

Abstract
L-edge spectroscopy of 3d transition metals provides important electronic structure information and has been used in many fields. However, the use of this method for studying dilute aqueous systems, such as metalloenzymes, has not been prevalent because of severe radiation damage and the lack of suitable detection systems. Here we present spectra from a dilute Mn aqueous solution using a high-transmission zone-plate spectrometer at the Linac Coherent Light Source (LCLS). The spectrometer has been optimized for discriminating the Mn L-edge signal from the overwhelming O K-edge background that arises from water and protein itself, and the ultrashort LCLS X-ray pulses can outrun X-ray induced damage. We show that the deviations of the partial-fluorescence yield-detected spectra from the true absorption can be well modeled using the state-dependence of the fluorescence yield, and discuss implications for the application of our concept to biological samples.
SECTION: Spectroscopy, Photochemistry, Excited States
Recent developments of X-ray free electron lasers (XFELs) open up opportunities for studying the chemical and structural dynamics of a wide variety of systems beyond what is possible at synchrotron radiation (SR) sources. Ultrabright femtosecond X-ray pulses allow data collection before the onset of radiation damage, enabling studies under functional conditions. This is particularly beneficial for biological materials and samples in aqueous solution, e.g., to follow chemical dynamics during catalytic reactions and electron transfer processes. The data collection of biological systems are traditionally carried out under cryogenic temperature at SR sources by trapping intermediate states to minimize chemical relaxation, and to avoid fast X-ray-induced radiation damage at room temperature. The “probe-before-destroy” approach with femtosecond (fs) pulses at XFELs has been demonstrated for structural determination by serial X-ray diffraction of microcrystals1–3 as well as X-ray emission spectroscopy of Photosystem II (PS II)4,5 using hard X-rays.
To probe the chemical interactions of 3d transition metal catalysts in metalloenzymes and bioinspired materials, metal Kedge spectroscopy (1s → np transitions in the hard X-ray energy range) has been widely used at SR sources (for example, ref 6 for Mn X-ray absorption spectroscopy (XAS) in PS II). The preference of K-edge over L-edge spectroscopy (2p → 3d transitions in the soft X-ray energy range) is mainly due to the advantages arising from 2 orders of magnitude smaller absorption cross sections at the K-edges. This reduces radiation damage, guarantees inherent bulk sensitivity due to the larger penetration depth, and often results in comparatively simpler experimental setups compared to L-edge spectroscopy experiments that typically require an ultrahigh vacuum environment. Despite the experimental difficulties, it is highly desirable to use L-edge spectroscopy for studying metal catalysts, as (i) it provides significantly higher resolution since the natural line widths at the L2 and L3 edges are approximately one-fourth of those at the K-edge, and (ii) the frontier orbitals are directly accessible under dipole selection rules (2p → 3d transitions), while only s → p transitions are allowed at the K-edge (note that the 1s → 3d transitions are quadrupole-allowed at the Kedge and are – together with dipole transitions to hybridized states that have gained some p-character, visible as a pre-edge feature in the K-edge spectra). Direct probing of the 3d electronic structure at higher resolution gives greater chemical sensitivity to, e.g., oxidation states, symmetry, and covalency of the complex studied.7–11 Furthermore, (iii) the experimental Ledge data can be complemented by theoretical calculations of the 3d electronic structure using the crystal-field multiplet approach11 and recently developed state-of-the-art ab initio treatments based on quantum-chemical methods.12–14
Even at cryogenic temperatures, biological spectroscopy in the soft X-ray regime is hampered mainly by severe radiation damage caused by radical diffusion taking place in the picosecond time regime.15,16 Radiation damage can naturally be avoided if the sample is replaced faster than the rate of damage by circulating or injecting the sample into the X-ray interaction region. This also allows for probing the system at room temperature in its natural environment by preventing dehydration and therefore guaranteeing catalytic turnover. A drawback in this case is the necessity of fast sample replacement, requiring large quantities of protein samples. In this report, we describe our approach to biological L-edge XAS at free-electron lasers with a newly developed high-transmission energy-discriminating spectrometer using a reflection zone-plate. We discuss how the ‘probe-before-destroy’ approach using the ultrashort fs X-ray pulses of XFELs can overcome radiation damage guaranteeing radiation-damage free detection of L-edge XAS. The design of the spectrometer is optimized for the collection of transition metal L-edge spectra from dilute systems that are relevant to metalloenzymes in biology. The spectrometer described here is designed for Mn L-edge XAS with the aim of using it for data collection from Mn-containing metalloenzymes such as PS II, Mn catalase,17,18 and MnFe ribonucleotide reductase,19,20 by injecting solution samples into the X-ray interaction point. However, we emphasize that the idea of the reflection zone-plate spectrometer is quite general and is applicable to other dilute metalloenzyme systems containing metals such as Fe, Cu, Mo, and others.
The most direct way of measuring XAS is to perform a transmission measurement, and numerous approaches for collecting transmission XAS of liquid samples at soft X-ray energy have been introduced recently.16,21–24 Due to the low concentration (~ mM regime) of biological samples in solution, however, transmission measurements are impossible as the sample would be either too thin for appreciable solute absorption at acceptable solvent transmission or too thick with unacceptable solvent transmission at appreciable solute absorption. An alternative way is to collect Auger electrons or fluorescence photons resulting from the core-hole decay while scanning through the absorption edge to indirectly measure the absorption cross section. Auger electrons represent, by far, the dominant decay channel in the soft X-ray regime and measuring the Auger electron yield (or the inelastically scattered background via partial or total electron-yield detection) can provide the true absorption cross section. Although electron detection for biological samples in solution has been employed,25 the surface sensitivity of electron-detected XAS (1 nm) complicates the analysis. Furthermore, for time-resolved experiments at XFELs, electrons suffer from space-charge effects due to strong X-ray or optical fields.26,27
Fluorescence-detected 3d transition metal L-edge XAS is bulk-sensitive with a probing depth on the order of 1 μm. However, total-fluorescence yield (TFY) detection without discrimination of solute and solvent signals often suffers from severe artifacts that render the collected signal different from the true absorption cross section, e.g., state-dependent decay probabilities28 and concentration effects.29–31 These influences might be small at low concentrations, and TFY L-edge XAS of biological samples in solution in the mM regime at SR sources have been reported.32–35 In the case of Mn in biological samples like PS II, an additional challenge arises from the presence of an overwhelming oxygen Kα fluorescence signal, i.e., detecting Mn L-fluorescence at ~640 eV from 4 Mn atoms out of the dominant O K-fluorescence at ~525 eV from ~25000 O-atoms in the protein of ~350 kDa, in addition to the overwhelming 55 molar oxygen from water. Therefore, collecting partial fluorescence yield (PFY)-detected XAS instead of TFY by energy-discriminating other fluorescence signals is mandatory. So far, and to the best of our knowledge, the only approach for separating the Mn L- and the O K-fluorescence for PFY XAS was done with a superconducting solid state detector with 20 eV resolution36 (see Supporting Information Figure S1). This detector, however, is not suitable for our aims to measure XAS at an XFEL, because it operates in single-photon counting mode with low repetition rates. Thus, it would be swamped by the excess O K-fluorescence photons which arrive within the short (fs) length of the XFEL pulse.
Our approach to Mn L-edge PFY XAS at XFELs is to use a high-transmission spectrometer optimized for discrimination of Mn L- and O K-fluorescence. A schematic depiction of the experimental setup is shown in Figure 1. With its bulk sensitivity and the absence of space-charge effects, PFY detection enables time-resolved X-ray spectroscopy of biological samples at XFELs.
Figure 1.

Schematic depiction of the experimental setup with the high-transmission X-ray spectrometer showing (a) overview and (b) top view with a front view of the CCD camera. (b) Left: CCD front view with schematic depiction of Mn L-fluorescence focused at the top of the CCD (left in the figure), and O K-fluorescence defocused at the bottom of the CCD (right in the figure) and the measured CCD images depicting the fluorescence of a solid MnO sample as diffracted off one zone plate taken at an incident photon energy below (600 eV) and above (653 eV) the Mn L-edge (intensity encoded in color: red high, blue low). The axis of linear polarization of the incident beam is indicated.
We used a detection scheme with a single optical element based on total reflection zone plates (RZPs), optimized for high-transmission PFY XAS. The spectrometer has high transmission due to its ability to disperse and focus the fluorescence signal in one optical element with a large acceptance (solid) angle. It consists of three zone-plates on a single Si substrate (Figure 1)37 and was designed to select the Mn L-edge fluorescence at 640 eV with a bandwidth of 20 eV (fwhm), sufficient to separate the Mn L- from the O K-edge fluorescence at ~525 eV. The three reflection zone plates are placed at 90 mm from the sample jet to collect the maximum possible solid angle of the fluorescence. The spectrometer is optimized such that the negative first order diffracted light is used to record the X-ray spectra (Figure 1a). O K- and Mn Ledge fluorescence are focused to different focal planes along the ray and dispersed to different vertical positions at the detection plane (Figure 1b). By placing the CCD detector at the appropriate focus position, the Mn L-edge fluorescence can be efficiently separated from the O K-edge fluorescence. A test measurement on solid MnO performed at the BESSYII SR source with incident photon energies below and above the Mn L-edges, shown in Figure 1b, confirmed the feasibility of this concept.
Our concept provides a solid angle larger by 2 orders of magnitude compared to more conventional high-resolution X-ray spectrometers with grazing-incidence gratings in Rowland geometry (ref 38 and references therein). The spectrometer was designed for an optimum of both tangential angular acceptance and diffraction efficiency at the working wavelength of 1.94 nm (640 eV). Choosing the negative first order (Figure 1a) allows for optimized grazing incidence geometry with incidence angles of 1.5° to 3.9° and corresponding diffraction angles of 1.18° to 0.96°. The depth of profile was chosen to be 17 nm, thus resulting in a maximum diffraction efficiency of 16% for the middle of the zone plate. Compared to the normally chosen positive first order, the use of the negative first order provides several times higher integral diffraction efficiency for the Mn fluorescence (for details, see Figure S2 and Supporting Information). As the horizontal aperture of a single RZP is limited by its smallest useful structures (about 80 nm in our spectrometer), we used three separate RZPs on one substrate in order to maximize the solid angle.
To quantify RZP misalignment tolerances and to develop a fast and reliable adjustment strategy, we conducted extensive ray tracing simulations utilizing the ‘Ray’ code (for details, see Supporting Information, Figure S3).39 In short, the RZPs are prealigned ex situ with respect to the optical axis of the spectrometer using visible laser light. Alignment of the whole spectrometer, including the RZPs and the detector with respect to the source of fluorescence, is done in situ by manipulating the three translational degrees of freedom (horizontal and vertical displacement of the RZP and RZP-source distance). We used an iKon-L 936 X-ray CCD (Andor Ltd.) as a 2D detector to ensure high quantum yield and single photon sensitivity.
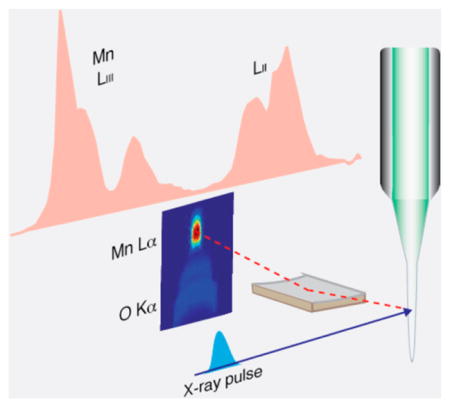
As a proof-of-principle of detecting Mn L-edge PFY XAS in an aqueous biological system, we measured solvated Mn2+ from an aqueous solution of 500 mM MnCl2 at LCLS. An electrospun liquid jet40 was used to deliver the sample at the X-ray intersection point, with a flow rate of ~0.2 μL/min (~5 μm diameter). Representative CCD images and the integrated spectrum are shown in Figure 2.
Figure 2.

Mn L-edge partial fluorescence yield (PFY) X-ray absorption spectrum (red line) of MnCl2 (500 mM) aqueous solution measured with the PFY detector. The panels above the spectrum depict CCD images in the region of the Mn L-edge fluorescence taken below (at 636 eV) and above (at 640 eV) the Mn L3-absorption edge (intensity encoded in color: red high, blue low). The dashed lines mark the three areas on the CCD with Mn L-edge fluorescence from the three zone plates. Photon energies were calibrated at the maximum of the L3 edge to 639.7 eV41 with a reference spectrum of solid MnO (see Supporting Information, Figure S4).
The monochromator of the SXR instrument at LCLS42 was scanned from 634.5 to 658.5 eV in steps of 0.24 eV, and the CCD images were acquired with an acquisition time of 10 s per step, yielding a total measurement time of 17 min for the spectrum. The three areas on the CCD comprising the Mn L-fluorescence as diffracted from the three RZPs can easily be identified by comparing the CCD images taken at incident photon energies below and above the Mn L3 absorption edge (dashed areas in Figure 2). The PFY Mn2+ (aq.) L-edge XAS was obtained by plotting the integrated intensity of these three areas versus the incident photon energy. The average count rate in the integrated area was approximately 2000 counts per pixel (per 10 s) at the maximum of the L3 absorption (639.7 eV) with single pixels recording up to 20 000 counts (per 10 s). The conversion factor is about 50 counts/photon for the Andor CCD. The measurement was virtually background-free as the combined counts arising from both dark noise counts and stray light, which arises from remaining O K-fluorescence, were less than 20 counts per pixel (on average), <1% of the average signal. In Figure 2, this background has been subtracted, and the data were normalized to the maximum intensity. This measurement demonstrates that the fluorescence signal from 3d transition metal atoms can be separated from ligand and solvent signals with unprecedented efficiency with the concept presented here. We also obtained two additional spectra under slightly varied experimental conditions (see Figure S5) to confirm the reproducibility of this approach. From an extrapolation of the present count rates to measurements on PS II at a 100 times lower concentration, we expect count rates of 20 counts per pixel (on average) compared to an expected background of 20 counts per pixel. Based on these numbers, we expect a collection time of about 30–36 h for a spectrum from PS II. This demonstrates the feasibility of biological spectroscopy at low concentrations with the current zone-plate design, and shows that significant reductions in total signal levels or signal-to-background levels expected from alternative lower solid angle detection schemes or lower energy discrimination detectors would render the experiments unfeasible.
In the following, we address the deviations in the experimental PFY detected L-edge XAS from the true absorption cross-section. We confirmed using simulations that self-absorption does not influence the PFY spectrum in the 500 mM Mn solution sample (Supporting Information Figure S6), and therefore such influence will not affect biological samples with much lower metal concentration. Like TFY, PFY L-edge XAS of 3d transition metals has been shown to deviate from the true absorption coefficient due to state-dependent fluorescence yields.28,43–45 In order to apply PFY-detected XAS collected with XFEL pulses for the analysis of chemical states in dilute biological samples, it is important to address the influence of state-dependent fluorescence yield on the PFY spectrum. In Figure 3 (top panel), we compare the Mn L-edge PFY spectrum from Figure 2 with a measurement of the true absorption cross section of Mn2+ (aq.) as obtained in transmission mode at BESSYII. We note that peak A is strongly suppressed in the PFY spectrum, while peaks C–G are enhanced compared to the transmission measurement (both spectra are normalized to one at peak B). In the bottom panel of Figure 3, we show the theoretical spectra evaluated with ligand-field theory11 for a Mn2+ ion in a hexa–aqua complex in cubic symmetry (6A1 state, crystal field parameter 10Dq = 1.2 eV) excluding (XAS) and including (PFY) state dependent X-ray emission cross sections. State-dependent fluorescence yield is an intrinsic atomic property of the system under investigation and thus represents an intrinsic property of PFY XAS measurements.28,45 Polarization effects are taken into account in the calculated spectra28 in accordance with the experimental conditions, i.e., the axis of linear polarization of the incident radiation was parallel to the scattering plane (image plane in Figure 1b) defined by the propagation direction of the incident radiation and the optical axis of the spectrometer that is at an angle of 90° (Figure 1). An overall excellent agreement is obtained, with only small deviations apparent in peaks D and G. In particular, the suppression of A and the enhancement of C-G, when going from PFY to the true absorption cross section, is quantitatively reproduced. We thus conclude that the PFY Mn L-edge XAS measured at LCLS can be quantitatively described, taking into account the intrinsic state-dependent fluorescent yield.
Figure 3.

Top: Mn L-edge partial fluorescence yield spectrum (PFY, red, same as in Figure 2) and X-ray absorption spectrum measured in transmission mode (Trans., blue) of Mn2+ (aq.). Bottom: Calculated Mn L-edge PFY (pink) and X-ray absorption (XAS, violet) spectra of Mn2+ as calculated with the crystal-field multiplet approach (taken from ref 28). State-dependent fluorescence and polarization effects were taken into account for the calculated PFY spectrum (see text). All spectra are normalized to one at the maximum.
The comparison of measured and calculated spectra in Figure 3 reveals an additional important aspect. The LCLS PFY experiment was done at a flux of 1011 photons per pulse for ~100 fs pulses, while the transmission measurement at BESSYII was performed at a photon flux of approximately 108 photons/s (corresponding to 3 × 105 per pulse) with a pulse duration of 100 ps. The beam diameter on the sample was 200 (vertical) μm × 20 (horizontal) μm at LCLS and it was 100 μm at BESSYII (round spot, fwhm). The photon power density was thus several orders of magnitude higher at LCLS than at BESSYII. Since the deviations of the PFY Mn L-edge spectrum from the true absorption cross section is quantitatively captured by taking state-dependent fluorescence yield into account, sequential ionization/excitation or other unwanted processes due to the ultrabright femtosecond X-ray pulses at LCLS do not seem to significantly contribute to the spectrum under the flux used in our experiment.
In conclusion, we have shown how PFY-detected 3d transition metal L-edge XAS from Mn aqueous solution samples can be collected with the ultrabright femtosecond X-ray pulses at the XFEL, by discriminating the solute from the solvent fluorescence with a novel fluorescence-yield detector with high efficiency. We expect that this approach will be suitable for collecting XAS of dilute solvated species in the mM range efficiently at XFELs. We also demonstrated how state-dependent fluorescence yield causes the PFY-detected XAS to deviate from the true absorption cross section for the model case of Mn2+ ions in aqueous solution, and discussed implications for the application of our concept to biological samples. We expect that the approach presented here will enable us to gain insights into the chemical interactions of 3d transition metal catalysts in metalloenzymes and biomolecules, and to pave the way for their investigation with time-resolved L-edge spectroscopy at XFELs.
EXPERIMENTAL SECTION
Materials
MnCl2 was purchased from Sigma-Aldrich and used without further purification. MnIICl2, 500 mM Mn, was dissolved in a glycerol/water (35%, w/v) mixture. Thin solid MnOx films with various thicknesses (10, 20, and 50 nm MnOX coated on Si wafer) were purchased from Luxel Corporation (Friday Harbor, WA, USA).
Data Collection and Analysis
The PFY measurements were performed at the SXR instrument at the LCLS.42,46 The MnCl2 aqueous solution samples (500 mM) were injected into the X-ray beam in the liquid jet end station (LJE) (for details, see the work of Kunnus et al.38) by means of an electro-hydrodynamic-focused liquid jet of several micrometers in diameter in the cone-jet mode.40 X-ray pulses of ~100 fs pulse length at a repetition rate of 120 Hz were used for the data collection. The SXR monochromator was scanned from 634.5 to 658.5 eV using a bandwidth of 300 meV.42 The X-ray flux was estimated to be 1.3 × 1010 photons per pulse on the sample (using 1 mJ per pulse before the beamline, and an overall beamline transmittance including the monochromator and the Kirkpatrick-Baez (KB) optics of about 0.2%), and the KB optics were bent to produce a beam size of 200 (v) × 20 (h) μm2 at the sample. The Mn fluorescence signals on the three areas of the CCD camera (iKon-L 936, Andor Ltd., UK) were integrated, background-corrected, and normalized to the total fluorescence signal (extracted from the CCD images utilizing the zero-order diffraction of the zone plates).
The transmission measurements of aqueous MnIICl2 solutions (1000 mM) were carried out at the synchrotron radiation facility BESSYII of Helmholtz-Zentrum Berlin, Berlin, Germany, at the dipole beamline PM3 in single bunch operation mode with the specialized sample holder optimized for soft X-ray transmission measurements of liquid samples described in more detail in Schreck et al.24 The coordination number of Mn2+ ion in water at ~1000 mM concentration is close to six, with the hexa-aqua complex [Mn(H2O)6]2+ as the dominant entity.47,48
The total electron yield (TEY) measurements of thin solid MnOX films (see Supporting Information) were performed at the Stanford Synchrotron Radiation Light source (SSRL) at the SLAC National Accelerator Laboratory, Stanford, USA at beamline 10.1. The wiggler beamline is equipped with a spherical grating monochromator which was operated at 0.2 eV resolution. The TEY was collected by the sample drain current and normalized by a freshly evaporated gold grid intercepting the beam upstream.
Supplementary Material
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences (OBES), Division of Chemical Sciences, Geosciences, and Biosciences (CSGB) of the Department of Energy (DOE) under Contract DE-AC02-05CH11231 for X-ray spectroscopy instrumentation (J.Y. and V.K.Y.), a LBNL Laboratory Directed Research and Development award (N.K.S.) for data processing methods, and the NIH Grant GM50781 (A.B.) and GM055302 (V.K.Y.) for Mn inorganic chemistry and spectroscopy. Support from the Alexander von Humboldt Foundation (J.K.) and the Ruth L. Kirschstein National Research Service Award (F32GM100595, R.T.) are acknowledged. The injector work was supported by OBES, CSGB of the DOE under contract DE-AC02-76SF00515 (H.L. and M.J.B), LCLS (M.J.B.), and a SLAC Laboratory Directed Research and Development award (H.L. and M.J.B.). S.T. and K.K. are grateful to SFB755-DFG and the Volkswagenstiftung Peter Paul Ewald fellowship for financial support. Support by the Swedish Energy Agency (Energimyndigheten), K&A Wallenberg Foundation (Artificial Leaf Umeå) and Umeå University (Solar Fuels Umeå) to J.M. is acknowledged. Portions of this research were carried out on the SXR Instrument at the Linac Coherent Light Source (LCLS), a division of SLAC National Accelerator Laboratory and an Office of Science user facility operated by Stanford University for the U.S. Department of Energy. The SXR Instrument is funded by a consortium whose membership includes the LCLS, Stanford University through the Stanford Institute for Materials Energy Sciences (SIMES), Lawrence Berkeley National Laboratory (LBNL), University of Hamburg through the BMBF priority program FSP 301, and the Center for Free Electron Laser Science (CFEL). We are grateful to the LCLS and BESSYII staff for their valuable support. We gratefully thank the Helmholtz Virtual Institute “Dynamic Pathways in Multidimensional Landscapes” as well as the BMBF project “Next generation instrumentation for ultrafast X-ray science at accelerator-driven photon source” (project no. 05K12CB4) and a Marie Curie FP7-Reintegration-Grant within the 7th European Community Framework Program (project no. PCIG10-GA-2011-297905) for support and funds for the spectrometer. We also thank Dr. Stephan Friedrich, Lawrence Livermore National Laboratory, for providing the spectra from a superconducting tunnel junction detector.
Footnotes
The authors declare no competing financial interest.
Spectra from a super conducting tunnel junction detector (Figure S1), the simulation of the diffraction efficiency of the −1 and +1 order of the zone plate (Figure S2), the design parameters and optimization of the zone plate (Figure S3), the Mn L-edge spectra using different detection methods (Figure S4), the MnCl2 spectra collected at LCLS under different conditions (S5), and the effects of saturation on the spectra (Figure S6). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, et al. Femtosecond X-Ray Protein Nanocrystallography. Nature. 2011;470:73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boutet S, Lomb L, Williams GJ, Barends TRM, Aquila A, Doak RB, Weierstall U, DePonte DP, Steinbrener J, Shoeman RL, et al. High-Resolution Protein Structure Determination by Serial Femtosecond Crystallography. Science. 2012;337:362–364. doi: 10.1126/science.1217737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kern J, Alonso-Mori R, Hellmich J, Tran R, Hattne J, Laksmono H, Glöckner C, Echols N, Sierra RG, Sellberg J, et al. Room Temperature Femtosecond X-ray Diffraction of Photosystem II Microcrystals. Proc Natl Acad Sci US A. 2012;109:9721–9726. doi: 10.1073/pnas.1204598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alonso-Mori R, Kern J, Gildea RJ, Sokaras D, Weng TC, Lassalle-Kaiser B, Tran R, Hattne J, Laksmono H, Hellmich J, et al. Energy-Dispersive X-ray Emission Spectroscopy Using an X-ray Free-Electron Laser in a Shot-by-Shot Mode. Proc Natl Acad Sci US A. 2012;109:19103–19107. doi: 10.1073/pnas.1211384109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kern J, Alonso-Mori R, Tran R, Hattne J, Gildea RJ, Echols N, Glöckner C, Hellmich J, Laksmono H, Sierra RG, et al. Simultaneous Femtosecond X-ray Spectroscopy and Diffraction of Photosystem II at Room Temperature. Science. 2013;340:491–495. doi: 10.1126/science.1234273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yano J, Yachandra VK. Where Water Is Oxidized to Dioxygen: Structure of the Photosynthetic Mn4Ca Cluster from X-ray Spectroscopy. Inorg Chem. 2008;47:1711–1726. doi: 10.1021/ic7016837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hocking RK, George SD, Gross Z, Walker FA, Hodgson KO, Hedman B, Solomon EI. Fe L- and K-Edge XAS of Low-Spin Ferric Corrole: Bonding and Reactivity Relative to Low-Spin Ferric Porphyrin. Inorg Chem. 2009;48:1678–1688. doi: 10.1021/ic802248t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson SA, Kroll T, Decreau RA, Hocking RK, Lundberg M, Hedman B, Hodgson KO, Solomon EI. Iron LEdge X-ray Absorption Spectroscopy of Oxy-Picket Fence Porphyrin: Experimental Insight into Fe-O2 Bonding. J Am Chem Soc. 2013;135:1124–1136. doi: 10.1021/ja3103583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hocking RK, Wasinger EC, Yan YL, deGroot FMF, Walker FA, Hodgson KO, Hedman B, Solomon EI. Fe L-Edge X-ray Absorption Spectroscopy of Low-Spin Heme Relative to Non-Heme Fe Complexes: Delocalization of Fe d-Electrons into the Porphyrin Ligand. J Am Chem Soc. 2007;129:113–125. doi: 10.1021/ja065627h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Degroot FMF, Hu ZW, Lopez MF, Kaindl G, Guillot F, Tronc M. Differences between L3 and L2 X-ray-Absorption Spectra of Transition-Metal Compounds. J Chem Phys. 1994;101:6570–6576. [Google Scholar]
- 11.de Groot F. Multiplet Effects in X-ray Spectroscopy. Coord Chem Rev. 2005;249:31–63. [Google Scholar]
- 12.Josefsson I, Kunnus K, Schreck S, Foehlisch A, de Groot F, Wernet P, Odelius M. Ab Initio Calculations of X-ray Spectra: Atomic Multiplet and Molecular Orbital Effects in a Multiconfigurational SCF Approach to the L-Edge Spectra of Transition Metal Complexes. J Phys Chem Letts. 2012;3:3565–3570. doi: 10.1021/jz301479j. [DOI] [PubMed] [Google Scholar]
- 13.Roemelt M, Maganas D, DeBeer S, Neese F. A Combined DFT and Restricted Open-Shell Configuration Interaction Method Including Spin-Orbit Coupling: Application to Transition Metal LEdge X-ray Absorption Spectroscopy. J Chem Phys. 2013;138:204101. doi: 10.1063/1.4804607. [DOI] [PubMed] [Google Scholar]
- 14.Suljoti E, Garcia-Diez R, Bokarev SI, Lange KM, Schoch R, Dierker B, Dantz M, Yamamoto K, Engel N, Atak K. Direct Observation of Molecular Orbital Mixing in a Solvated Organometallic Complex. Angew Chem, Int Ed Engl. 2013 doi: 10.1002/anie.201303310. [DOI] [PubMed] [Google Scholar]
- 15.Yano J, Kern J, Irrgang KD, Latimer MJ, Bergmann U, Glatzel P, Pushkar Y, Biesiadka J, Loll B, Sauer K, et al. X-ray Damage to the Mn4Ca Complex in Single Crystals of Photosystem II: A Case Study for Metalloprotein Crystallography. Proc Natl Acad Sci US A. 2005;102:12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wernet P, Gavrila G, Godehusen K, Weniger C, Nibbering ETJ, Elsaesser T, Eberhardt W. Ultrafast Temperature Jump in Liquid Water Studied by a Novel Infrared Pump–X-ray Probe Technique. Appl Phys A. 2008;92:511–516. [Google Scholar]
- 17.Whittaker JW. Non-Heme Manganese Catalase - The ‘Other’ Catalase. Arch Biochem Biophys. 2012;525:111–120. doi: 10.1016/j.abb.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoder DW, Hwang J, Penner-Hahn JE. Manganese Catalases. In: Sigel A, Sigel H, editors. Manganese and Its Role in Biological Processes. Vol. 37. Marcel Dekker, Inc; New York: 2000. pp. 527–557. [PubMed] [Google Scholar]
- 19.Leidel N, Popovic-Bijelic A, Havelius KGV, Chernev P, Voevodskaya N, Graslund A, Haumann M. High-Valent MnFe and FeFe Cofactors in Ribonucleotide Reductases. Biochim Biophys Acta: Bioenergetics. 2012;1817:430–444. doi: 10.1016/j.bbabio.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 20.Bollinger JM, Jr, Jiang W, Green MT, Krebs C. The Manganese(IV)/Iron(III) Cofactor of Chlamydia Trachomatis Ribonucleotide Reductase: Structure Assembly Radical Initiation and Evolution. Curr Opin Struct Biol. 2008;18:650–657. doi: 10.1016/j.sbi.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 21.Huse N, Wen H, Nordlund D, Szilagyi E, Daranciang D, Miller TA, Nilsson A, Schoenlein RW, Lindenberg AM. Probing the Hydrogen-Bond Network of Water via Time-Resolved Soft X-ray Spectroscopy. Phys Chem Chem Phys. 2009;11:3951–3957. doi: 10.1039/b822210j. [DOI] [PubMed] [Google Scholar]
- 22.Nagasaka M, Hatsui T, Horigome T, Hamamura Y, Kosugi N. Development of a Liquid Flow Cell to Measure Soft X-ray Absorption in Transmission Mode: A Test for Liquid Water. J Electron Spectrosc Relat Phenom. 2010;177:130–134. [Google Scholar]
- 23.Lange LM, Aziz EF. Electronic Structure of Ions and Molecules in Solution: A View from Modern Soft X-ray Spectroscopies. Chem Soc Rev. 2013:6840–6859. doi: 10.1039/c3cs00008g. [DOI] [PubMed] [Google Scholar]
- 24.Schreck S, Gavrila G, Weniger C, Wernet P. A Sample Holder for Soft X-ray Absorption Spectroscopy of Liquids in Transmission Mode. Rev Sci Instrum. 2011;82:103101. doi: 10.1063/1.3644192. [DOI] [PubMed] [Google Scholar]
- 25.Schwartz CP, Uejio JS, Duffin AM, England AH, Kelly DN, Prendergast D, Saykally RJ. Investigation of Protein Conformation and Interactions with Salts via X-ray Absorption Spectroscopy. Proc Natl Acad Sci US A. 2010;107:14008–14013. doi: 10.1073/pnas.1006435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hellmann S, Rossnagel K, Marczynski-Buhlow M, Kipp L. Vacuum Space-Charge Effects in Solid-State Photoemission. Phys Rev B. 2009;79:035402. [Google Scholar]
- 27.Pietzsch A, Fohlisch A, Beye M, Deppe M, Hennies F, Nagasono M, Suljoti E, Wurth W, Gahl C, Dobrich K, et al. Towards Time Resolved Core Level Photoelectron Spectroscopy with Femtosecond X-ray Free-Electron Lasers. New J Phys. 2008;10:033004. [Google Scholar]
- 28.Kurian R, Kunnus K, Wernet P, Butorin SM, Glatzel P, de Groot FMF. Intrinsic Deviations in Fluorescence Yield Detected X-ray Absorption Spectroscopy: The Case of the Transition Metal L2, L3 Edges. J Phys : Condens Matter. 2012;24:452201. doi: 10.1088/0953-8984/24/45/452201. [DOI] [PubMed] [Google Scholar]
- 29.Regier TZ, Achkar AJ, Peak D, Tse JS, Hawthorn DG. Dark Channel Fluorescence Observations Result from Concentration Effects Rather Than Solvent-Solute Charge Transfer. Nat Chem. 2012;4:765–766. doi: 10.1038/nchem.1430. [DOI] [PubMed] [Google Scholar]
- 30.Eisebitt S, Boske T, Rubensson JE, Eberhardt W. Determination of Absorption-Coefficients for Concentrated Samples by Fluorescence Detection. Phys Rev B. 1993;47:14103–14109. doi: 10.1103/physrevb.47.14103. [DOI] [PubMed] [Google Scholar]
- 31.Achkar AJ, Regier TZ, Wadati H, Kim YJ, Zhang H, Hawthorn DG. Bulk Sensitive X-ray Absorption Spectroscopy Free of Self-Absorption Effects. Phys Rev B. 2011;83:081106(R). [Google Scholar]
- 32.Lange KM, Golnak R, Bonhommeau S, Aziz EF. Ligand Discrimination of Myoglobin in Solution: An Iron L-Edge X-ray Absorption Study of the Active Centre. Chem Commun. 2013;49:4163–4165. doi: 10.1039/c3cc37973f. [DOI] [PubMed] [Google Scholar]
- 33.Panzer D, Beck C, Hahn M, Maul J, Schoenhense G, Decker H, Aziz EF. Water Influences on the Copper Active Site in Hemocyanin. J Phys Chem Lett. 2010;1:1642–1647. [Google Scholar]
- 34.Bergmann N, Bonhommeau S, Lange KM, Greil SM, Eisebitt S, de Groot F, Chergui M, Aziz EF. On the Enzymatic Activity of Catalase: An Iron L-Edge X-ray Absorption Study of the Active Centre. Phys Chem Chem Phys. 2010;12:4827–4832. doi: 10.1039/b924245g. [DOI] [PubMed] [Google Scholar]
- 35.Aziz EF, Ottosson N, Bonhommeau S, Bergmann N, Eberhardt W, Chergui M. Probing the Electronic Structure of the Hemoglobin Active Center in Physiological Solutions. Phys Rev Lett. 2009;102:068103. doi: 10.1103/PhysRevLett.102.068103. [DOI] [PubMed] [Google Scholar]
- 36.Friedrich S, Hiller LJ, Frank M, le Grand JB, Mears CA, Niderost B, Labov SE, Barfknecht AT, LeGros M, Cramer SP. Superconducting High-Resolution X-ray Detectors for Metalloprotein L-Edge Spectroscopy. J Elec Spectr Rel Phenom. 1999;103:891–896. [Google Scholar]
- 37.Firsov A, Erko A, Senf F, Rehanek J, Brzhezinskaya M, Mitzner R, Wernet P, Foehlisch A. Novel Wavelength-Dispersive X-ray Fluorescence Spectrometer. J Phys Conf Ser. 2013;425:152013. [Google Scholar]
- 38.Kunnus K, Rajkovic I, Schreck S, Quevedo W, Eckert S, Beye M, Suljoti E, Weniger C, Kalus C, Gruebel S, et al. A Setup for Resonant Inelastic Soft X-ray Scattering on Liquids at Free Electron Laser Light Sources. Rev Sci Instrum. 2012;83:123109. doi: 10.1063/1.4772685. [DOI] [PubMed] [Google Scholar]
- 39.Schäfers F. The Bessy Raytrace Program Ray. In: Erko A, Krist T, Idir M, Michette A, editors. Modern Developments in X-ray and Neutron Optics. Springer; Berlin: 2008. pp. 9–39. [Google Scholar]
- 40.Sierra RG, Laksmono H, Kern J, Tran R, Hattne J, Alonso-Mori R, Lassalle-Kaiser B, Glockner C, Hellmich J, Schafer DW, et al. Nanoflow Electrospinning Serial Femtosecond Crystallography. Acta Crystallogr D. 2012;68:1584–1587. doi: 10.1107/S0907444912038152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert B, Frazer BH, Belz A, Conrad PG, Nealson KH, Haskel D, Lang JC, Srajer G, De Stasio G. Multiple Scattering Calculations of Bonding and X-ray Absorption Spectroscopy of Manganese Oxides. J Phys Chem A. 2003;107:2839–2847. [Google Scholar]
- 42.Heimann P, Krupin O, Schlotter WF, Turner J, Krzywinski J, Sorgenfrei F, Messerschmidt M, Bernstein D, Chalupsky J, Hajkova V, et al. Linac Coherent Light Source Soft X-ray Materials Science Instrument Optical Design and Monochromator Commissioning. Rev Sci Instrum. 2011;82:093104. doi: 10.1063/1.3633947. [DOI] [PubMed] [Google Scholar]
- 43.de Groot FMF, Arrio MA, Sainctavit P, Cartier C, Chen CT. Fluorescence Yield Detection - Why It Does Not Measure the X-ray-Absorption Cross-Section. Solid State Commun. 1994;92:991–995. [Google Scholar]
- 44.de Groot FMF. Dips and Peaks in Fluorescence Yield X-ray Absorption Are Due to State-Dependent Decay. Nat Chem. 2012;4:766–767. doi: 10.1038/nchem.1431. [DOI] [PubMed] [Google Scholar]
- 45.Wernet P, Kunnus K, Schreck S, Quevedo W, Kurian R, Techert S, de Groot FMF, Odelius M, Foehlisch A. Dissecting Local Atomic and Intermolecular Interactions of Transition-Metal Ions in Solution with Selective X-ray Spectroscopy. J Phys Chem Lett. 2012;3:3448–3453. doi: 10.1021/jz301486u. [DOI] [PubMed] [Google Scholar]
- 46.Schlotter WF, Turner JJ, Rowen M, Heimann P, Holmes M, Krupin O, Messerschmidt M, Moeller S, Krzywinski J, Soufli R, et al. The Soft X-ray Instrument for Materials Studies at the Linac Coherent Light Source X-ray Free-Electron Laser. Rev Sci Instrum. 2012;83:043107. doi: 10.1063/1.3698294. [DOI] [PubMed] [Google Scholar]
- 47.Beagley B, McAuliffe CA, Smith SPB, White EW. EXAFS Studies of Aqueous-Solutions of Manganese(II) Chloride and Bromide - Structural Variations with Concentration and Interactions with Solvent. J Phys : Condens Matter. 1991;3:7919–7930. [Google Scholar]
- 48.Waizumi K, Kouda T, Tanio A, Fukushima N, Ohtaki H. Structural Studies on Saturated Aqueous Solutions of Manganese(II), Cobalt(II), and Nickel(II) Chlorides by X-ray Diffraction. J Solution Chem. 1999;28:83–100. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.