

Abstract
Infection by the parasite Toxoplasma gondii (tg) can lead to toxoplasmosis in immunocompromised patients such as organ transplant, cancer, and HIV/AIDS patients. The bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) enzyme is crucial for nucleotide synthesis in T. gondii and represents a potential target to combat T. gondii infection. While species selectivity with drugs has been attained for DHFR, TS is much more conserved across species, and specificity is significantly more challenging. We discovered novel substituted-9H-pyrimido[4,5-b]indoles 1–3 with single-digit nanomolar Ki for tgTS, two of which, 2 and 3, are 28- and 122-fold selective over human TS (hTS). The synthesis of these compounds, and their structures in complex with tgTS-DHFR are presented along with binding measurements and cell culture data. These results show, for the very first time, that, in spite of the high degree of conservation of active site residues between hTS and the parasite TS, specificity has been accomplished via novel structures and provides a new target (TS) for selective drug development against parasitic infections.
Keywords: Opportunistic infection, Toxoplasma gondii, crystal structure, thymidylate-synthase inhibitors, active-site inhibitors
According to the World Health Organization, the parasite Toxoplasma gondii (T. gondii, tg) has infected up to a third of the world’s population.1 Though the advent of antiretroviral therapy (ART) has decreased the incidence of T. gondii infection in AIDS patients, the infections still arise because of noncompliance with therapy, discontinuation of therapy because of toxicities with current treatments, and the emergence of drug-resistance strains. Late diagnosis of HIV and the rise of untreated HIV in developing countries also contributes to the persistence of T. gondii infections as a clinical issue.2T. gondii has been defined by the Centers for Disease Control and Prevention as an AIDS defining infection and a major cause of morbidity and indeed mortality in HIV patients as recently as 20093 and 2013.4 Toxoplasma encephalitis (TE) is the principal opportunistic infection (OI) caused by T. gondii in AIDS patients. In addition to AIDS, other immune-compromised patients such as those with organ transplants and those undergoing cancer chemotherapy are also highly susceptible to T. gondii infections.
Sulfadiazine (SDZ) with pyrimethamine (PM) (Figure 1) is the principal drug combination for T. gondii infection along with folinic acid for rescue of host bone marrow toxicity from PM.2 Alternate first line therapy with sulfamethoxazole (SMX) and trimethoprim (TMP) (Figure 1) is also used.2 Problems in chemoprophylaxis, treatment failures, high rates of adverse drug reactions, and drug intolerance to these very similar first-line agents, particularly PM and the sulfonamide component,5 indicate an urgent need for new agents. The most clinically useful combinations, PM/SDZ (with folinic acid rescue) or TMP/SMX, act synergistically.6 The sulfonamide component inhibits dihydropteroate synthase (DHPS), which catalyzes an early step in the synthesis of folic acid. DHPS catalyzes the synthesis of de novo folate biosynthesis, a pathway absent in the human host. PM and TMP inhibit dihydrofolate reductase (DHFR). DHFR catalyzes the synthesis of tetrahydrofolate (THF) from folic acid as well as dihydrofolate (DHF). Thus, the most successful drugs for T. gondii inhibit folate biosynthesis and metabolism at two separate steps to afford inhibition of purine and pyrimidine synthesis. Since Toxoplasma cannot salvage thymidine from the host,7 indirect inhibition of thymidylate synthase (TS) via DHFR inhibition is possible, but the direct inhibition of TS, the enzyme that catalyzes the conversion of dUMP to dTMP for DNA synthesis would be most logical. However, no selective inhibitors of T. gondii TS (tgTS) are known, up to now, that do not also potently inhibit human TS (hTS) as well.8 Unlike DHFR, where the human and parasite enzymes are somewhat divergent in amino acid sequence and structure allowing for selective inhibition of the parasite DHFR by 100-fold or greater, such divergence is much less in TS from parasites and humans.9 There is significant homology between the active site residues of tgTS and hTS. The significance of selective inhibition of tgTS is that this would inhibit the replication of T. gondii cells since T. gondii, unlike human, lacks salvage of thymidine, and thus, TS function in T. gondii is obligatory for survival.10 Such selective inhibitors could be used alone or in combination with PM, TMP, SMX, and other sulfonamides.
Figure 1.

DHFR inhibitors (PM and TMP) in the clinic for T. gondii infections and compounds 1–3.
On the basis of the potent bicyclic hTS inhibitor nolatrexed reported by Webber et al.,11 we designed tricyclic compounds 1–3 using as a novel scaffold for hTS inhibition and as potential anticancer agents. Compounds 1–3 (Figure 1) contain a tricyclic pyrimido[4,5-b]indole scaffold connected to a phenyl or a naphthalene moiety via a sulfur bridge. Compounds 1–3 were synthesized using Scheme 1. Compound 4 was prepared by a method developed by Gangjee et al.12 Ullmann coupling13 of 4 with aryl thiols 5–7 in a microwave apparatus (Initiator from Biotage) in the presence of potassium carbonate provided target compounds 1–3, respectively, in yields of 28–40%.
Scheme 1. Synthesis of 1–3.

Inhibition assays were performed on isolated T. gondii TS-DHFR and hTS. Compounds 1–3 exhibited single-digit nanomolar Ki values (Table 1). Surprisingly, compounds 2 and 3 showed unprecedented selectivity for tgTS over hTS of 28- and 122-fold, respectively.
Table 1. Kinetic Evaluation of 1–3 Reveals Species Selectivitya.
| IC50 (nM) |
Ki (nM)b |
||||
|---|---|---|---|---|---|
| compd | tgTS | hTS | tgTS | hTS | selectivity (hTS Ki/tgTS Ki) |
| 1 | 21 ± 13 | 28.8 ± 21 | 1.8 ± 1.1 | 3.3 ± 2.4 | 1.83 |
| 2 | 23 ± 6.6 | 481 ± 162 | 2.0 ± 0.6 | 55.7 ± 18.8 | 27.9 |
| 3 | 35.8 ± 2.9 | 3250 ± 184 | 3.1 ± 0.25 | 378 ± 21 | 121.9 |
T. gondii TS-DHFR (25 nM) and human TS (50 nM) were preincubated with 100 μM dUMP and inhibitor. The reaction was initiated with 100 μM methyleneTHF.
Because of the concentrations of enzyme required for assay and the limits of detection, for those compounds having Ki values in the lower nanomolar range, the values reported represent an upper limit.
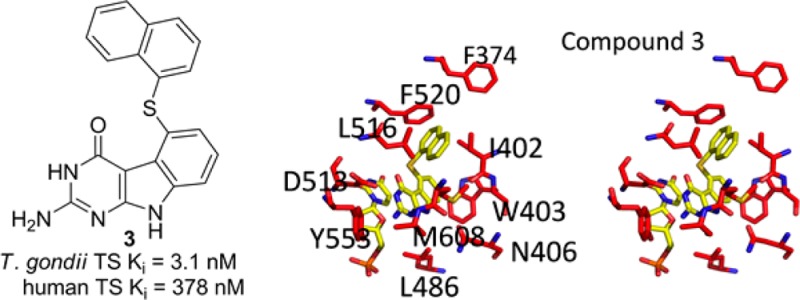
To determine the structural basis for the observed activity and selectivity, we determined the crystal structures of inhibitors 2 and 3 with T. gondii TS-DHFR (Figure 2). By superimposing the tgTS and hTS (Figure S3, Supporting Information), almost all of the residues in the active site were identical. It was surprising that a remarkable difference of 28- and 122-fold in the Ki values of 2 and 3, respectively, were observed in spite of almost identical architectures of the active sites of TS of the two organisms. As shown in Figure 2, the crystal structure of the tgTS-DHFR with 2 and 3 revealed extensive base stacking interactions between the inhibitor and the nucleotide dUMP as expected. Using cocrystal structures of 2 and 3 with tgTS-DHFR, we generated ligand interactions using MOE 2011.10.14 Compound 2 is bound to the active site by aromatic stacking of the pyrimido[4,5-b]indole scaffold and the dUMP pyrimidine ring. Additionally, hydrogen bonds are formed between the N9 of 2 and carboxyl of Asn406, the Asp513 carboxylic oxygen with N3, and the 2-NH2 group. The 2-NH2 group forms an additional hydrogen bond with the backbone oxygen of Ala609. The scaffold is also stabilized by hydrophobic interactions of the C-ring with Trp403. The phenyl ring is oriented almost at a right angle to the tricyclic scaffold and interacts with Ile402, Leu516, Phe520, and Met608. Compound 3 retains the interactions of the tricyclic scaffold and the hydrogen bonds with Asp513, Ala609, and Asn409. Similar to 2, the 1′-naphthyl ring of 3 is oriented almost at a right angle to the tricyclic scaffold and interacts with Ile402, Leu516, Phe520, and Met608.
Figure 2.
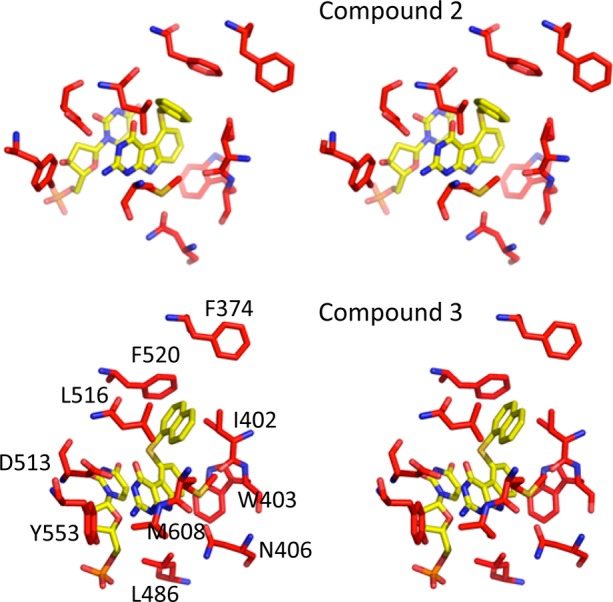
T. gondii TS active site with key residues highlighted in red. The inhibitors 2 and 3 and dUMP are colored yellow.
The structure of 3 in the TS active site suggested that the 1′-naphthalene moiety of 3 must be a key determinant of higher selectivity. The position of the compound in the active site orients the 1′-naphthalene moiety close to the C-terminal tail of the T. gondii TS domain. In fact, the residues of the C-terminal tail are well ordered only in T. gondii and not in the known X-ray crystal structures of human TS. The 1′-naphthalene substituent of 3 also makes hydrophobic interactions with M608 only in tgTS since the C-terminal tail shields the active site from solvent; the tail could be a possible structural determinant that facilitates additional hydrophobic interactions with tgTS conferring selectivity for the parasite (Figure 2).
Compounds 1–3 have also been evaluated against T. gondii cells in culture (Table 2), and 3, at 5 μM, was found to be equivalent to PM at 3 μM indicating a significant effect on T. gondii in culture comparable to a clinically used agent PM. The culture model requires that the experimental compounds rapidly kill exposed tachyzoites as they are released from cells and/or that the experimental compounds penetrate the cell and the vacuole containing tachyzoites where the compounds may kill the organism or prevent its replication.
Table 2. Toxoplasma Cell Culture Study.
| condition | T. gondii in media at day 4 | T. gondii in cells at day 4 | estimated IC50 value (μM)a |
|---|---|---|---|
| no drug | 3710000 | 1140000 | |
| 1 (5 μM) | 2350000 | 340000 | 2.6 |
| 2 (5 μM) | 100000 | 140000 | 3.7 |
| 3 (5 μM) | <25000 | <2500 | 0.6 |
| PM (3 μM) | <25000 | <2500 | 0.57 |
Dose–response curves using data from the three concentrations of test compounds gave estimates of IC50 values.
We have discovered, for the first time, highly potent (single-digit nM) tgTS inhibitors 1–3 with up to a remarkable 122-fold selectivity for tgTS over hTS (Ki in Table 1). This level of selectivity is unprecedented for any parasite with regard to TS. For comparison, TMP selectively inhibits tgDHFR and is used extensively with SMX to treat Toxoplasma infection but has an IC50 = 2.7 μM and a selectivity of only 49-fold for tgDHFR over hDHFR.15 Further PM, a widely used gold standard tgDHFR inhibitor for TE and other T. gondii infections, only has a potency of IC50 = 3.9 μM and a selectivity of only 5.9 for tgDHFR over mammalian DHFR. In addition, we have also, for the very first time, solved the X-ray crystal structure of the bifunctional tgTS-DHFR enzyme with compounds 2 and 3.16 In cell cuture study, compound 3 showed similar potency to PM, a highly effective standard drug used as a control in these studies. We have thus demonstrated with 2 and 3 that structure variation affords increased selectivity while maintaining potency (compared to 1). Figure S3, Supporting Information, shows dUMP and compound 3 and the superposition of key active site residues (using tg numbering) in both human and tgTS. While all residues are identical between the two species, only M608 is present in the tgTS structure. Future studies will be directed toward solving the structure of compound 3 with human TS to more fully discern possible differences. TS is a completely new and novel target for T. gondii. There is no report, to our knowledge, of selective and potent inhibitors of tgTS as anti-OI agents. The ability to provide agents that are selective for tgTS on the basis of structure-based rational design is a major step toward providing novel clinically useful agents with tgTS inhibition.
Glossary
Abbreviations
- tg
T. gondii
- TS
thymidylate synthase
- DHFR
dihydrofolate reductase
- TE
toxoplasma encephalitis
- OI
opportunistic infection
- SDZ
sulfadiazine
- PM
pyrimethamine
- SMX
sulfamethoxazole
- TMP
trimethoprim
Supporting Information Available
Synthetic procedures, compound characterization, biological assays, and X-ray structure determination. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
The atomic coordinates of T.gondii TS-DHFR/Compound 2 and T.gondii TS-DHFR/Compound 3 described here have been deposited in the RCSB Protein Data Bank (www.pdb.org) with accession codes 4KY4 and 4KYA, respectively.
Author Contributions
§ These authors (N.Z. and H.S.) contributed equally to this work.
This work was supported, in part, by NIH grant CA152316 (to A.G.), the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence (to A.G.), and AI083146 (to K.S.A.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Montoya J. G.; Liesenfeld O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [DOI] [PubMed] [Google Scholar]
- Kaplan J. E.; Benson C.; Holmes K. K.; Brooks J. T.; Pau A.; Masur H.. Guidelines for Prevention and Treatment of Opportunistic Infections in HIV-Infected Adults and Adolescents. Recommendations from CDC, The National Institutes of Health and the HIV Medicine Association of the Infectious Diseases Society of America. CDC, MMWR, April 10, 2009, Vol. 58, NoRR-4. [PubMed] [Google Scholar]
- CDC. http://www.cdc.gov/parasites/toxoplasmosis/ (accessed May 2013).
- Dudrand C. M.; Flexner C. HIV Cure: Knocking on the Door. Clin. Pharmacol. Ther. 2013, 93, 382–384. [DOI] [PubMed] [Google Scholar]
- Aspinall T. V.; Joynson D. H.; Guy E.; Hyde J. E.; Sims P. F. The Molecular Basis of Sulfonamide Resistance in Toxoplasma gondii and Implications for the Clinical Management of Toxoplasmosis. J. Infect. Dis. 2002, 185, 1637–1643. [DOI] [PubMed] [Google Scholar]
- Chulay J. D.; Watkins W. M.; Sixsmith D. G. Synergistic Antimalarial Activity of Pyrimethamine and Sulfadoxine against Plasmodium falciparum in vitro. Am. J. Trop. Med. Hyg. 1984, 33, 325–330. [DOI] [PubMed] [Google Scholar]
- Hyde J. E. Targeting Purine and Pyrimidine Metabolism in Human Apicomplexan Parasites. Curr. Drug Targets 2007, 8, 31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang C. K.; De S. K.; White J.; Buckner F. S.; Varani G.; Rathod P. K. Differential Drug Binding by the Highly Conserved Plasmodium falciparum Thymidylate Synthase. Mol. Biochem. Parasitol. 2005, 143, 121–124. [DOI] [PubMed] [Google Scholar]
- Ferrari S.; Lasasso V.; Costi M. P. Sequence-Based Identification of Specific Drug Target Regions in the Thymidylate Synthase Enzyme Family. ChemMedChem 2008, 3, 392–401. [DOI] [PubMed] [Google Scholar]
- Fox B. A.; Bzik D. J. De Novo Pyrimidine Biosynthesis is Required for Virulence of Toxoplasma gondii. Nature 2002, 415, 926–929. [DOI] [PubMed] [Google Scholar]
- Webber S. E.; Bleckman T. M.; Attard J.; Deal J. G.; Kathardekar V.; Welsh K. M.; Webber S.; Janson C. A.; Matthews D. A.; Smith W. W.; Freer S. T.; Jordan S. R.; Bacquet R. J.; Howland E. F.; Booth C. L. J.; Ward R. W.; Hermann S. M.; White J.; Morse C. A.; Hilliard J. A.; Bartlett C. A. Design of Thymidylate Synthase Inhibitors Using Protein Crystal Structures: The Synthesis and Biological Evaluation of a Novel Class of 5-Substituted Quinazolinones. J. Med. Chem. 1993, 36, 733–746. [DOI] [PubMed] [Google Scholar]
- Gangjee A.; Zaware N.; Raghavan S.; Ihnat M.; Shenoy S.; Kisluik R. L. Single Agents with Designed Combination Chemotherapy Potential: Synthesis and Evaluation of Substituted Pyrimido[4,5-b]indoles as Receptor Tyrosine Kinase and Thymidylate Synthase Inhibitors and as Antitumor Agents. J. Med. Chem. 2010, 53, 1563–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperotto E.; van Klink G. P. M.; de Vries J. G.; van Koten G. Ligand-Free Copper-Catalyzed C-S Coupling of Aryl Iodides and Thiols. J. Org. Chem. 2008, 73, 5625–5628. [DOI] [PubMed] [Google Scholar]
- Molecular Operating Environment (MOE 2011.10); Chemical Computing Group, Inc.: Montreal, Quebec, Canada, 2008; www.chemcomp.com.
- Piper J. R.; Johnson C. A.; Krauth C. A.; Carter R. L.; Hosmer C. A.; Queener S. F.; Borotz S. E.; Pfefferkorn E. R. Lipophilic Antifolates as Agents against Opportunistic Infections. 1. Agents Superior to Trimetrexate and Piritrexim against Toxoplasma gondii and Pneumocystis carinii in in Vitro Evaluations. J. Med. Chem. 1996, 39, 1271–1280. [DOI] [PubMed] [Google Scholar]
- Sharma H.; Landau M. J.; Vargo M. A.; Spasov K. A.; Anderson K. S.. First Three-Dimensional Structure of Toxoplasma gondii Thymidylate Synthase-Dihydrofolate Reductase: Insights for Catalysis, Interdomain Interactions, and Substrate Channeling. Biochemistry 2013, submitted [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.