

Abstract
Aims: The response of AMP-activated protein kinase (AMPK) to oxidative stress has been recently reported but the downstream signals of this response are largely unknown. Meanwhile, the upstream events for the activation of nuclear factor erythroid-2-related factor-2 (Nrf2), a critical transcriptional activator for antioxidative responses, remain unclear. In the present study, we investigated the relationship between AMPK and Nrf2 signal pathways in lipopolysaccharide (LPS)-triggered inflammatory system, in which berberine (BBR), a known AMPK activator, was used for inflammation suppression. Results and Innovation: In inflammatory macrophages, BBR attenuated LPS-induced expression of inflammatory genes (inducible nitric oxide synthase [iNOS], cyclooxygenase-2 [COX2], interleukin [IL]-6), and the generation of nitric oxide and reactive oxygen species, but increased the transcription of Nrf2-targeted antioxidative genes (NADPH quinone oxidoreductase-1 [NQO-1], heme oxygenase-1 [HO-1]), as well as the nuclear localization and phosphorylation of Nrf2 protein. Importantly, we found BBR-induced activation of Nrf2 is AMPK-dependent, as either pharmacologically or genetically inactivating AMPK blocked the activation of Nrf2. Consistent with in vitro experiments, BBR down-regulated the expression of proinflammatory genes but upregulated those of Nrf2-targeted genes in lungs of LPS-injected mice, and these effects were attenuated in Nrf2-deficient mice. Moreover, the effect of BBR on survival time extension and plasma redox regulation in endotoxin-shocked mice was largely weakened when Nrf2-depleted. Conclusions: Our results demonstrate convergence between AMPK and Nrf2 pathways and this intersection is essential for anti-inflammatory effect of BBR in LPS-stimulated macrophages and endotoxin-shocked mice. Uncovering this intersection is significant for understanding the relationship between energy homeostasis and antioxidative responses and may be beneficial for developing new therapeutic strategies against inflammatory diseases. Antioxid. Redox Signal. 20, 574–588.
Introduction
Inflammation is a complicated response that not only protects host organisms from external injuries and pathogens, but also contributes to the development of metabolic disorders and cancers (4, 43). Cellular events occurring during inflammatory responses are always associated with redox balance (10). As a major component of bacterial cell walls and a ligand of Toll-like receptor 4 (TLR4), lipopolysaccharide (LPS) dramatically increases the levels of reactive oxygen species (ROS) in various cell types, and results in proinflammatory responses (9). Correspondingly, specifically inhibiting the production of intracellular ROS is a general way to suppress intracellular proinflammatory signals (52). Therefore, the modulators for redox balance are taken for the key regulators of inflammatory responses, and the signal pathways with regard to ROS production and clearance have become major targets for inflammation research.
Innovation.
Our findings revealed that the functional intersection of AMP-activated protein kinase pathway and nuclear factor erythroid-2-related factor-2 pathway is necessary for the anti-inflammatory effect of berberine. Uncovering the connection of these two signal pathways sheds a light on the way to further explore the relationship between energy homeostasis and antioxidative response. A better understanding of such relationship is undoubtedly important in searching new therapeutic solutions for human diseases.
The signal pathways involved in inflammatory response are grouped into proinflammatory pathways and anti-inflammatory pathways. The former includes NF-κB pathway and the latter includes nuclear factor erythroid-2-related factor-2 (Nrf2) pathway (21, 45). NF-κB pathway promotes the production of ROS and proinflammatory cytokines, particularly from immunocytes, such as macrophages (44). In contrast, Nrf2 pathway acts protectively against inflammation by activating antioxidant cascades (13). As a basic-leucine zipper transcription factor, Nrf2 has been shown to regulate gene expression by binding to the antioxidant responsive element (ARE). The target genes of Nrf2 include NADPH quinone oxidoreductase-1 (NQO-1), heme oxygenase-1 (HO-1) and glutathione S-transferase (GST) (47). It is generally believed that the activation of Nrf2 signaling is an adaptive response to the environmental and endogenous stresses (12). The transcriptional activity of Nrf2 protein is suppressed by association with Kelch-like ECH-associated protein 1 (Keap1) under homeostatic conditions, but is activated when cells are exposed to oxidative or electrophilic stress, which results in the dissociation of Nrf2 from Keap1, the nucleus translocation of Nrf2 and its binding to ARE on the promoter region of a group of genes that act to combat oxidative stress (47). In addition, some studies have demonstrated that protein phosphorylation is a potential mechanism for activating Nrf2-ARE mediated pathways (35). To date, several cytosolic kinases, including protein kinase C (17), phosphatidylinositol 3-kinase (23), and mitogen-activated protein kinase (57), have been shown to modify Nrf2 and to potentially play a role in Nrf2-mediated signal transduction at AREs.
The involvement of cellular energy metabolism in inflammation suppression has recently become an area of intense interest, although the exact mechanisms involved are still poorly characterized (54, 55). As a sensor of intracellular energy status, AMP-activated protein kinase (AMPK) is an attractive target for inflammation control. Indeed, there is emerging evidence showing that AMPK activation can decrease the oxidative stress and inhibit inflammation (16, 25, 38). However, mechanistic connections between AMPK and inflammation have been limited to links with NF-κB pathway (40). It has been shown that chemical activators of AMPK decreased NF-κB-mediated transcription (5, 14), and that constitutively active AMPK suppressed NF-κB signaling and fatty acid-induced inflammation in macrophages (54), although NF-κB subunits are not the direct targets of AMPK (40). Therefore, expanding the investigation to signaling axes other than NF-κB pathway is necessary. Consistent with the unequivocal action of Nrf2-pathway on ROS clearance and the suppression of inflammation, the potential for the crosstalk between Nrf2 and AMPK pathways has been noted. The relationships between AMPK and Nrf2 cascades in Caenorhabditis elegans and in human endothelial cells have been reported (31, 37). However, no information about the potential for convergence between AMPK and Nrf2 pathways in the mammalian inflammatory system exists.
The suppressive role of berberine (BBR) in bacteria-induced inflammation has been recognized for years (11). Even so its beneficial effects on metabolic disorders were reported only recently (28). BBR suppresses the expression of proinflammatory cytokines likely due to its capacity of AMPK activation (19). BBR-induced activation of AMPK is related to an increase of AMP/ATP ratio (49). Similar to other chemical activators of AMPK, such as 5-aminoimidazole-4-carboxamide ribonucleoside (AICAR) and metformin, BBR has been increasingly used for studies for AMPK signaling (29, 49). The impact of BBR on Nrf2 pathway was recently reported in motor neuron-like cells and in astrocytes (6, 15).
To investigate the potential for a functional interaction between Nrf2 and AMPK pathways in an inflammatory system, we examined the effect of BBR on inflammatory stress by utilizing LPS-stimulated macrophages and LPS-shocked mice in the present study. For clarifying the interactions and temporal relationships between these two pathways, we conducted series of blocking experiments, via both chemical and genetic strategies in vitro. In vivo, the effects of BBR in wild-type mice and Nrf2 deficient mice were compared. Our results demonstrate the importance of Nrf2 pathway for the anti-inflammatory role of BBR, and provide the evidence showing that AMPK pathway is connected with Nrf2 pathway.
Results
Anti-inflammatory role of BBR in macrophages in vitro
BBR prevented LPS-induced production of inflammatory cytokines and NF-κB activation
Since the elevation of inflammatory gene expression is a well-known response of macrophages to LPS stimulation (2), we looked at the effect of BBR on the expression of representative inflammatory genes. Similar to previous studies (26), LPS upreglated the transcription of proinflammatory genes (inducible nitric oxide synthase [iNOS], cyclooxygenase-2 [COX-2] and interleukin [IL]-6) in RAW264.7 cells, primary macrophages, as well as phorbol 12-myristate 13-acetate (PMA)-differentiated THP-1 cell. As the result, nontoxic dosages of BBR (5 and 10 μM) significantly suppressed LPS-induced expression of these genes (Fig. 1A–C). Similar changes in tumor necrosis factor-α (TNF-α) and IL-1β protein abundance were detected in RAW264.7 cells and primary macrophages (Fig. 1D, E). Given that the expression of inflammatory cytokines is generally NF-κB dependent (53), we checked the phosphorylation status of NF-κB in RAW264.7 cells. As shown in Figure 1F, LPS robustly increased the phosphorylation of NF-κB, while BBR significantly suppressed LPS-induced NF-κB phosphorylation. These results showed that BBR can inhibit LPS-induced, NF-κB-dependent inflammatory cytokine production in both murine and human macrophages.
FIG. 1.

BBR reduced the expression of proinflammatory genes and NF-κB activation in LPS-stimulated macrophages. RAW264.7 cells (A), peritoneal macrophages (B) and PMA-differentiated THP-1 cells (C) plated in six-well cell culture plates were preincubated with BBR (5 μM and 10 μM) for 2 h, and then stimulated by LPS (10 ng/ml) for 24 h. The expression of proinflammatory genes (iNOS, COX2 and IL-6) was checked by quantitative real-time (qRT)-PCR. To assess the expression of IL-1β and TNF-α at protein level, RAW264.7 cells (D) and mouse peritoneal macrophages (E) were treated as described above and culture supernatants were collected followed by ELISA assay. In addition, total protein was extracted from RAW264.7 cells and used for Western-blot analysis against p-NF-κB and NF-κB (F). Data were presented as mean±SEM of three separate experiments. *p<0.05 and **p<0.01, compared with LPS group. BBR, berberine; COX2, cyclooxygenase-2; ELISA, enzyme-linked immunosorbent assay; iNOS, inducible nitric oxide synthase; IL, interleukin; LPS, lipopolysaccharide; PMA, phorbol 12-myristate 13-acetate; qRT-PCR, quantitative RT-polymerase chain reaction; TNF-α, tumor necrosis factor-α.
BBR inhibited inflammatory reactions in LPS-stimulated macrophages
To confirm the anti-inflammatory role of BBR in LPS-stimulated macrophages, nitric oxide (NO) and ROS productions were detected, since a burst of production of these free radicals is known to accompany the inflammatory response (30). In RAW264.7 cells, LPS caused a considerable release of NO, but BBR alone did not. However, BBR treatment markedly inhibited the NO production elicited by LPS (Fig. 2A). As shown in Figure 2B and C, the impact of BBR on ROS production is similar to NO release, that is, BBR alone did not affect ROS production in the cell population but greatly reduced the LPS-induced ROS production. These results indicate that BBR can inhibit LPS-induced inflammatory response in macrophages.
FIG. 2.
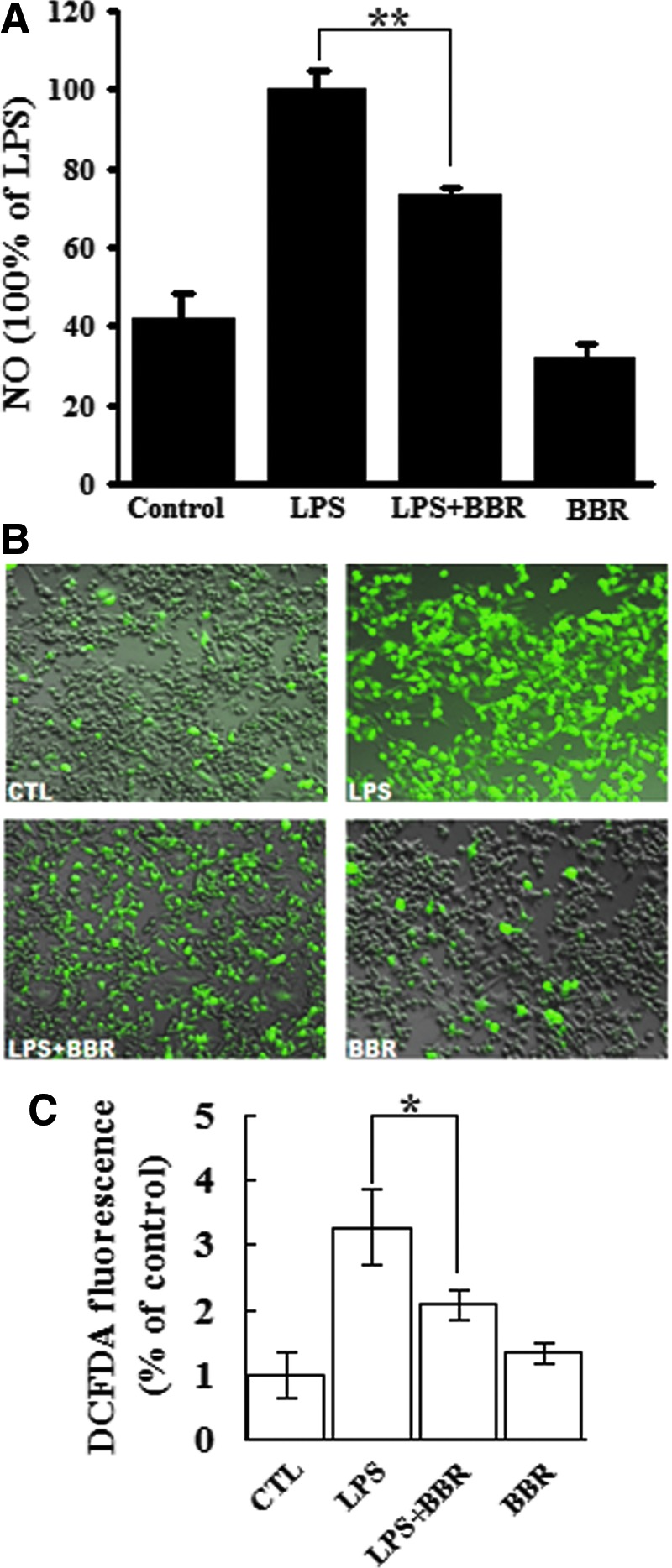
BBR inhibited NO release and ROS production in LPS-stimulated macrophages. (A) RAW264.7 cells were treated with LPS (10 ng/ml), BBR (5 μM, 1 h earlier when used in combination) or both for 24 h. NO concentration in culture supernatant was measured and expressed as percentage values, taking the LPS treatment group as 100%. (B) After treatment with LPS or/and BBR, cells were washed and labeled with H2-DCFH-DA probe. A set of representative images showing fluorescent ROS was shown. (C) Quantitative measurement of cellular ROS was performed by flow cytometry after H2-DCFH-DA staining. Similar results were obtained from three independent experiments. *p<0.05 and **p<0.01. DCFH-DA, 2′, 7′-dichlorodihydrofluorescein diacetate; ROS, reactive oxygen species. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
BBR activated anti-inflammatory reactions in LPS-stimulated macrophages
Mitochondria are the main organelles targeted by intracellular free radicals (39); therefore, protecting mitochondria from those radicals is an important aspect of the anti-oxidative and anti-inflammatory responses. To address the effect of BBR on mitochondria protection, we first assayed mitochondria membrane potential (MMP) using a fluorescent mitochondrial dye, JC-1. As shown in Fig. 3A and B, MMP was greatly degraded by LPS, showing obvious green fluorescence, while BBR effectively restored the MMP. Next, we measured the activity of GST, a major antioxidant enzymes located in mitochondria. As shown in Fig. 3C, BBR addition increased GST activity in LPS-treated cells, but BBR alone had no this effect. In addition, we examined particle-engulfing activity of macrophages as it plays an essential role for the defensive function of macrophages (42). The results showed that there were more engulfed neutral red particles in LPS-treated cells when BBR was present (Fig. 3D, E). Together, these results suggest that BBR activated the anti-inflammatory response in LPS-stimulated macrophages.
FIG. 3.

BBR activated anti-inflammatory and antioxidant reactions in LPS-stimulated macrophages. Raw267.4 cells were treated as described in Fig 2A. (A and B), cell MMP was monitored by JC-1 method. Fluorescence was recorded by fluorescence microscopy (A) and flow cytometry (B), respectively. (C) Cellular GST activity was measured as described in Materials and Methods. (D and E), Pinocytosis capacity of macrophages was evaluated by neutral red assay. Representative images were recorded by light microscopy (D), whole lysates from neutral red stained cells were measured at 570 nm with a Spectra Reader (E). Data were represented as mean±SEM of three independent determinations. *p<0.05 and **p<0.01. GST, glutathione S-transferase; MMP, mitochondria membrane potential. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The involvement of Nrf2 signaling in the effects of BBR in LPS-stimulated macrophages
BBR elevated the expression Nrf2 downstream genes in inflammatory macrophages
Antioxidation is important for the anti-inflammatory response, while Nrf2-modulated signaling is vital for the antioxidative response (23). To see if the anti-inflammatory role of BBR is Nrf2 signaling relevant, we measured the expression of two Nrf2 target genes, NQO-1 and HO-1. As shown in Fig. 4A, LPS or BBR alone had almost no effect on the transcription of the NQO-1 and HO-1 genes in RAW264.7 cells, but BBR significantly activated the transcription of these two genes in the presence of LPS. The similar phenomenon induced by BBR can also be observed in peritoneal macrophages (Fig. 4B). These results, together with those shown in Fig. 3C (GST activity) indicate that BBR activated Nrf2-mediated antioxidative cascades in LPS-stimulated macrophages.
FIG. 4.

BBR induced Nrf2 activation in LPS-stimulated macrophages. Cells were treated as described in Fig. 2A. (A and B) Relative mRNA levels of NQO-1 and HO-1 were analyzed by qRT-PCR in RAW 264.7 cells (A) and in peritoneal macrophages (B). (C) In RAW264.7 cells transfected with pGFP-Nrf2 and pKeap1, subcellular localization of GFP-Nrf2 fusion protein in living cells was monitored under a fluorescent microscope after 24 h of transfection. (D) More than 100 GFP-positive cells were randomly selected and the percentage of cells with GFP in nucleus was expressed. (E) Cytoplasmic and nuclear fractions of RAW 264.7 cells were extracted and endogenious Nrf2 protein was probed with specific antibody via Western-blot assay. β-actin and H3 were probed as the controls for cytoplasm fraction and nucleus fraction, respectively. (F) Phosphorylated Nrf2 at Ser40 (p-Nrf2) and total Nrf2 in the whole cell lysates of RAW264.7 cells were probed by specific antibodies via Western-blot assay. Three separate experiments showed similar consequences, and one set of representative results are illustrated. *p<0.05 and **p<0.01, compared with control group in D. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
BBR induced nuclear translocation and phosphorylation of Nrf2 in inflammatory macrophages
Nrf2 translocation from the cytoplasm to the nucleus is an indispensable step for Nrf2 activation (47). The effect of BBR on Nrf2 translocation was therefore, investigated. We examined the distribution of fluorescent GFP-tagged Nrf2 (pGFP-Nrf2) and the presence of endogenous Nrf2 protein in cellular fractions. As shown in Fig. 4C, although LPS or BBR alone slightly induced the nuclear translocation of GFP-Nrf2, BBR significantly promoted the nuclear translocation of GFP-Nrf2 in LPS treated cells. The percentage of cells with nuclear GFP-Nrf2 in control group, LPS alone, BBR alone and LPS plus BBR samples were 8%, 10%, 15.6%, and 87.3%, respectively (Fig. 4D). The nuclear distribution of Nrf2 protein were accordingly displayed in Western-blot assay (Fig. 4E), which shows that the Nrf2 protein in cytoplasmic fraction were reduced by BBR alone or together with LPS when compared to control. In contrary, the Nrf2 protein in nuclear fraction was increased by BBR alone or together with LPS. The phosphorylation of Nrf2 was also investigated in parallel to above experiments, as it has been reported to play a critical role in the dissociation of Nrf2 from Keap1 and nuclear distribution of Nrf2 protein (17). As shown in Fig. 4F, compared to slightly increased Nrf2 phosphorylation in BBR alone or LPS treated cells, a significant level of Nrf2 phosphorylation occurred in the cells treated with BBR together with LPS. Above results together demonstrate that BBR promoted nuclear translocation and phosphorylation of Nrf2 protein in LPS-treated RAW264.7 cells.
Blocking Nrf2 signaling attenuated the protective effects of BBR on inflammatory macrophages
To ascertain whether BBR could activate Nrf2 signaling, blocking experiments were conducted. First, NQO-1 inhibitor dicoumarol (Dico) was used as the chemical inhibitor of Nrf2-mediated antioxidative cascades. Consistent with the results shown in Figure 1, BBR suppressed LPS-induced expression of proinflammatory genes to a great degree; however, this suppression was significantly abrogated when the cells were pretreated with Dico (Fig. 5A). Meanwhile, we measured the effect of Dico on BBR-suppressed ROS production. As shown in Figure 5B and C, pretreatment of Dico partly blocked the suppressive effect of BBR on ROS production.
FIG. 5.

Blockage of Nrf2 signaling abolished suppressive effects of BBR on inflammation and oxidative stress in RAW 264.7 cells. (A) Cells were preincubated with or without dicoumarol (Dico, 20 μM) and BBR (5 μM), then treated with LPS (10 ng/ml) for 24 h. mRNA levels of indicated genes were monitored by qRT-PCR. (B and C) Cells were treated as described in A. Intracellular ROS was labeled by H2-DCFH-DA probe. ROS level was assessed by fluorescence microscopy (B) and flow cytometry (C). (D) Cells with nonactivity Nrf2 expression vector (pDN-Nrf2) transfection were treated with BBR and LPS as described in A. mRNA levels of indicated genes were analyzed by qRT-PCR (D). *p<0.05 and **p<0.01. NS, no statistical difference. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Next, we engaged a genetic inactivation of Nrf2 gene to further ascertain the dependency of BBR on Nrf2 signaling. To this goal, RAW 264.7 cells were transfected with the expression construct for a dominant negative mutant of the Nrf2 gene (pDN-Nrf2) (36). Compared to empty vector, pDN-Nrf2 significantly reduced the suppressive effects of BBR on LPS-induced expression of proinflammatory gene (COX2), and impaired BBR-elevated expression of NQO-1 and HO-1 genes in LPS-stimulated macrophages (Fig. 5D). These results from blocking experiments would hence, support the notion that BBR exhibits its anti-inflammatory effect, at least in part, through an Nrf2-dependent manner.
The involvement of AMPK signaling in the effects of BBR in LPS-stimulated macrophages
BBR stimulated AMPK activation in inflammatory macrophages
BBR is capable of stimulating AMPK in macrophages (19). This capacity was confirmed in our experimental system. The phosphorylation of AMPKα1 subunit, which generally linked with AMPK activation, was first examined. As shown in Fig. 6A, both BBR alone and BBR together with LPS significantly enhanced the phosphorylation of AMPKα1 in RAW264.7 cells (lane 3 and lane 5). Moreover, pretreatment of cells with the AMPK inhibitor, compound C (CC), evidently prevented BBR-induced AMPK phosphorylation (lane 4 and lane 6). Given the AMP-dependency of AMPK activation (48), AMP-to-ATP ratio in cells was then measured. The results showed that BBR alone or together with LPS markedly elevated the AMP-to-ATP ratio in RAW 264.7 cells, whereas CC reduced the AMP-to-ATP ratio. These results show that BBR can activate AMPK in macrophages regardless of cellular inflammatory status.
FIG. 6.

Blockage of AMPK signaling abolished suppressive effects of BBR on inflammation and oxidative stress in RAW 264.7 cells. (A) Cells were preincubated with or without compound C (CC; 2 μM) and BBR (5 μM), then treated with LPS (10 ng/ml). Whole cell lysates were subjected to Western-blot analysis with antibodies against phosphoylate-AMPKα1 at Thr172 (p-AMPKα1) and total AMPKα1. (B) After treatments as in A, cellular concentrations of AMP and ATP were determined through high performance liquid chromatography. (C) After treatments as in A, mRNA levels of indicated genes were monitored by qRT-PCR. Results are means±SEM from three independent experiments. **p<0.01.
Blocking AMPK signaling attenuated the protective effects of BBR on inflammatory macrophages
To determine if BBR-induced activation of AMPK is important for the anti-inflammatory and antioxidative effects of BBR, we examined the effect of AMPK inhibitor CC on the transcriptional regulation of proinflammatory genes, as well as antioxidative genes. As illustrated in Figure 6C, BBR clearly suppressed LPS-induced expression of the COX2 gene, but enhanced the expression of the NQO-1 and HO-1 genes. However, in the presence of CC, the effect of BBR on the expression of these genes was abrogated; indicating AMPK signaling plays an important role in anti-inflammatory effect of BBR in our system.
The crosstalk between AMPK and Nrf2 pathways functions in the anti-inflammatory effect of BBR
Blockage of AMPK signaling impaired the effect of BBR on Nrf2 activation
Although the results presented above demonstrated that BBR can activate both AMPK and Nrf2, little was known about the functional linkage between AMPK and Nrf2 signalings. To investigate this potential linkage, we first conducted a set of blocking experiments, in which Nrf2 activation was measured after AMPK was inhibited. As shown in Fig. 7A and B, when AMPK activity was inhibited by CC, BBR-induced nuclear translocation of GFP-Nrf2 in LPS-treated RAW264.7 cells was significantly reduced. In addition to use chemical inhibitor of AMPK, a genetic approach to address the AMPK involvement in BBR-induced Nrf2 activation was also employed, in which exogenous dominant negative form of AMPKα1 (DN-AMPKα1) was expressed in RAW246.7 cells (19). As shown in Figure 7C, comparing to the empty vector transfection, overexpression of DN-AMPKα1 abolished BBR-induced upregulation of NQO-1 and HO-1 genes. Consistently, DN-AMPKα1 transfection abolished the promoting effect on Nrf2 nuclear translocation (Fig. 7D). These results, based on AMPK blockage by either CC administration or dysfunctional AMPKα1 overexpression, suggest that BBR-induced Nrf2 activation was largely dependent on AMPK activity in LPS-treated macrophages.
FIG. 7.

Inactivation of AMPK relieved effect of BBR on Nrf2 activation in LPS-induced macrophages, while inactivation of Nrf2 did not affect BBR-triggered activation of AMPK. (A) RAW264.7 cells with pGFP-Nrf2 transfection were treated as described in Fig. 6. Subcellular localization of GFP-Nrf2 protein in living cells was monitored under a fluorescent microscope. (B) Quantitative analysis of GFP-Nrf2 nuclear distribution in transfected cells as described in Figure 4D. (C and D) RAW264.7 cells with nonactivity AMPKα1 (pDN-AMPKα1) transfection were treated as in A, mRNA abundance of Nrf2 target genes (C) and Nrf2 protein distribution in nucleus (D) were analyzed by qRT-PCR and Western blot, respectively. (E) RAW264.7 cells transfected with pDN-AMPKα1 (upper panel) or pDN-Nrf2 (lower panel) were treated by LPS and BBR, followed by the mRNA abundance of an AMPK target gene, CPT-1, was measured by qRT-PCR. Results are means±SEM from three independent experiments. **p<0.01. (F) Peritoneal macrophages from wild type mice (Nrf2+/+) or Nrf2 deficient mice (Nrf2−/−) were treated in vitro by LPS and BBR, phosphorylated-AMPKα1 (Thr172) and total AMPKα1 abundance in whole cell lysates were probed by specifical antibodies through Western blot. Representative results from three independent experiments are shown. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Blockage of Nrf2 signaling had no impact on BBR -induced AMPK activation
To determine the upstream-downstream relationship between AMPK and Nrf2, we tested the effect of Nrf2 blocking on AMPK activation. In this experiment, the transcription of the carnitine palmitoyl transferase 1 (CPT-1) gene was tested because it is a classic downstream gene of AMPK (58). The overexpression of DN-AMPKα1, but not of DN-Nrf2, effectively attenuated BBR-induced CPT-1 expression (Fig. 7E). The inefficiency of Nrf2 depletion in AMPK signaling activation was further confirmed by comparing the effect of Nrf2 on the phosphorylation of AMPK in peritoneal macrophages from both wild-type (Nrf2+/+) mice and Nrf2 deficient mice (Nrf2−/−). As shown in Figure 7F, BBR together with LPS promoted the phosphorylation of AMPK in both Nrf2+/+ mice and Nrf2−/− mice, meaning that Nrf2 depletion is not important for BBR-induced AMPK activation. Since Nrf2 blocking showed no impact on AMPK activation (Fig. 7E, F), while AMPK blocking attenuated Nrf2 activation (Fig. 7A–D), AMPK appears to be working upstream of Nrf2 in BBR-treated inflammatory macrophages.
Nrf2-dependent anti-inflammatory role of BBR in mice
Nrf2-dependency of the anti-inflammatory role of BBR was further assessed in a systemic acute inflammation mice model established by intraperitoneal (i.p.) injection of LPS, that is, endotoxin shock.
The effect of BBR on mice survival and Nrf2-dependency
Firstly, the survival time of mice after received LPS injection was measured. As shown in Fig. 8A, for Nrf2+/+ mice, the median survival times in control group and in the BBR-treated group were 36 and 84 h, respectively, showing significantly different. However, for Nrf2−/− mice, the difference of median survival time in control group and in BBR-treated group was much smaller, being 42 and 36 h, respectively. Secondly, the mortality at 60 h after LPS injection was measured. The results showed that the mortality of Nrf2+/+ mice was 62% in the control group but dropped to 25% in the BBR-treated group, whereas for Nrf2−/− mice, the mortality was 67% in the control group and 75% in the BBR-treated group. These results showed that Nrf2 depletion obliterated BBR-extended survival in LPS-shocked mice.
FIG. 8.

Suppressive effects of BBR on LPS-induced endotoxin shock and lung injury is Nrf2 dependent in mice. (A) Effect of BBR on the survival rate of Nrf2+/+ or Nrf2−/− mice. Mice were intraperitoneally injected with BBR (10 mg·kg−1) and/or LPS (35 mg·kg−1) as described in Materials and Methods (n=6). The survival test results are presented as Kaplan-Meier survival curves. (B) Effect of BBR on redox potential values (Eh) for reduced and oxidized GSH/GSSG in plasma. Mice were treated with BBR (10 mg·kg−1) and/or LPS (15 mg·kg−1) for 6 h (n=6). Eh values of plasma were calculated from GSH and GSSG concentrations using Nernst equation. A more negative Eh value corresponded to a more reduced redox state; likewise, the more positive Eh value, the more oxidized redox state. (C) Effects of BBR on the transcription of genes in mice. Mice were treated as in (B), and relative mRNA levels of indicated genes in lung tissue (lower lobe of right lungs) were analyzed by qRT-PCR. (D) Effects of BBR on inflammatory lung injury in mice. Mice were treated as in (B). Afterwards, upper lobe of right lungs was collected for HE staining, and representative images were displayed. Results are means±SEM from three independent experiments. *p<0.05 and **p<0.01. NS, no statistical difference. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The effect of BBR on blood redox potential and hemogram, and their Nrf2-dependency
Several studies have shown that endotoxin shock is associated with increased oxidative stress and depletion of plasma GSH (27, 50). Consistently, our study showed that redox potential value (Eh) was higher in the plasma from LPS-shocked mice, indicating that more oxidized GSH/GSSG exists (Fig. 8B). Additionally, we observed that Eh in LPS-treated mice was significantly reduced by BBR treatment in Nrf2+/+ mice, but not in Nrf2−/− mice (Fig. 8B).
Population distribution of white blood cells (WBCs) is another informative index upon systemic inflammation. As shown in Table 1, although LPS administration significantly increased the total counts of WBCs and of neutrophils in either Nrf2+/+ mice or Nrf2−/− mice, BBR treatment only reversed LPS-induced hemogram alterations in Nrf2+/+ mice. The total WBCs and neutrophils counts decreased remarkably in BBR treated Nrf2+/+ mice, but not in Nrf2−/− mice. All these results, from blood sample, suggest that Nrf2 signaling is significant for BBR in its protection role played in LPS-shocked mice.
Table 1.
Total Counts and Population Counts of White Blood Cells
| Mice type | Group | WBC (X109/L) | Neutrophil (%) | Lymphocyte (%) |
|---|---|---|---|---|
| Nrf2+/+ | Control | 7.98±0.72 | 24.00±6.01 | 74.33±6.51 |
| LPS | 14.07±1.45a | 66.67±3.51a | 30.67±3.05a | |
| LPS+BBR | 11.02±1.15b | 52.33±6.65b | 45.67±8.14b | |
| Nrf2−/− | Control | 3.00±0.47 | 38.33±4.04 | 55.67±5.03 |
| LPS | 8.63±0.23a | 64.67±4.73a | 30.33±3.06a | |
| LPS+BBR | 8.19±0.35 | 70.33±4.04 | 25.33±3.51 |
p<0.01 versus control group.
p<0.05 versus LPS-treated groups.
BBR, berberine; LPS, lipopolysaccharide; WBC, white blood cells.
The effect of BBR on LPS-induced lung injury and Nrf2 dependency
The inflammatory status of lungs was inspected next, since lung injury is remarkable in LPS-shocked mice. As shown in Figure 8C, BBR markedly upregulated transcriptional expression of Nrf2-target genes (NQO-1 and HO-1) in lung tissues of Nrf2+/+ mice (upper panel), but significantly down-regulated the transcription of proinflammatory genes (COX2 and iNOS) in the same tissue (lower panel). Importantly, both BBR-induced upregulation of Nrf2-target genes and down-regulation of proinflammatory genes were significantly abrogated in Nrf2−/− mice.
As shown in Figure 8D, histologic examination of the lungs from LPS-shocked Nrf2+/+ mice revealed a considerable amount of alveolar hemorrhage and infiltration of inflammatory cells, which are some typical physiological phenomenon of lung inflammation, are present. On the other hand, these inflammatory signs were significant reduced by BBR treatment. However, in lung tissue from Nrf2−/− mice, BBR on contrast did not reduce these LPS induced inflammatory signs.
Taken together, these results indicate that BBR can work as an efficient anti-inflammatory agent in endotoxemic mice in at least partly, Nrf2-dependent manner.
Discussion
In the present study, we found that the anti-inflammatory role of BBR is primarily involved in the activation of Nrf2 pathway and this activation of Nrf2 pathway is markedly AMPK-dependent in LPS-stimulated inflammatory responses. We also demonstrated the Nrf2-dependency of the suppressive effect of BBR on inflammation in vivo. Uncovering the functional relationship between AMPK and Nrf2 pathways is of significance because it reveals a novel link between energy homeostasis and inflammation suppression. This finding may substantially contribute to the development of new therapeutic approaches for inflammatory diseases. A schematic illustration for the relevant cascades is shown in Figure 9.
FIG. 9.

Schematic overview of the crosstalk between AMPK and Nrf2 pathways induced by BBR in LPS-stimulated inflammation. It was known that LPS activates NF-κB pathway and prompts inflammation through TLR4, and Nrf2 activation is crucial for antioxidative and anti-inflammation reaction. One the other hand, comparing to its homeostatic effect on energy metabolism, the anti-inflammatory role of AMPK is a lately finding with mechanistic unknowns. Based on the present study, we propose a cascade-flow initiated by berberine in LPS-treated macrophages, which starts from AMP/ATP ratio elevation and AMPK activation, but goes to the phosphorylation and nuclear distribution of Nrf2 protein, the up-regulation of Nrf2-targeted antioxidative genes, the suppression of NF-κB-mediated inflammatory response, the reduced ROS production and even alleviated in vivo inflammation, sequentially. Thus, the interaction between AMPK and Nrf2 pathways plays an important role in inflammation suppression. ↓, activation; ⊥, inhibition.
Comparing to proinflammatory signaling cascades, we know less about anti-inflammatory signal pathways (8, 34), particularly about the network around Nrf2-mediated antioxidative pathway. In addition, as a new but probably central pathway for inflammation suppression, the downstream partner of AMPK is also undefined. Given this situation and the promising importance of the relationship between energy metabolism and redox equilibrium, we hence, planned this line of study, by asking specific questions: Is there a functional link between Nrf2 pathway and AMPK pathway? If there is, how do they work together for inflammation suppression? The work presented in this report essentially addresses the first question.
Our study identified the AMPK-Nrf2 association in the TLR-mediated inflammatory circumstance, a common status actually present in both infectious and noninfectious diseases. It has previously reported that TLR-mediated proinflammatory pathways, particularly NF-κB signaling, are correlated with the inflammation inhibitory role of AMPK (40). In our study, BBR-treatment not only suppressed the expression of NF-κB-targeted cytokines and the phosphorylation of NF-κB (a sign of NF-κB activation, Fig. 1), but also elevated the expression of Nrf2-targeted genes and increased the nuclear translocation and phosphorylation of Nrf2 (signs of Nrf2 activation, Fig. 4). More significantly, the anti-inflammatory role of BBR was clearly attenuated in Nrf2 inactivated macrophages (Fig. 5) and in Nrf2 deficient mice (Fig. 8). Together with previous reports, our results support the new notion that is, the inhibitory role of AMPK in inflammation is based on, at least partially, a mechanism underlying the suppression of TLR-mediated proinflammatory cascades on one hand, and of the activation of Nrf2-mediated antioxidative cascades on the other hand. With regard to the connection between AMPK and Nrf2 signaling, some results from previous studies should be mentioned here. For example, Hsu et al. (15) found that BBR promoted Nrf2 nuclear translocation in a motor neuron-like cell line, and Chen et al. (6) reported that BBR induced transcriptional upregulation of HO-1 gene in rat primary astrocytes. A few other reports simply mentioned the role of the AMPK activator in LPS-induced endotoxemic mice (24, 38). We noticed, however, that these previous studies have not successfully establish associations between AMPK activation, inflammation protection and Nrf2 signaling, because they either examined in a noninflammation circumstance when inspecting the association between AMPK and Nrf2 signals, or did not determine the Nrf2-dependency of AMPK-mediated inflammation protection. In other words, the present study provides novel evidence important for establishing the connection between of AMPK activation, inflammation protection and Nrf2 signaling.
In previous studies, upregulated expression of the HO-1 gene is mostly used as evidence for Nrf2 signaling activation (1). However, other information suggests that this gene can be targeted by upstream signals other than Nrf2, such as PPARγ (51) or hypoxia-inducible factor-1α (HIF-1α) (56). To determine the role of BBR was Nrf2-dependent, we provided more evidence from different angles. At first, in addition to the HO-1 gene, the NQO-1 gene was analyzed for its expression, because it is a specific target gene of Nrf2 (46). Then, Nrf2 signaling block experiments were conducted by utilizing chemical inhibitor Dico and the expression of an Nrf2 mutant protein. Finally, the assays based on in vivo experiments were conducted, including the use of Nrf2 deficient mice. Our results from both in vitro and in vivo experiments coherently demonstrate that the anti-inflammatory role of BBR in LPS-treated macrophages and mice is Nrf2-dependent and Nrf2 pathway-relevant.
Although it is known that BBR is an activator of AMPK pathway, there has been no evidence on the relationship between AMPK and Nrf2 when triggered by BBR in its anti-inflammatory role. To this end, different approaches were considered. First, BBR-induced activation of AMPK was shown by checking the phosphorylation level of AMPKα1 (Fig. 6). Second, AMPK-dependency of Nrf2 activation was revealed by the pretreatment of cells with AMPK specific inhibitor CC, and by the transfection of an AMPK mutant gene construct into cells (19, 33). Furthermore, two sets of signal blocking experiments, against Nrf2 or AMPK, were directly compared. The relationship, in terms of up- and downstream, between Nrf2 and AMPK became clear, that is, AMPK acts upstream of Nrf2. The third approach was to determine whether other AMPK activators have a role similar to BBR in respect of activating Nrf2. We therefore, tested metformin and AICAR, two other known AMPK activators, and similar results were obtained (data not shown). Coincident with our results, the involvement of metformin and AICAR in the antioxidative response has been reported before. For example, metformin was shown to have a role in activating the conserved oxidative stress-responsive transcription factor SKN-1/Nrf2 in C. elegans (37), and AICAR stimulated HO-1 expression in human ECs via Nrf2/ARE signaling pathway (31).
It is clear there is a functional association between AMPK and Nrf2 pathways in our system. However, the details about this interaction have yet fully characterized, and we still do not know whether Nrf2 protein is a direct molecular target of AMPK. In our view, to identify the protein(s) that play a critical role for the interaction is indispensable, and to expand the experimental system to other inflammatory models that more close to metabolic disorder is urgently needed. Nevertheless, the present work has made a significant step forward in exploring the crosstalk between energy metabolism and oxidative homeostasis in macrophages and made an entry point for working in organs and even in whole bodies. We of course realized that AMPK-Nrf2 crosstalk may not be the only way that is responsible for Nrf2 activation, and other mechanisms may be involved in other situations. For example, Itoh et al. reported COX-2-mediated intracellular accumulation of 15d-PGJ2, which induced Nrf2 activation and consequently in turn regulates the expression antioxidative stress enzymes in inflammatory macrophages (18).
In conclusion, our results for the first time demonstrate that BBR coupled AMPK pathway together with Nrf2 pathway in both LPS-shocked macrophages and mice, and that AMPK works upstream of Nrf2. This finding provides an innovative platform to further explore the mechanism underlying inflammation suppression and the crosstalk between energy homeostasis and oxidative clearance. Understanding and rationally utilizing this functional relationship should be helpful to establish effective therapies for inflammatory diseases.
Materials and Methods
Reagents
BBR was obtained from CDMUST Biotech and dissolved in phosphate-buffered saline (PBS). LPS (E.coli, serotype 055:B5), phorbol 12-myristate 13-acetate (PMA) and H2-2′, 7′-dichlorodihydrofluorescein diacetate (DCFH-DA) were purchased from Sigma. Dico and CC was purchased from Calbiochem. Anti-Nrf2, Anti-actin and Anti-H3 antibodies were from Santa Cruz Biotechnologies. Phospho-NF-κB p65 (Ser536), NF-κB p65, phospho-AMPKα (Thr172) and AMPK antibodies were purchased from Cell Signaling Technology. Anti-phospho-Nrf2 (Ser40) antibody was from Bioss Biotechnologies.
Cells
RAW264.7 cells (mouse macrophages line) were cultured in RPMI-1640 medium supplemented with 10% FCS. THP-1 cells (human monocyte line) were maintained in RPMI 1640 medium supplemented with 10% FBS and 2 mM L-glutamine. The differentiation of THP-1 monocytes toward macrophages was induced by 10 ng/ml PMA treatment for 72 h. Differentiated macrophage-like THP-1 cells were then washed twice and cultured in fresh medium before used in following experiments. For primary macrophage preparation, mice were stimulated firstly by a single i.p. injection of 4% thioglycollate solution (1 ml per mouse) after 12 h fasting; peritoneal macrophages were harvested 4 days later by washing peritoneal cavity with PBS (5 ml per mouse), followed by seeding in RPMI-1640 medium with 10% FCS. Nonadherent cells were removed 3 h after seeding with medium change. Adherent cells were maintained in a humidified 5% CO2 atmosphere at 37°C and used for experiments within 7 days (2, 32).
Animals
Normal ICR mice (Nrf2+/+) and Nrf2 deficient mice (Nrf2−/−) with ICR background were provided by Jiling Hospital Laboratory center (Nanjing, China) with permission from Dr. Thomas W. Kensler (Johns Hopkins University, Baltimore, MD). All animals were raised under SPF-condition and had free accessible to food and water. All experiments were conducted following the instructions of the University Committee on Use and Care of Laboratory Animals at Sichuan University.
Quantitative real-time polymerase chain reaction analysis
Total RNA were extracted from cell lines or lung tissues by using Trizol Reagent (Invitrogen). cDNAs were reverse-transcribed from 1 μg of RNA with reverse transcription kit (TaKaRa) and used as templates in quantitative real-time polymerase chain reactions (PCRs) using SYBR Green Supermix kit (Bio-Rad). The sequences of primers used for PCR amplification are shown in Table 2. 18S served as internal normalization control.
Table 2.
Primers Used for Quantitative Real-Time Polymerase Chain Reaction
| Gene | Forward primer | Reverse primer |
|---|---|---|
| (m)NQO-1 | 5′-GCC TGA GCC CAG ATA TTG TG-3′ | 5′-GGA AAG GAC CGT TGT CGT-3′ |
| (m)HO-1 | 5′-GAG ATA GAG CGC AAC AAG CAG-3′ | 5′-CTT GAC CTC AGG TGT CAT CTC-3′ |
| (m)iNOS | 5′-CAG CAC AGG AAA TGT TTC AGC-3′ | 5′-TAG CCA GCG TAC CGG ATG A-3′ |
| (h) iNOS | 5′-ACA AGC CTA CCC CTC CAG AT-3′ | 5′-CTT GGA TGG TTG ACT GCC CT-3′ |
| (m)IL-6 | 5′-GTT GCC TTC TTG GGA CTG AT-3′ | 5′-CTG GCT TTG TCT TTC TTG TTA T-3′ |
| (h)IL-6 | 5′-AAA TGC CAG CCT GCT GAC GAA C-3′ | 5′-AAC AAC AAT CTG AGG TGC CCA TGC TAC-3′ |
| (m)COX2 | 5′-CCC TGA AGC CGT ACA CAT CA-3′ | 5′-TGT CAC TGT AGA GGG CTT TCA ATT-3′ |
| (h) COX2 | 5′-GGG CAA AGA CTG CGA AGA AG-3′ | 5′- CCC ATG TGA CGA AAT GAC TG-3′ |
| (m)IL-10 | 5′-GCT CTT ACT GAC TGG CAT GAG-3′ | 5′-CGC AGC TCT AGG AGC ATG TG-3′ |
| (h)IL-10 | 5′-CTT TAA GGG TTA CCT GGG TTG C-3′ | 5′- CCT TGA TGT CTG GGT CTT GGT-3′ |
| (m)CPT-1 | 5′-ATG GCA GAG GCT CAC CAA GC-3′ | 5′-GAT GAA CTT CCA GGA GTG C-3′ |
| 18S | 5′-TTG ACG GAA GGG CAC CAC CAG-3′ | 5′-GCA CCA CCA CC ACG GAA TCG-3′ |
Western blot analysis
Whole cell lysates were prepared by directly denaturing cell pellets and homogenized lung tissues in SDS loading buffer, and immediately boiling for 10 min. Nuclear and cytoplasmic protein fragmentation was performed as per the manufacturer's instructions (MBI Fermentas). Western blotting assays were done as described previously (16).
Enzyme-linked immunosorbent assay
Macrophage cultural supernatants were used for ELISA assay to measure TNF-α and IL-1β proteins as per the manufacturer's instructions (BD Biosciences).
Free radical assay
For intracellular ROS measurement, the cells treated with BBR and/or LPS for 24 h were washed with PBS twice and incubated with 10 μM H2-DCFH-DA for 30 min at 37°C in the dark. Then cells were washed again and collected by scraping. Collected cells were resuspended in PBS and their fluorescence intensity corresponding to intracellular DCFH-DA levels was immediately read on a Becton Dickinson FACS. The data was analyzed with Cell Quest software (BD Biosciences). DCFH-DA levels were also visualized using fluorescence microscope (Nikon, TE2000). For NO measurements, the level of NO derivative nitrite in culture medium was determined by Griess reaction as per the kit's manual (Beyotime).
Pinocytosis assay
Culture media were removed and 200 μl/well of 0.1% neutral red was added. After 3 h, cells were washed twice with PBS, and then imaged with an optical microscope or lysed in 200 μl of lysis solution (1:1 of 0.1 M acetic acid and 100% ethanol) for 4 h for quantification. Absorbance was measured at 570 nm with a Spectra (Shell) Reader.
MMP assay
MMP was measured using a mitochondrial-specific dual-fluorescence probe, JC-1 (Beyotime). At the end of the experiments, cells were washed twice with PBS and loaded with JC-1-containing solution for 30 min at 37°C. After JC-1 solution was substituted with PBS, cells were imaged under a fluorescent microscope; alternatively, JC-1 fluorescence was quantified through flow cytometry, in which red JC-1 aggregates were gated in the FL2 channel and green JC-1 monomers in the FL1 channel.
GST assay
RAW264.7 cells were treated with BBR (5 μM) or LPS (10 ng/ml) for 24 h as above. GST activity was determined spectrophotometrically at 340 nm as per the kit's instructions (Jiancheng).
AMP: ATP ratio measurement
After treatment, cells were washed with ice-cold PBS and scraped with 1.6 M HClO4. Cell lysates were centrifuged at 4°C (10 min, 2000 g). After centrifugation, supernatants were collected and neutralized by 0.1 M K2CO3. The concentrations of ATP and AMP in supernatants were determined by high performance liquid chromatography (7).
Plasma redox state assay
Reduced (GST) and oxidized (GSSG) glutathione levels in mouse plasma were measured through colorimetric determination by using GSH and GSSG Assay Kit (Beyotime) (19,20). Redox states (Eh) of GSH/GSSG were calculated from the concentrations of GSH, GSSG in molar units with following forms of Nernst equation for pH 7.4: GSH/GSSG, Eh=− 264+30 log([GSSG]/[GSH]2)(22).
DNA transfection
Endoxin-free cDNA expression plasmid DNAs were transfected into cells using Lipofectamine 2000 reagent (Invitrogen). For Nrf2 overexpression, pGFP-Nrf2 (pGFP/pOB125) and pKeap1(Keap1/pcDNA3.1) plasmid DNAs were cotransfected as described previously (36). For dominant-negative Nrf2 expression, pDN-Nrf2 (Nrf2(S40A)/pcDNA3.1) was cotransfected in conjunction with pKeap1/pcDNA3.1 (36). For dominant-negative AMPKα1 expression, cells were transfected with the pDN-AMPKα1 (T172A) plasmid DNA. pDN-AMPKα1 vector was a gift from Dr Jae Bum Kim (19).
Animal experiments
Mice (female, 7–8 weeks) were divided randomly into three groups: (i) saline; (ii) LPS; and (iii) LPS+BBR. To evaluate the effect of BBR on the survival of mice, endotoxemia was induced by a single i.p. injection of bacterial endotoxin (LPS, 35 mg·kg−1), with three i.p. injections of BBR (10 mg·kg−1), at 2 h before and 12 h and 24 h after the injection of LPS. Survival rates of animals were monitored up to 96 h after LPS challenge. To examine the effects of BBR on LPS-induced lung injuries, mice were injected intraperitoneally with either saline or BBR (10 mg·kg−1) at the same time as LPS injection (15 mg kg−1). Six hours after LPS injection, mice were killed under chloral hydrate anesthesia and the lungs were removed. Lung tissues were subsequently used for quantitative real-time PCR assay, western blot assay, and hematoxylin-eosin (HE) staining.
Population analysis of WBCs
After mice were anesthetized, peripheral blood was collected from retro-orbital cavity into a tube with decoagulant, and then diluted 1:10 in 1% glacial acetic acid. Total WBC counting was performed using a hemocytometer. Additionally, blood smears were made, followed by Wright-Giemsa staining and population analysis of WBCs. Neutrophils, lymphocytes and monocytes were counted separately.
Statistical analysis
Results represent data from multiple (at least three) independent experiments. Data are presented as means±SE, and p-values were calculated from the Student's t-test or one-way ANOVA followed by a Turkey comparisons test, using SPSS 17 software (SPSS, Inc.). The differences were considered to be significant when p<0.05 and highly significant when p<0.01. The survival test was presented as Kaplan-Meier survival curves using the SPSS software (41).
Abbreviations Used
- AMPK
AMP-activated protein kinase
- ARE
antioxidant responsive element
- BBR
berberine
- CC
compound C
- COX2
cyclooxygenase-2
- CPT-1
carnitine palmitoyl transferase 1
- DCFH-DA
2′, 7′-dichlorodihydrofluorescein diacetate
- Dico
dicoumarol
- ELISA
enzyme-linked immunosorbent assay
- GST
glutathione S-transferase
- HO-1
heme oxygenase-1
- i.p.
intraperitoneal
- IL-1β
interleukin-1 beta
- iNOS
inducible nitric oxide synthase
- LPS
lipopolysaccharide
- MMP
mitochondria membrane potential
- NO
nitric oxide
- NQO-1
NADPH quinone oxidoreductase-1
- Nrf2
nuclear factor erythroid-2-related factor-2
- PBS
phosphate-buffered saline
- PMA
phorbol 12-myristate 13-acetate
- qRT-PCR
quantitative real-time polymerase chain reaction
- ROS
reactive oxygen species
- TLR4
toll-like receptor 4
- TNF-α
tumor necrosis factor-α
- WBCs
white blood cells
Acknowledgments
We thank Prof. Jae Bum Kim for providing plasmids expressing pDN-AMPKα1, thank Meiyi Pu, and Prof. Anthony N. Imbalzano for critical reading of the manuscript and great contribution to the English improvement. This work was supported by National Natural Science Foundation of China (Grant Number 81273224), National 973 Basic Research Program of China (Nos. 2013CB911300), and National S&T Major project (Grant Number 2012ZX09501001-003). The authors thank Dr. Ping Lin, Xiujie Wang and Yi Chen for general supports.
Author Disclosure Statement
There is no conflict of interest.
References
- 1.Anwar AA, Li FYL, Leake DS, Ishii T, Mann GE, and Siow RCM. Induction of heme oxygenase 1 by moderately oxidized low-density lipoproteins in human vascular smooth muscle cells: Role of mitogen-activated protein kinases and Nrf2. Free Radic Biol Med 39: 227–236, 2005 [DOI] [PubMed] [Google Scholar]
- 2.Aung HT, Schroder K, Himes SR, Brion K, van Zuylen W, Trieu A, Suzuki H, Hayashizaki Y, Hume DA, Sweet MJ, and Ravasi T. LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J 20: 1315–1327, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Aziz M, Jacob A, Matsuda A, Wu R, Zhou M, Dong W, Yang W-L, and Wang P. Pre-Treatment of Recombinant Mouse MFG-E8 Downregulates LPS-Induced TNF-α Production in Macrophages via STAT3-Mediated SOCS3 Activation. PLoS One 6: e27685, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks-Worrell B. and Palmer JP. Immunology in the Clinic Review Series; focus on metabolic diseases: development of islet autoimmune disease in type 2 diabetes patients: potential sequelae of chronic inflammation. Clin Exp Immunol 167: 40–46, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cacicedo JM, Yagihashi N, Keaney JF, Jr, Ruderman NB, and Ido Y. AMPK inhibits fatty acid-induced increases in NF-κB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun 324: 1204–1209, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Chen J-H, Huang S-M, Tan T-W, Lin H-Y, Chen P-Y, Yeh W-L, Chou S-C, Tsai C-F, Wei IH, and Lu D-Y. Berberine induces heme oxygenase-1 up-regulation through phosphatidylinositol 3-kinase/AKT and NF-E2-related factor-2 signaling pathway in astrocytes. Int Immunopharmacol 12: 94–100, 2012 [DOI] [PubMed] [Google Scholar]
- 7.Cheng Z, Pang T, Gu M, Gao A-H, Xie C-M, Li J-Y, Nan F-J, and Li J. Berberine-stimulated glucose uptake in L6 myotubes involves both AMPK and p38 MAPK. Biochim Biophys Acta 1760: 1682–1689, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Cho H-Y, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, and Kleeberger SR. Nrf2-regulated PPARγ expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med 182: 170–182, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Souza LF, Barreto F, da Silva EG, Andrades ME, Guimarães ELM, Behr GA, Moreira JCF, and Bernard EA. Regulation of LPS stimulated ROS production in peritoneal macrophages from alloxan-induced diabetic rats: involvement of high glucose and PPARγ. Life Sci 81: 153–159, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Dyson A, Bryan NS, Fernandez BO, Garcia-Saura M-F, Saijo F, Mongardon N, Rodriguez J, Singer M, and Feelisch M. An integrated approach to assessing nitroso-redox balance in systemic inflammation. Free Radic Biol Med 51: 1137–1145, 2011 [DOI] [PubMed] [Google Scholar]
- 11.Gibbs PJ. and Seddon KR. Berberine. Altern Med Rev 5: 175–177, 2000 [PubMed] [Google Scholar]
- 12.Giudice A. and Montella M. Activation of the Nrf2–ARE signaling pathway: a promising strategy in cancer prevention. Bioessays 28: 169–181, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Guo RF. and Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal 9: 1991–2002, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Hattori Y, Suzuki K, Hattori S, and Kasai K. Metformin inhibits cytokine-induced nuclear factor κB activation via AMP-activated protein kinase activation in vascular endothelial cells. Hypertension 47: 1183–1188, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Hsu Y-Y, Chen C-S, Wu S-N, Jong Y-J, and Lo Y-C. Berberine activates Nrf2 nuclear translocation and protects against oxidative damage via a phosphatidylinositol 3-kinase/Akt-dependent mechanism in NSC34 motor neuron-like cells. Eur J Pharm Sci 46: 415–425, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Hu J-P, Nishishita K, Sakai E, Yoshida H, Kato Y, Tsukuba T, and Okamoto K. Berberine inhibits RANKL-induced osteoclast formation and survival through suppressing the NF-κB and Akt pathways. Eur J Pharmacol 580: 70–79, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Huang H-C, Nguyen T, and Pickett CB. Phosphorylation of Nrf2 at Ser-40 by Protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem 277: 42769–42774, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, Kelly V, Sekizawa K, Uchida K, and Yamamoto M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-Deoxy-Δ12,14-Prostaglandin J2. Mol Cell Biol 24: 36–45, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jeong HW, Hsu KC, Lee J-W, Ham M, Huh JY, Shin HJ, Kim WS, and Kim JB. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab 296: E955–E964, 2009 [DOI] [PubMed] [Google Scholar]
- 20.Jia J-H, Wang Y, Cao Y-B, Gao P-H, Jia X-M, Ma Z-P, Xu Y-G, Dai B-D, and Jiang Y-Y. CaIPF7817 is involved in the regulation of redox homeostasis in Candida albicans. Biochem Biophys Res Commun 359: 163–167, 2007 [DOI] [PubMed] [Google Scholar]
- 21.Jin W, Wang H, Yan W, Xu L, Wang X, Zhao X, Yang X, Chen G, and Ji Y. Disruption of Nrf2 enhances upregulation of nuclear factor-κB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Mediat Inflamm 2008: 725174, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones DP, Carlson JL, Mody VC, Jr, Cai J, Lynn MJ, and Sternberg P., JrRedox state of glutathione in human plasma. Free Radic Biol Med 28: 625–635, 2000 [DOI] [PubMed] [Google Scholar]
- 23.Kang KW, Choi SH, and Kim SG. Peroxynitrite activates NF-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide 7: 244–253, 2002 [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Cha Y-N, and Surh Y-J. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat Res Fundam Mol Mech Mutagen 690: 12–23, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Kim K-W, Ha K-T, Park C-S, Jin U-H, Chang HW, Lee I-S, and Kim C-H. Polygonum cuspidatum, compared with baicalin and berberine, inhibits inducible nitric oxide synthase and cyclooxygenase-2 gene expressions in RAW 264.7 macrophages. Vasc Pharmacol 47: 99–107, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Kim Y-O, Lee S-W, Oh C-H, and Rhee Y-H. Hericium erinaceus suppresses LPS-induced pro-inflammation gene activation in RAW264.7 macrophages. Immunopharmacol Immunotoxicol 34: 504–512, 2012 [DOI] [PubMed] [Google Scholar]
- 27.Ko K, Yang H, Noureddin M, Iglesia-Ara A, Xia M, Wagner C, Luka Z, Mato JM, and Lu SC. Changes in S-adenosylmethionine and GSH homeostasis during endotoxemia in mice. Lab Invest 88: 1121–1129, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong WJ, Wei J, Abidi P, Lin MH, Inaba S, Li C, Wang YL, Wang ZZ, Si SY, Pan HN, Wang SK, Wu JD, Wang Y, Li ZR, Liu JW, and Jiang JD. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med 10: 1344–1351, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Lee YS, Kim WS, Kim KH, Yoon MJ, Cho HJ, Shen Y, Ye J-M, Lee CH, Oh WK, Kim CT, Hohnen-Behrens C, Gosby A, Kraegen EW, James DE, and Kim JB. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes 55: 2256–2264, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Lipton SA, Gu Z, and Nakamura T. Inflammatory mediators leading to protein misfolding and uncompetitive/fast off—rate drug therapy for neurodegenerative disorders. In: International Review of Neurobiology, edited by Giacinto Bagetta MTC. and Stuart AL. San Diego, CA: Academic Press, 2007, pp. 1–27 [DOI] [PubMed] [Google Scholar]
- 31.Liu X-m, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, and Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am J Physiol Heart Circ Physiol 300: H84–H93, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z, Swindall AF, Kesterson RA, Schoeb TR, Bullard DC, and Bellis SL. ST6Gal-I regulates macrophage apoptosis via α2–6 sialylation of the TNFR1 death receptor. J Biol Chem 286: 39654–39662, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu D-Y, Tang C-H, Chen Y-H, and Wei IH. Berberine suppresses neuroinflammatory responses through AMP-activated protein kinase activation in BV-2 microglia. J Cell Biochem 110: 697–705, 2010 [DOI] [PubMed] [Google Scholar]
- 34.Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med 10: 369–373, 2010 [DOI] [PubMed] [Google Scholar]
- 35.Nguyen T, Sherratt PJ, Huang H-C, Yang CS, and Pickett CB. Increased Protein Stability as a Mechanism That Enhances Nrf2-mediated Transcriptional Activation of the Antioxidant Response Element: DEGRADATION OF Nrf2 BY THE 26 S PROTEASOME. J Biol Chem 278: 4536–4541, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Numazawa S, Ishikawa M, Yoshida A, Tanaka S, and Yoshida T. Atypical protein kinase C mediates activation of NF-E2-related factor 2 in response to oxidative stress. Am J Physiol Cell Physiol 285: C334–C342, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Onken B. and Driscoll M. Metformin induces a dietary restriction–like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1. PLoS One 5: e8758, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peairs A, Radjavi A, Davis S, Li L, Ahmed A, Giri S, and Reilly CM. Activation of AMPK inhibits inflammation in MRL/lpr mouse mesangial cells. Clin Exp Immunol 156: 542–551, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin L, Liu Y, Wang T, Wei S-J, Block ML, Wilson B, Liu B, and Hong J-S. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279: 1415–1421, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Salminen A, Hyttinen J, and Kaarniranta K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: impact on healthspan and lifespan. J Mol Med 89: 667–676, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siaperas P, Pefanis A, Iliopoulos D, Katsarolis I, Kyroudi-Voulgari A, Donta I, Karayiannakos P, and Giamarellou H. Evidence of less severe aortic valve destruction after treatment of experimental staphylococcal endocarditis with vancomycin and dexamethasone. Antimicrob Agents Chemother 45: 3531–3537, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silva MT. Macrophage phagocytosis of neutrophils at inflammatory/infectious foci: a cooperative mechanism in the control of infection and infectious inflammation. J Leukoc Biol 89: 675–683, 2011 [DOI] [PubMed] [Google Scholar]
- 43.Stolp HB. and Dziegielewska KM. Review: Role of developmental inflammation and blood–brain barrier dysfunction in neurodevelopmental and neurodegenerative diseases. Neuropathol Appl Neurobiol 35: 132–146, 2009 [DOI] [PubMed] [Google Scholar]
- 44.Sundaresan NR, Vasudevan P, Zhong L, Kim G, Samant S, Parekh V, Pillai VB, Ravindra PV, Gupta M, Jeevanandam V, Cunningham JM, Deng CX, Lombard DB, Mostoslavsky R, and Gupta MP. The sirtuin SIRT6 blocks IGF-Akt signaling and development of cardiac hypertrophy by targeting c-Jun. Nat Med 18: 1643–1650, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Surh Y. NF-kappa B and Nrf2 as potential chemopreventive targets of some anti-inflammatory and antioxidative phytonutrients with anti-inflammatory and antioxidative activities. Asia Pac J Clin Nutr 17: 269, 2008 [PubMed] [Google Scholar]
- 46.Tanigawa S, Fujii M, and Hou D-X. Action of Nrf2 and Keap1 in ARE-mediated NQO1 expression by quercetin. Free Radic Biol Med 42: 1690–1703, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Tkachev V, Menshchikova E, and Zenkov N. Mechanism of the Nrf2/Keap1/ARE signaling system. Biochemistry (Moscow) 76: 407–422, 2011 [DOI] [PubMed] [Google Scholar]
- 48.Towler MC. and Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res 100: 328–341, 2007 [DOI] [PubMed] [Google Scholar]
- 49.Turner N, Li J-Y, Gosby A, To SWC, Cheng Z, Miyoshi H, Taketo MM, Cooney GJ, Kraegen EW, James DE, Hu L-H, Li J, and Ye J-M. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I. Diabetes 57: 1414–1418, 2008 [DOI] [PubMed] [Google Scholar]
- 50.Villa P, Saccani A, Sica A, and Ghezzi P. Glutathione protects mice from lethal sepsis by limiting inflammation and potentiating host defense. J Infect Dis 185: 1115–1120, 2002 [DOI] [PubMed] [Google Scholar]
- 51.von Knethen A, Neb H, Morbitzer V, Schmidt MV, Kuhn A-M, Kuchler L, and Brüne B. PPARγ stabilizes HO-1 mRNA in monocytes/macrophages which affects IFN-β expression. Free Radic Biol Med 51: 396–405, 2011 [DOI] [PubMed] [Google Scholar]
- 52.Wang H, Khor TO, Saw CLL, Lin W, Wu T, Huang Y, and Kong A-NT. Role of Nrf2 in suppressing LPS-Induced inflammation in mouse peritoneal macrophages by polyunsaturated fatty acids docosahexaenoic acid and eicosapentaenoic acid. Mol Pharm 7: 2185–2193, 2010 [DOI] [PubMed] [Google Scholar]
- 53.Yamamoto M, Yamazaki S, Uematsu S, Sato S, Hemmi H, Hoshino K, Kaisho T, Kuwata H, Takeuchi O, Takeshige K, Saitoh T, Yamaoka S, Yamamoto N, Yamamoto S, Muta T, Takeda K, and Akira S. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein I[kappa] B[zeta]. Nature 430: 218–222, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Yang Z, Kahn BB, Shi H, and Xue B-z. Macrophage α1 AMP-activated Protein Kinase (α1AMPK) Antagonizes Fatty Acid-induced Inflammation through SIRT1. J Biol Chem 285: 19051–19059, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ye J. and Keller JN. Regulation of energy metabolism by inflammation: a feedback response in obesity and calorie restriction. Aging 2: 361–368, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yeligar SM, Machida K, and Kalra VK. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1α and Nrf2 to attenuate inflammatory cytokine expression. J Biol Chem 285: 35359–35373, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu R, Chen C, Mo Y-Y, Hebbar V, Owuor ED, Tan T-H, and Kong A-NT. Activation of mitogen-activated protein kinase pathways induces antioxidant response element-mediated gene expression via a Nrf2-dependent mechanism. J Biol Chem 275: 39907–39913, 2000 [DOI] [PubMed] [Google Scholar]
- 58.Zammit VA. and Arduini A. The AMPK-Malonyl-CoA-CPT1 Axis in the control of hypothalamic neuronal function. Cell Metab 8: 175, 2008 [DOI] [PubMed] [Google Scholar]