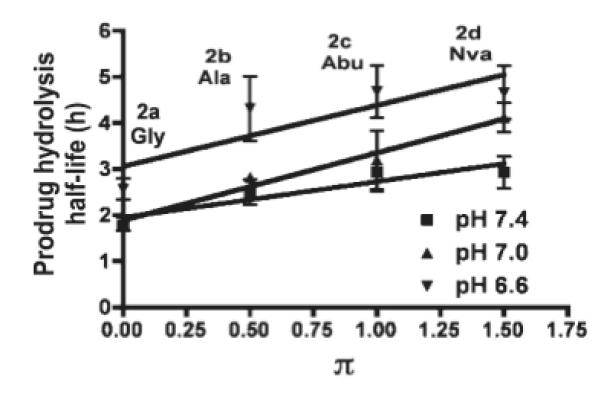
Correlation of 2a-d hydrolysis half-life (see steps b and d, Scheme 2) and hydrophobicity constants (π) of amino acid. The hydrolysis half-lives of 2a-d in PB (pH 6.6, 7.0, and 7.4) at 37°C are reported as the mean ± SD (n=3). Slopes: pH 7.4 = 0.7788 (r2= 0.8493), pH 7.0 = 1.465 (r2= 0.9731), pH 6.6 = 1.322 (r2= 0.7161). The slopes at pH 6.6 and 7.0 are approximately twice as large as the slope at pH 7.4 suggesting that the prodrug reconversion in cancerous tissues or in the lung (the target organ) would be expected to be twice as fast.
Figure 7.
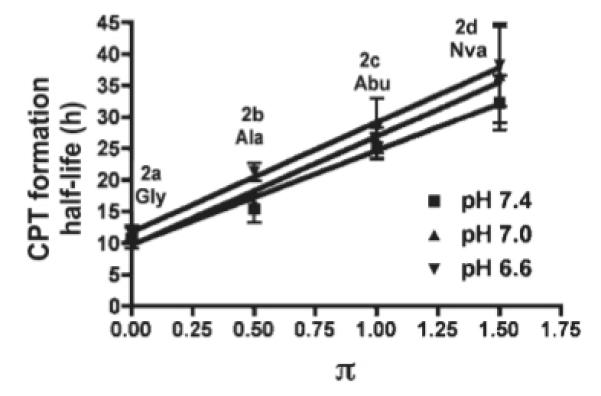
Correlation of CPT formation half-life (see step e, Scheme 2) at three pH values and hydrophobicity constant (π) of amino acid. The reported half-life of CPT formation in PB in three pH values at 37°C represents the mean ± SD (n=3). Even though the rate of prodrug hydrolysis is dependent upon pH (Figure 6), once prodrug hydrolysis occurs the rate of CPT formation is independent of pH. Slopes: pH 7.4 = 14.89 (r2= 0.9837), pH 7.0 = 17.26 (r2= 0.9783), pH 6.6 = 17.42 (r2= 0.9954).
Increasing aliphatic chain length generally increases prodrug reconversion time because of the effect of steric hindrance. In the case of 2a, steric hindrance was not suggested. Nevertheless, the rate of hydrolysis is directly proportional to the hydrophobic amino acid chain length for 2b, 2c and 2d. 2d contained the largest aliphatic side chain length and was more slowly hydrolyzed as compared to 2c and 2b. Steric hindrance decreased the hydrolysis rate thereby, slowing the formation of CPT via the conversion of I-1 to I-2. The rate of 2a-d hydrolysis at pH 6.6 and 7.0 was 1.7- and 1.9-fold slower, respectively, than at pH 7.4. On the other hand, the formation of CPT post-prodrug hydrolysis was independent of pH. Since 2a-d will eventually be immobilized on a passive lung targeting microparticle, selective prodrug hydrolysis in the lung and at lung cancer sites is required and appears to be readily achievable using the bulkier amino acid CPT prodrugs.
Cytotoxicity Studies
CPT is known to become nontoxic after esterification of the hydroxyl group at the C-20 position.48, 49 For this series of prodrugs, the rank order of hydrolysis rate and CPT formation rate is 2a > 2b > 2c > 2d. As expected, the IC50 value of 2a is greater than that of 2b; however, the IC50 of 2d and 2c were much lower than that for 2a. The IC50 was statistically the same as CPT alone at pH 7.4 suggesting that the toxicity of the 2a-d was not solely dependent on the stability (i.e., prodrug hydrolysis and CPT formation), but also on the physicochemical properties of the drugs.
There are several possible reasons that in vitro hydrolysis rates might not be indicative of in vivo biological efficacy including: passive permeation or active transport of CPT analogues into cells, differences in extracellular and/or intracellular hydrolysis kinetics of 2a-d due to endogenous esterases and proteases, increased toxicity of intermediates, or a possible synergistic effect between the hydrolysis products (i.e., CPT and amino acid).
In addition to 2a-d hydrolysis and CPT formation rates, the effectiveness of 2a-d at various concentrations was tested. Unlike IC50 values, which are indicative of concentrations required for optimal cell death, the interpretation of the shape of the dose-response curve and the importance of the Hill slope factor to the biological effect are still not clear.50 It is generally believe to be closely related to the duration of drug exposure to the cells.50, 51 The estimated Hill slopes of 2a-c and CPT were similar. The Hill slope of 2d was significantly (P < 0.05) less than other tested treatments (Figure 8, Table 2), suggesting a higher cell death at lower drug concentrations than CPT and its other prodrug analogues at the same concentration. This could be a potential benefit, as systemic side effects would be reduced. In addition, although all of the treatments in this study had the same drug exposure time, there was no definitive means of stopping cell apoptosis during the MTT assay. This was due to residual drug remaining inside the cell after removing the extracellular source of the drug.
Figure 8.
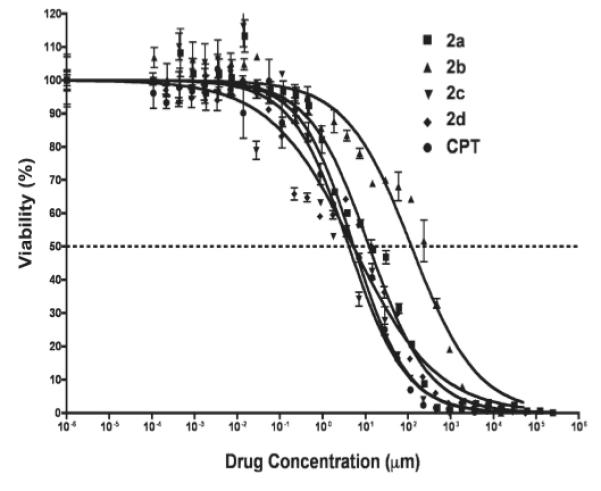
Cytotoxicity of CPT and 2a-d in A549 human lung carcinoma cells. The data were fit using the Hill equation.
Therefore, it is reasonable to suggest that 2d had differential extracellular and intracellular hydrolysis kinetics, as demonstrated by the hydrolysis study, or a possible synergistic effect of released Nva with CPT. Nva is commonly used in pharmacological studies as an inhibitor of arginase, which degrades arginine irreversibly to ornithine.52, 53 Even though Nva is not cytotoxic, it was found to suppress cell growth in mouse leukemia cells (L1210) and in a human cervical cancer cell line (HeLa cells).53 Therefore, the distinguishing shape of the dose-response curve of 2d was possibly due to the combination of the stabilization effect of Nva and the cytotoxicity of CPT. Moreover, a Nva containing tri-peptide analogue (MF13) of an akylating anti-cancer agent, m-L-sarcolysin, showed the most potent and selective anti-cancer activity in vitro54, 55 to a panel of cancer cell lines, as well as in vivo54, 56 in studies of more than 100 newly synthesized di- and tri-peptides with a variety of amino acids analogues of m-L-sarcolysin. The molecular mechanism of selective apoptosis in tumor cells of this Nva containing tripeptide was unclear, but it was proven to be S-phase specific and act through the bcl-2/bax balance pathway.54 Unfortunately, the role of Nva to improved anti-cancer activity of the tripeptide analogue to its parent compound, m-L-sarcolysin, was not clear and more investigations were necessary for elucidation of the mechanism.
Conclusions
A series of α-amino acid ester prodrugs of CPT with increasing aliphatic chain lengths has been prepared. The hydrolysis of 2a-d at three PB pH values, (pH 6.6, 7.0 and 7.4 corresponding to tumor, lung and extracellular/physiological pH, respectively) were determined by HPLC, 1H NMR and mass spectroscopy. Essentially complete hydrolysis of prodrugs containing terminal amines occurred, resulting in the formation of hydrolysis intermediates (I-1, I′-1 and I-2) and eventually the release of active CPT. The hydrolysis of the ester bond was found to be pH dependent and proportional to the size of the of aliphatic amino acid side chain length. The formation of CPT was independent of pH. The cytotoxicity of 2a-d to cancer cells was not solely dependent on the stability of the ester bond. While the hydrolysis rates of 2d and 2c were similar, the favorable Hill slope and toxicity profile of 2d suggest that it is the lead candidate for immobilization on passively targeted, lung delivery microparticles.
Experimental Section
Materials
Boc-glycine, Boc-alanine, Boc-aminobutanoic acid and Boc-norvaline were purchased from EMD Biosciences (Gibbstown, NJ). CPT, 4-dimethylaminopyridine (DMAP), dimethyl sulfoxide (DMSO), trifluoroacetic acid (TFA), and N,N’-diisopropylcarbodiimide (DIPC) were purchased from Sigma-Aldrich (St. Louis, MO). For in vitro assays, human lung carcinoma cells (A549 cells) were purchased from American Type Culture Collection (ATCC) (Manassas, VA). All purchased reagents were used without further purification. High-purity water was provided by a Milli-Q Plus purification system (Millipore, Billerica, MA). Flash column chromatography was carried out using silica gel (35-70 μm, 6 nm pore diameter, Fisher Scientific, Boston, MA). TLC analysis was conducted on alumina precoated with silica gel 60 (EMD Chemicals Inc., Gibbstown, NJ). Apparent purity was measured by HPLC (Waters, Milford, MA) with a scanning fluorescence detector (Shimadzu Scientific Instruments Inc., Columbia, MO). 1H NMR spectra were recorded in DMSO-d6, CDCl3, on a Varian 500 or 400 MHz instrument. Signals of solvents were used as internal references. Chemical shifts (δ) and coupling constants (J) are given in ppm and hertz, respectively. HRMS was performed by the Washington University Mass Spectrometry Resource, an NIH Research Resource.
Methods
Synthesis of 2a-d Prodrugs
General procedure (A)
The 1a-d was synthesized as shown in Figure 2. In general, CPT (1 eq), Boc-amino acid (1.2 eq) and DMAP (1 eq) were dissolved in DCM (15 mL). The reaction mixture was cooled to 0°C. DIPC (1.3 eq) was added drop wise into the reaction mixture. The reaction was kept in an ice bath and stirred continuously for one hour and then stirred at RT to avoid side reactions. The progress of the reaction was monitored using TLC. After completion of the reaction, the solvent was removed and crude products were purified through flash column chromatography (10% MeOH/CHCl3).
General procedure (B)
The product obtained by procedure A, was then dissolved in 30% TFA in DCM (10 mL) and continuously stirred for four hours at RT to cleave the Boc group on the amino acids. After removal of 30% TFA in DCM, the 2a-d was precipitated by adding ice cold anhydrous diethyl ether (20 mL). The precipitate was collected by centrifugation and the product was washed twice using ice-cold diethyl ether (20 mL). The crude product was purified using flash column chromatography (10% MeOH/CHCl3).
All four prodrugs are pure (~ 99%). The purity of four prodrugs was measured using NMR, ESI-MS and HRMS (Supporting information).
CPT-Gly-Boc (1a)
General Procedure A. Yield: 96 %; 1H(400 MHz, DMSO-d6): δ = 8.65 (1H, s, H-7), 8.13 (2H, m, H-9 and H-12), 7.84 (1H, t, J = 8.2 Hz, H-11), 7.66 (1H, d, J = 7.8 Hz, H-10), 7.40 (1H, t, J = 6.0 Hz, NH), 7.21 (1H, s, H-14), 5.47 (2H, s, H-17), 5.40 (2H, s, H-5), 3.91 (1H, dd, J = 18.0 Hz and J = 6.0Hz,, Gly-α), 3.78 (1H, dd, J = 18.0 Hz and J = 6.0 Hz, Gly-α), 2.12 (2H, m, H-19), 1.36 (9H, s, Boc-CH3), and 0.91 (3H, m, H-18). ESI-MS (MeOH) (m/z): 519 (M + H)+, 542.1(M + Na)+.
CPT-Ala-Boc (1b)
General Procedure A. Yield: 94 %; 1H(400 MHz, DMSO-d6) δ = 8.66 (1H, d, J = 6.0 Hz, H-7), 8.08 (2H, m, H-9 and H-12), 7.85 (1H, t, J = 8.0 Hz, H-11), 7.69 (1H, t, J = 5.8 Hz, H-10), 7.57 (1H, d, J = 6.4 Hz, CONH), 7.42 (1H, d, J = 6.8 Hz, CONH), 7.32 (1H, d, J = 19.5 Hz, H-14), 5.46 (2H, s, H-17), 5.27 (2H, s, H-5), 4.06 (2H, m, Ala-α), 3.55 (2H, m, H-19), 1.32 (3H, m, Ala-β), 1.0 (9H, m, Boc-CH3), and 0.94 (3H, m, H-18). ESI-MS (MeOH) (m/z): 507 (M + H)+.
CPT-Abu-Boc (1c)
General Procedure A. Yield: 97 %; 1H(400 MHz, DMSO-d6): δ = 8.64 (1H, s, H-7), 8.07 (2H, m, H-9 and H-12), 7.85 (1H, t, J = 6.8 Hz, H-11), 7.70 (1H, t, J = 6.8 Hz, H-10), 7.52 (1H, d, J = 6.6 Hz, CONH), 7.40 (1H, d, J = 7.0 Hz, CONH), 7.30 (1H, d, J = 11.9 Hz, H-14), 5.47 (2H, s, H-17), 5.26 (2H, s, H-5), 4.06 (2H, m, Abu-α), 3.28 (2H, m, H-19), 2.09 (1H, m, Abu-β), 1.84 (1H, m, Abu-β), 1.4 (3H, s, Abu-γ), 1.24 (9H, m, Boc-CH3), and 0.95 (3H, m, H-18). ESI-MS (MeOH) (m/z): 534.0 (M + H)+, 1067 (2M + H)+.
CPT-Nva-Boc (1d)
General Procedure A. Yield: 98 %; 1H(400 MHz, DMSO-d6) δ = 8.65 (1H, s, H-7), 8.07 (2H, m, H-9 and H-12), 7.85 (1H, t, J = 7.0 Hz, H-11), 7.68 (1H, t, J = 6.8 Hz, H-10), 7.51 (1H, d, J = 7.2 Hz, CONH), 7.41 (1H, d, J = 7.2 Hz, CONH), 7.26 (1H, d, J = 15.6 Hz, H-14), 5.47 (2H, s, H-17), 5.26 (2H, s, H-5), 4.07 (2H, m, Nva-α), 2.10 (2H, m, H-19), 1.63 (2H, m, Nva-β), 1.47 (3H, m, Nva-γ), 1.4 (3H, s, Nva-δ), 0.96 (9H, m, Boc-CH3), and 0.90 (3H, m, H-18). ESI-MS (MeOH) (m/z): 548.0 (M + H) +, 1094.0 (2M + H) +.
Prodrug 2a
General Procedure B. Yield: 87 %; 1H(400 MHz, DMSO-d6): δ = 8.70 (1H, s, H-7), 8.29 (2H, s, Gly-NH2), 8.13 (2H, dd, J = 7.9 Hz & J = 3.0 Hz, H-9 and H-12), 7.85 (1H, t, J = 7.9 Hz, H-11), 7.71 (1H, d, J = 7.0 Hz, H-10), 7.27 (1H, s, H-14), 5.53 (2H, s, H-17), 5.31 (2H, s, H-5), 4.32 (1H, d, J = 18.0 Hz,, Gly-α), 4.08 (1H, d, J = 18.0 Hz, Gly-α), 2.16 (2H, m, H-19), and 0.93 (3H, m, H-18). ESI-MS (MeOH) (m/z): 406.0 (M + H)+, 810.0 (2M + H)+. HRMS (m/z): observed 406.1389, calculated 406.140.
Prodrug 2b
General Procedure B. Yield: 84 %; 1H(400 MHz, DMSO-d6) δ = 8.70 (1H, s, H-7), 8.48 (2H, br. d, NH2), 8.13 (2H, q, J = 8.6 Hz, H-9 and H-12), 7.85 (1H, t, J = 8.1 Hz, H-11), 7.71 (1H, t, J = 8.0 Hz, H-10), 7.21 (1H, s, H-14), 5.53 (2H, s, H-17), 5.28 (2H, d, J = 15.8 Hz, H-5), 4.40 (1H, q, J=6 Hz, Ala-α), 2.24 (2H, m, H-19), 2.24 (2H, dd, J = 7.0 Hz and J = 7.2 Hz, H-19), and 0.95 (3H, m, H-18). ESI-MS (MeOH) (m/z): 420.0 (M + H) +, 839.0 (2M + 1)+. HRMS (m/z): observed 420.1554, calculated 420.155.
Prodrug 2c
General Procedure B. Yield: 85 %; 1H(400 MHz, DMSO-d6) δ = 8.70 (1H, s, H-7), 8.49 (2H, d, NH2), 8.13 (2H, d, J = 8.4 Hz, H-9 and H-12), 7.85 (1H, dd, J = 8.5 Hz J = 1.3 Hz, H-11), 7.71 (1H, t, J = 7.2 Hz, H-10), 7.25 (1H, d, J = 13 Hz, H-14), 5.52 (2H, s, H-17), 5.30 (2H, s, H-5), 4.31 (1H, dd, J = 5.4 Hz, Abu-α), 2.17 (2H, q, H-19), 1.98 (2H, m, Abu-β), 1.0 (3H, m, Abu-γ), and 0.94 (3H, m, H-18). ESI-MS (MeOH) (m/z): 434.0 (M + H)+, 867.0 (2M + H)+. HRMS (m/z): observed 434.1700, calculated 434.171.
Prodrug 2d
General Procedure B. Yield: 81 %; 1H(400 MHz, DMSO-d6) δ = 8.7 (1H, s, H-7), 8.48 (2H, d, NH2), 8.10 (2H, m, H-9 and H-12), 7.85 (1H, dd, J = 8.5 Hz and J = 1.3 Hz, H-11), 7.70 (1H, dd, J = 8.2 Hz and J = 0.9 Hz, H-10), 7.25 (1H, d, J = 9.7 Hz, H-14), 5.51 (2H, s, H-17), 5.3 (2H, s, H-5), 4.4 (1H, dd, J = 5.6 Hz, Nva-α), 2.19 (2H, m, H-19), 1.9 (2H, m, Nva-β), 1.52 (3H, m, Nva-γ), 1.05 (3H, m, Nva-δ), and 0.94 (3H, m, H-18). ESI-MS (MeOH) (m/z): 448.0 (M + H)+, 895.0 (2M + H)+. HRMS (m/z): observed 448.1855, calculated 448.187.
Analysis of CPT
HPLC methods have been used for the quantitative analysis of CPT by several groups.57, 58 The analysis of CPT formation and prodrug hydrolysis were performed using a C-18 reverse phase column (Waters Symmetry® C-18 5μm, 150 × 4.6 mm). HPLC parameters were as follows: 0.8% triethylamine/acetic acid (v/v) in a mixture of 20% acetonitrile and 80% water (mobile phase A) and acetonitrile (mobile phase B), linear gradient (t= 0 min, 90% A, 10% B; t= 10 min, 70% A, 30% B; t=15 min, 0% A, 100% B; t=25 min 0% A, 100% B; t=30 min, 90% A, 10% B; t=40 min stop), flow rate = 1 mL/min. The detection was performed using a fluorescence detector (Shimadzu Scientific Instruments Inc., Columbia, MO) with an excitation wavelength of 360 nm and an emission wavelength of 440 nm.
In vitro characterization of 2a-d prodrugs
Hydrolysis of 2a-d was carried out as follows. 2a-d (5 mg) were dissolved in PB (2 mL, 100 mM PB) and kept on a shaker plate at 37 °C. Studies were performed at three pH values (pH 6.6, 7.0 and 7.4). Aliquots (50 μL) were taken at predetermined time points. Samples aliquots were dried using a CentriVap™ concentrator (Labconco Corp., Kansas City, MO) and reconstituted in methanol and analyzed using HPLC. The concentration of 2a-d and CPT were analyzed using HPLC with fluorescence detection. Prodrug hydrolysis studies were performed in PB in triplicate at three different pH values (pH 6.6, 7.0, or 7.4).
Cytotoxicity of 2a-d prodrugs
Cell culture
The human lung carcinoma cell line A549 was cultured as recommended. The cells were cultured in Dulbecco’s Modified Eagle Medium: Nutrient Mixture F-12 1:1 Mixture (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U penicillin and 100 μg streptomycin, and grown at 37 °C in a humidified atmosphere of 5% CO2 in air. Cells were passages when they reached 90% confluency by using 0.25% (w/v) trypsin-0.53 mM EDTA solution (Invitrogen, Carlsbad, CA).
Cytotoxicity Assay
The cytotoxicity of CPT and its amino acid prodrugs on A549 cells were assessed using a modified MTT assay as previously described.59 A549 cells were harvested with trypsin-EDTA solution and seeded on 96-well polystyrene cell culture plates at a density of 10,000 cells per well. The cells were allowed to attach for 24 hours. Various treatments with a wide range of concentrations (30 dilutions) and a control (blank) were applied to the cells in triplicate for 48 hours. At the end of incubation, the media was removed, and the cells were rinsed three times with phosphate buffered saline (PBS) to remove any unspecificly bound CPT. Cell viability was determined by the addition of 25 μL of MTT solution (5 mg/mL in PBS) for 5 hours at 37 °C in the dark. The crystals were then dissolved by addition of sodium dodecyl sulfate (SDS) in DMF:water (20.9 g SDS in 1:1 DMF:water). The absorption wavelength 570±10 nm of each well was determined in a plate reader (Tecan U.S. Inc., Durham, NC) with a reference wavelength of 690±10 nm.
Data Analysis
GraphPad Prism v.4.0.1 for Windows (GraphPad Software, San Diego, CA) was used for the analysis of cytotoxicity data. Cells incubated with drug-free medium were the negative control, and considered to be 100% viable. The cell viability for each drug treatment was calculated by comparing the absorption intensity to the average value of the control wells. The data were fit to a nonlinear regression sigmoidal curve. The log value of IC50 for each treatment was estimated as the concentration at which 50% of the cells were viable.
Statistical analysis
Statistical analyses were performed using Microsoft Excel v.9.0 (Microsoft Corp.). The experimental values are expressed by mean ± standard deviation. Differences between experimental groups were tested using a t-test at α = 0.05. The significance of a single factor in groups was tested by analysis of variance (ANOVA) at α = 0.05.
Acknowledgments
This research is supported by the Parke-Davis Endowed Chair in Pharmaceutics and Controlled Drug Delivery and National Institutes of Health CounterACT Program through the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Award #U54AR055073). This research content is solely the responsibility of the authors and do not necessarily represent the official views of the federal government. The NSF Integrative Graduate Education and Research Traineeship (IGERT) #0504497 and American Foundation for Pharmaceutical Education (AFPE) are acknowledged for providing graduate fellow support to Hilliard Kutscher. We would like to thank Sujata Sundara Rajan and Scott Pfeil for their help.
Abbreviations
- CPT
camptothecin
- Gly
glycine
- Ala
alanine
- Abu
aminobutyric acid
- Nva
norvaline
- DMAP
4-dimethylaminopyridine
- DIPC
N,N’-Diisopropylcarbodiimide
- DMF
dimethyl formamide
- DMSO
dimethylsulfoxide
- MeOH
methanol
- DCM
dichloromethane
- TFA
trifluoroacetic acid
- MTT
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
- SDS
sodium dodecyl sulfate
- Boc
di-tert-butoxycarbonyl
- EDTA
ethylenediaminetetraacetic acid
- HRMS
high-resolution mass spectrometry
- NMR
nuclear magnetic resonance spectroscopy
- ESI-MS
electrospray ionization mass spectrometry
- TLC
thin layer chromatography
- PB
sodium phosphate buffer
- PBS
phosphate buffered saline
- RT
room temperature
Footnotes
Supporting Information Available. Analytical data for all compounds and intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.i
References
- 1.Spataro A, Kessel D. Studies on camptothecin-induced degradation and apparent reaggregation of DNA from L1210 cells. Biochem Biophys Res Commun. 1972;48:643–648. doi: 10.1016/0006-291x(72)90396-8. [DOI] [PubMed] [Google Scholar]
- 2.Li LH, Fraser TJ, Olin EJ, Bhuyan BK. Action of camptothecin on mammalian cells in culture. Cancer Res. 1972;32:2643–2650. [PubMed] [Google Scholar]
- 3.Horwitz SB, Chang CK, Grollman AP. Studies on camptothecin. I. Effects of nucleic acid and protein synthesis. Mol Pharmacol. 1971;7:632–644. [PubMed] [Google Scholar]
- 4.Hertzberg RP, Caranfa MJ, Hecht SM. On the Mechanism of Topoisomerase I Inhibition by Camptothecin: Evidence for Binding to an Enzyme-DNA Complex. Biochemistry. 1989;28:4629–4638. doi: 10.1021/bi00437a018. [DOI] [PubMed] [Google Scholar]
- 5.Yurkovetskiy AV, Fram RJ. XMT-1001, a novel polymeric camptothecin pro-drug in clinical development for patients with advanced cancer. Adv Drug Deliv Rev. 2009 doi: 10.1016/j.addr.2009.01.007. in press., doi:10.1016/j.addr.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Hsiang YH, Hertzberg R, Hecht S, Liu LF. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem. 1985;260:14873–14878. [PubMed] [Google Scholar]
- 7.Hsiang YH, Liu LF. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cencer Res. 1988;48:1722–1726. [PubMed] [Google Scholar]
- 8.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat. Rev. Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 9.Wang JL, Wang X, Wang H, Illakis G, Wang Y. CHK1-regulated S-phase checkpoint response reduces campthothecin cytotoxicity. Cell. 2002;1:267–272. [PubMed] [Google Scholar]
- 10.Shah MA, Schwartz GK. Cell cycle-mediated drug resistance: an emerging concept in cancer therapy. Clin. Cancer Res. 2001;7:2168–2181. [PubMed] [Google Scholar]
- 11.Li Y, Lin B, Agadir A, Liu R, Dawson MI, Reed JC, Fontana JA, Bost F, Hobbs PD, Zheng Y, Chen GQ, Shroot B, Mercola D, Zhang XK. Molecular determinants of AHPN (CD437)-induced growth arrest and apoptosis in human lung cancer cell lines. Mol Cell Biol. 1998;18:4719–4731. doi: 10.1128/mcb.18.8.4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burke TG, Bom D. Camptothecin design and delivery approaches for elevating anti-topoisomerase I activities in vivo. Ann N Y Acad Sci. 2000;922:36–45. doi: 10.1111/j.1749-6632.2000.tb07023.x. [DOI] [PubMed] [Google Scholar]
- 13.Zunino F, Pratesi G. Camptothecins in clinical development. Expert Opin Investig Drugs. 2004;13:269–284. doi: 10.1517/13543784.13.3.269. [DOI] [PubMed] [Google Scholar]
- 14.Fassberg J, Stella VJ. A kinetic and mechanistic study of the hydrolysis of camptothecin and some analogues. J Pharm Sci. 1992;81:676–684. doi: 10.1002/jps.2600810718. [DOI] [PubMed] [Google Scholar]
- 15.Pantazis P, Hinz HR, Mendoza JT, Kozielski AJ, Williams LJ, Jr., Stehlin JS, Jr., Giovanella BC. Complete inhibition of growth followed by death of human malignant melanoma cells in vitro and regression of human melanoma xenografts in immunodeficient mice induced by camptothecins. Cancer Res. 1992;52:3980–3987. [PubMed] [Google Scholar]
- 16.Moertel CG, Schutt AJ, Reitemeier RJ, Hahn RG. Phase II study of camptothecin (NSC-100880) in the treatment of advanced gastrointestinal cancer. Cancer Chemother Rep. 1972;56:95–101. [PubMed] [Google Scholar]
- 17.Mross K, Richly H, Schleucher N, Korfee S, Tewes M, Scheulen ME, Seeber S, Beinert T, Schweigert M, Sauer U, Unger C, Behringer D, Brendel E, Haase CG, Voliotis D, Strumberg D. A phase I clinical and pharmacokinetic study of the camptothecin glycoconjugate, BAY 38-3441, as a daily infusion in patients with advanced solid tumors. Annals of Oncology. 2004;15:1284–1294. doi: 10.1093/annonc/mdh313. [DOI] [PubMed] [Google Scholar]
- 18.Muggia FM, Creaven PJ, Hansen HH, Cohen MH, Selawry OS. Phase I clinical trial of weekly and daily treatment with camptothecin (NSC-100880): correlation with preclinical studies. Cancer Chemother Rep. 1972;56:515–521. [PubMed] [Google Scholar]
- 19.Kumazawa E, Jimbo T, Ochi Y, Tohgo A. Potent and broad antitumor effects of DX-8951f, a water-soluble camptothecin derivative, against various human tumors xenografted in nude mice. Cancer Chemother Pharmacol. 1998;42:210–220. doi: 10.1007/s002800050807. [DOI] [PubMed] [Google Scholar]
- 20.Mitsui I, Kumazawa E, Hirota Y, Aonuma M, Sugimori M, Ohsuki S, Uoto K, Ejima A, Terasawa H, Sato K. A new water-soluble camptothecin derivative, DX-8951f, exhibits potent antitumor activity against human tumors in vitro and in vivo. Jpn J Cancer Res. 1995;86:776–782. doi: 10.1111/j.1349-7006.1995.tb02468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takiguchi S, Kumazawa E, Shimazoe T, Tohgo A, Kono A. Antitumor effect of DX-8951, a novel camptothecin analog, on human pancreatic tumor cells and their CPT-11-resistant variants cultured in vitro and xenografted into nude mice. Jpn J Cancer Res. 1997;88:760–769. doi: 10.1111/j.1349-7006.1997.tb00448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esteva FJ, Rivera E, Cristofanilli M, Valero V, Royce M, Duggal A, Colucci P, Dejager R, Hortobagyi GN. A phase II study of intravenous exatecan mesylate (DX-8951f) administered daily for 5 days every 3 weeks to patients with metastatic breast carcinoma. Cancer. 2003;98:900–907. doi: 10.1002/cncr.11557. [DOI] [PubMed] [Google Scholar]
- 23.Dai JR, Hallock YF, Cardellina IJ, Boyd MR. 20-O-beta-glucopyranosyl camptothecin from mostuea brunonis: A potential camptothecin pro-drug with improved solubility. J Nat Prod. 1999;62:1427–1429. doi: 10.1021/np990100m. [DOI] [PubMed] [Google Scholar]
- 24.Yurkovetskiy AV, Hiller A, Syed S, Yin M, Lu XM, Fischman AJ, Papisov MI. Synthesis of a macromolecular camptothecin conjugate with dual phase drug release. Mol Pharm. 2004;1:375–382. doi: 10.1021/mp0499306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmid B, Chung DE, Warnecke A, Fichtner I, Kratz F. Albumin-binding prodrugs of camptothecin and doxorubicin with an Ala-Leu-Ala-Leu-linker that are cleaved by cathepsin B: synthesis and antitumor efficacy. Bioconjug Chem. 2007;18:702–716. doi: 10.1021/bc0602735. [DOI] [PubMed] [Google Scholar]
- 26.Warnecke A, Kratz F. Maleimide-oligo(ethylene glycol) derivatives of camptothecin as albumin-binding prodrugs: synthesis and antitumor efficacy. Bioconjug Chem. 2003;14:377–387. doi: 10.1021/bc0256289. [DOI] [PubMed] [Google Scholar]
- 27.Gopin A, Ebner S, Attali B, Shabat D. Enzymatic activation of second-generation dendritic prodrugs: Conjugation of self-immolative dendrimers with poly(ethylene glycol) via click chemistry. Bioconjug Chem. 2006;17:1432–1440. doi: 10.1021/bc060180n. [DOI] [PubMed] [Google Scholar]
- 28.Leu YL, Roffler SR, Chern JW. Design and synthesis of water-soluble glucuronide derivatives of camptothecin for cancer prodrug monotherapy and antibody-directed enzyme prodrug therapy (ADEPT) J Med Chem. 1999;42:3623–3628. doi: 10.1021/jm990124q. [DOI] [PubMed] [Google Scholar]
- 29.Schmid B, Warnecke A, Fichtner I, Jung M, Kratz F. Development of albumin-binding camptothecin prodrugs using a Peptide positional scanning library. Bioconjug Chem. 2007;18:1786–1799. doi: 10.1021/bc0700842. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Lynn BC, Zhang J, Song L, Bom D, Du W, Curran DP, Burke TG. A versatile prodrug approach for liposomal core-loading of water-insoluble camptothecin anticancer drugs. J Am Chem Soc. 2002;124:7650–7651. doi: 10.1021/ja0256212. [DOI] [PubMed] [Google Scholar]
- 31.Liu Z, Robinson JT, Sun X, Dai H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J Am Chem Soc. 2008;130:10876–10877. doi: 10.1021/ja803688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caiolfa VR, Zamai M, Fiorino A, Frigerio E, Pellizzoni C, d’Argy R, Ghiglieri A, Castelli MG, Farao M, Pesenti E, Gigli M, Angelucci F, Suarato A. Polymer-bound camptothecin: initial biodistribution and antitumour activity studies. J Control Release. 2000;65:105–119. doi: 10.1016/s0168-3659(99)00243-6. [DOI] [PubMed] [Google Scholar]
- 33.Singer JW, Bhatt R, Tulinsky J, Buhler KR, Heasley E, Klein P, de Vries P. Water-soluble poly-(L-glutamic acid)-Gly-camptothecin conjugates enhance camptothecin stability and efficacy in vivo. J Control Release. 2001;74:243–247. doi: 10.1016/s0168-3659(01)00323-6. [DOI] [PubMed] [Google Scholar]
- 34.Singer JW, De Vries P, Bhatt R, Tulinsky J, Klein P, Li C, Milas L, Lewis RA, Wallace S. Conjugation of camptothecins to poly-(L-glutamic acid) Ann N Y Acad Sci. 2000;922:136–150. doi: 10.1111/j.1749-6632.2000.tb07032.x. [DOI] [PubMed] [Google Scholar]
- 35.Conover CD, Greenwald RB, Pendri A, Gilbert CW, Shum KL. Camptothecin delivery systems: enhanced efficacy and tumor accumulation of camptothecin following its conjugation to polyethylene glycol via a glycine linker. Cancer Chemother Pharmacol. 1998;42:407–414. doi: 10.1007/s002800050837. [DOI] [PubMed] [Google Scholar]
- 36.Okuno S, Harada M, Yano T, Yano S, Kiuchi S, Tsuda N, Sakamura Y, Imai J, Kawaguchi T, Tsujihara K. Complete regression of xenografted human carcinomas by camptothecin analogue-carboxymethyl dextran conjugate (T-0128) Cancer Res. 2000;60:2988–2995. [PubMed] [Google Scholar]
- 37.Minko T, Paranjpe PV, Qiu B, Lalloo A, Won R, Stein S, Sinko PJ. Enhancing the anticancer efficacy of camptothecin using biotinylated poly(ethylene glycol) conjugates in sensitive and multidrug-resistant human ovarian carcinoma cells. Cancer Chemother Pharmacol. 2002;50:143–150. doi: 10.1007/s00280-002-0463-1. [DOI] [PubMed] [Google Scholar]
- 38.Paranjpe PV, Chen Y, Kholodovych V, Welsh W, Stein S, Sinko PJ. Tumor-targeted bioconjugate based delivery of camptothecin: design, synthesis and in vitro evaluation. J Control Release. 2004;100:275–292. doi: 10.1016/j.jconrel.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 39.Song L, Bevins R, Anderson BD. Kinetics and mechanisms of activation of alpha-amino acid ester prodrugs of camptothecins. J Med Chem. 2006;49:4344–4355. doi: 10.1021/jm060016l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee ES, Na K, Bae YH. Polymeric micelle for tumor pH and folate-mediated targeting. J Control Release. 2003;91:103–113. doi: 10.1016/s0168-3659(03)00239-6. [DOI] [PubMed] [Google Scholar]
- 41.Schanker LS, Less MJ. Lung pH and pulmonary absorption of nonvolatile drugs in the rat. Drug Metab Dispos. 1977;5:174–178. [PubMed] [Google Scholar]
- 42.Palumbo M, Sissi C, Gatto B, Moro S, Zagotto G. Quantitation of camptothecin and related compounds. J Chromatogr B Biomed Sci Appl. 2001;764:121–140. doi: 10.1016/s0378-4347(01)00345-0. [DOI] [PubMed] [Google Scholar]
- 43.Conover CD, Greenwald RB, Pendri A, Shum KL. Camptothecin delivery systems: the utility of amino acid spacers for the conjugation of camptothecin with polyethylene glycol to create prodrugs. Anticancer Drug Des. 1999;14:499–506. [PubMed] [Google Scholar]
- 44.Kim DK, Ryu DH, Lee JY, Lee N, Kim YW, Kim JS, Chang K, Im GJ, Kim TK, Choi WS. Synthesis and biological evaluation of novel A-ring modified hexacyclic camptothecin analogues. J Med Chem. 2001;44:1594–1602. doi: 10.1021/jm0004751. [DOI] [PubMed] [Google Scholar]
- 45.Blagosklonny MV, Darzynkiewicz Z. Cyclotherapy: Protection of normal cells and unshielding of cancer cells. Cell Cycle. 2002;1:375–382. doi: 10.4161/cc.1.6.259. [DOI] [PubMed] [Google Scholar]
- 46.Bocci G, Falcone A, Fioravanti A, Orlandi P, Di Paolo A, Fanelli G, Viacava P, Naccarato AG, Kerbel RS, Danesi R, Del Tacca M, Allegrini G. Antiangiogenic and anticolorectal cancer effects of metronomic irinotecan chemotherapy alone and in combination with semaxinib. Br J Cancer. 2008;98:1619–1629. doi: 10.1038/sj.bjc.6604352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu X, Zhang J, Song L, Lynn BC, Burke TG. Degradation of camptothecin-20(S)-glycinate ester prodrug under physiological conditions. J Pharm Biomed Anal. 2004;35:1113–1125. doi: 10.1016/j.jpba.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 48.Liehr JG, Harris NJ, Mendoza J, Ahmed AE, Giovanella BC. Pharmacology of camptothecin esters. Ann N Y Acad Sci. 2000;922:216–223. doi: 10.1111/j.1749-6632.2000.tb07040.x. [DOI] [PubMed] [Google Scholar]
- 49.Wall ME, Wani MC. Water soluble esters of camptothecin compounds. 6,040,313 US Patent. 2000 Mar 21;
- 50.Gardner SN. A mechanistic, predictive model of dose-response curves for cell cycle phase-specific and -nonspecific drugs. Cancer Res. 2000;60:1417–1425. [PubMed] [Google Scholar]
- 51.Hassan SB, Jonsson E, Larsson R, Karlsson MO. Model for time dependency of cytotoxic effect of CHS 828 in vitro suggests two different mechanisms of action. J Pharmacol Exp Ther. 2001;299:1140–1147. [PubMed] [Google Scholar]
- 52.van Rijn J, van den Berg J, Teerlink T, Kruyt FA, Schor DS, Renardel de Lavalette AC, van den Berg TK, Jakobs C, Slotman BJ. Changes in the ornithine cycle following ionising radiation cause a cytotoxic conditioning of the culture medium of H35 hepatoma cells. Br J Cancer. 2003;88:447–454. doi: 10.1038/sj.bjc.6600700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wheatley DN, Philip R, Campbell E. Arginine deprivation and tumour cell death: arginase and its inhibition. Mol Cell Biochem. 2003;244:177–185. [PubMed] [Google Scholar]
- 54.Hu QY, Li JN, Song DQ, Wang YL, Bekesi G, Weisz I, Jiang JD. Inhibition of human hepatocellular carcinoma by L-proline-m-bis (2-chloroethyl) amino-L-phenylalanyl-L-norvaline ethyl ester hydrochloride (MF13) in vitro and in vivo. Int J Oncol. 2004;25:1289–1296. [PubMed] [Google Scholar]
- 55.Roboz J, Jiang J, Holland JF, Bekesi JG. Selective tumor apoptosis by MF13, L-prolyl-L-m-[bis(chloroethyl)amino]-phenylalanyl-L-norvaline ethyl ester, a new sarcolysin containing tripeptide. Cancer Res. 1997;57:4795–4802. [PubMed] [Google Scholar]
- 56.Jiang JD, Zhang H, Li JN, Roboz J, Qiao WB, Holland JF, Bekesi G. High anticancer efficacy of L-proline-m-bis (2-chloroethyl) amino-L-phenylalanyl-L-norvaline ethyl ester hydrochloride (MF13) in vivo. Anticancer Res. 2001;21:1681–1689. [PubMed] [Google Scholar]
- 57.Ahmed F, Vyas V, Saleem A, Li XG, Zamek R, Cornfield A, Haluska P, Ibrahim N, Rubin EH, Gupta E. High-performance liquid chromatographic quantitation of total and lactone 20(S)camptothecin in patients receiving oral 20(S)camptothecin. J Chromatogr B Biomed Sci Appl. 1998;707:227–233. doi: 10.1016/s0378-4347(97)00615-4. [DOI] [PubMed] [Google Scholar]
- 58.Warner DL, Burke TG. Simple and versatile high-performance liquid chromatographic method for the simultaneous quantitation of the lactone and carboxylate forms of camptothecin anticancer drugs. J Chromatogr B Biomed Sci Appl. 1997;691:161–171. doi: 10.1016/s0378-4347(96)00426-4. [DOI] [PubMed] [Google Scholar]
- 59.Minko T, Kopeckova P, Pozharov V, Kopecek J. HPMA copolymer bound adriamycin overcomes MDR1 gene encoded resistance in a human ovarian carcinoma cell line. J Control Release. 1998;54:223–233. doi: 10.1016/s0168-3659(98)00009-1. [DOI] [PubMed] [Google Scholar]