

Summary:
The immune response to cancer is critically determined by the way in which tumor cells die. As necrotic, stress-associated death can be associated with activation of antitumor immunity, whole tumor cell antigen loading strategies for dendritic cell (DC)-based vaccination have commonly used freeze-thaw “necrotic” lysates as an immunogenic source of tumor-associated antigens. In this study, the effect of such lysates on the ability of DCs to mature in response to well-established maturation stimuli was examined, and methods to enhance lysate-induced DC activation explored. Freeze-thaw lysates were prepared from murine tumor cell lines and their effects on bone marrow-derived DC maturation and function examined. Unmodified freeze-thaw tumor cell lysates inhibited the toll-like receptor-induced maturation and function of bone marrow-derived DCs, preventing up-regulation of CD40, CD86, and major histocompatibility complex class II, and reducing secretion of inflammatory cytokines [interleukin (IL)-12 p70, tumor necrosis factor-α, and IL-6]. Although IL-10 secretion was increased by lysate-pulsed DCs, this was not responsible for the observed suppression of IL-12. Although activation of the nuclear factor-κB pathway remained intact, the kinase activity of phosphorylated p38 mitogen-activated protein kinase was inhibited in lysate-pulsed DCs. Lysate-induced DC suppression was partially reversed in vitro by induction of tumor cell stress before lysis, and only DCs loaded with stressed lysates afforded protection against tumor challenge in vivo. These data suggest that ex vivo freeze-thaw of tumor cells does not effectively mimic in vivo immunogenic necrosis, and advocates careful characterization and optimization of tumor cell-derived vaccine sources for cancer immunotherapy.
Keywords: dendritic cell, necrosis, lysate, maturation, TLR
Antitumor vaccination seeks to present tumor-associated antigens (TAA) in a context that overcomes the prevailing tolerance of the immune system to tumor growth. Various formulations have been tried as sources of TAA for vaccination, including defined peptides or TAA derived from the entire tumor proteome.1 Whole cells have the advantage of providing a full complement of TAA, including both major histocompatibility complex (MHC) class I and class II-restricted epitopes, thus reducing the risk of immune escape by antigen loss variants.
Dendritic cells (DCs) are key orchestrators of the immune system and have been widely used for cancer immunotherapy. At rest, immature DCs reside in peripheral tissues, where they sample their environment and acquire antigens. In the presence of appropriate “danger” signals, DCs undergo a programmed cell event known as maturation, characterized by the up-regulation of cell surface molecules involved in antigen presentation and costimulation (eg, CD80, CD86, CD40, and MHC class II), the release of proinflammatory cytokines [eg, interleukin (IL)-12 p70], and migration to lymphoid tissue. Agonists of the toll-like receptor (TLR) family, which represent a broad range of microbial motifs, are particularly potent triggers for DC maturation and immune priming.2 Once in the lymph node, DCs activate cognate CD4 and CD8 T cells, which recognize antigenic epitopes on their surface presented by appropriate MHC molecules.3
Many investigators have loaded DCs with TAA as a means of stimulating an antitumor response. Approaches based on whole tumor cells have included the use of apoptotic cells,4-6 fused tumor cell-DC hybrids,7 and tumor-derived exosomes.8 Perhaps the commonest strategy, however, has been to load DCs with ostensibly “necrotic” tumor cell lysates, generated by repeated cycles of freezing and thawing, with the solid debris removed by centrifugation. DCs loaded with such lysates have provided modest protection in various animal models,9-11 and stimulated limited responses in a range of clinical trials.12-14 Most encouragingly, freeze-thaw lysates injected without DCs [derived from autologous tumor cells pretreated with interferon (IFN)-γ and α-tocopherol], prolonged progression-free survival in renal cancer patients within a large, randomized clinical trial.15
There has been considerable controversy concerning the relative immunogenicity of different modes of cell death (necrotic vs. apoptotic). For example, several authors have suggested that apoptosis is inhibitory to DC activation,16-19 others have shown the reverse,6,20,21 and some have shown broad equivalence between different types of cell death.22,23 Much of this confusion may derive from the difficulty in isolating pure necrotic and “apoptotic” populations, the differences between physiologic apoptosis/necrosis and modes of cell death generated in vitro, and the importance of the cellular stress response in determining immune outcomes. We have previously argued24 that the passive “necrosis” generated by repeated freeze-thaw cycles in vitro is distinct from true pathologic necrosis in vivo, where a variety of immunologic stress signals can be generated. One family of such signals is the heat shock proteins (HSP), which act as both nonspecific immune adjuvants and as chaperones for successful delivery of TAA to antigen presenting cells such as DCs.25
Although some studies have shown that stressed apoptotic cells, with induced HSP production, have enhanced immunogenicity,26,27 the clinically attractive and simple strategy of freeze-thaw, unmodified lysate-loaded DCs has been widely applied in early trials. This has been despite limited data assessing the interaction between such lysates and DCs, and a failure to address the question of whether their preparation is indeed optimally immunogenic.
As the immune response generated by DCs is related to their maturation state, we hypothesized that DCs, which had taken up TAA in the form of necrotic lysates, and were then matured using a range of TLR agonists, would represent a potent anticancer vaccine. In this study, we show that freeze-thaw lysates in fact inhibit the maturation of DCs in response to a range of TLR ligands, interfere with DC/T-cell interactions, and selectively target particular signaling pathways. To some extent, these effects are shown to be reduced by stressing cells before lysis. These data have important implications for the optimal preparation of tumor cell lysate-based vaccines for clinical application.
MATERIALS AND METHODS
Animals and Cell Lines
Female C57BL/6 and BALB/c mice (6 to 8 wk old) were purchased from Harlan (Bicester, UK). IL-10−/− mice (C57BL/6 background) were kindly provided by Prof Simon Carding (University of Leeds, Leeds, UK). B16.ovalbumin (OVA) murine melanoma cells (C57BL/6 background) were a gift from Edith Lord (Department of Microbiology and Immunology, University of Rochester Medical Center, Rochester, NY). B16.hsp70.GFP cells28 were a gift from Dr Stephen Todryk (University of Oxford, UK). The murine colorectal carcinoma lines MC38 (C57BL/6) and CT26 (BALB/c) were provided by Cancer Research UK Cell Services (Potters Bar, UK). B16.OVA and MC38 were grown in Dulbecco’s modified Eagle’s medium (Invitrogen, Paisley, UK) and the CT26 cells in Roswell Park Memorial Institute medium (RPMI) 1640 (Invitrogen). Both types of media were supplemented with heat-inactivated fetal calf serum (FCS) 10% v/v and 2-mM L-glutamine (Invitrogen). B16.OVA and B16.hsp70.GFP also required G418 selection (400 and 500 μg/mL, respectively). The B3Z hybridoma, which secretes IL-2 upon ligation of its Kb-SIINFEKL-specific receptor,29 was a gift to our unit from N. Shastri (Department of Molecular and Cell Biology, University of California, Berkeley), and was maintained in RPMI 1640 supplemented with 10% heat-inactivated FCS, 2-mM L-glutamine, 1-mM sodium pyruvate, and nonessential amino acids (Invitrogen). All cell lines were routinely tested and found to be free of mycoplasma.
Preparation of Necrotic Cell Lysates
Cells were harvested, washed in phosphate-buffered saline (PBS), and resuspended in PBS at 1×107 cells/mL, before 5 cycles of freezing and thawing (methanol with dry ice and 56°C). At this point, 100% of cells were seen to take up trypan blue by light microscopy. Solid debris was then spun down at 1700 g for 5 minutes. Lysates in this study constituted the supernatants, which were taken and stored at −80°C for later use. Uncentrifuged lysates were also kept for comparison in some experiments.
Preparation of Bone Marrow-derived DCs
Bone marrow was removed from the femur and tibia of female C57BL/6, BALB/c, and IL-10−/− mice by flushing into RPMI 1640. To enrich for DC precursors, cells were incubated with biotinylated antibodies to B220, I-A, Gr1, CD3, and CD4 (BD-Pharmingen, Oxford, UK), washed, incubated with streptavidin microbeads (Miltenyi-Biotech, Bisley, UK), and passed through a magnetic cell separation column (Miltenyi-Biotech). Eluted cells were cultured for 7 days, as previously described,30 at which point approximately 70% were CD11c+ by flow cytometry.
Coculture of DCs With Lysate
DCs were resuspended in fresh medium at a concentration of 1×106 cells/mL. Necrotic cell lysate or an equivalent volume of PBS control was added to DCs at a ratio of 3 tumor cell equivalents to 1 DC. Coculture continued for 8 hours before the addition of relevant maturation stimuli [lipopolysaccharide (LPS) 100 ng/mL; OK-432 10 μg/mL; and poly I:C 100 μg/mL in combination with CpG 2 μg/mL].
Analysis of Cell Phenotype
Cells were washed in staining buffer (sterile PBS, 1% FCS, and 0.1% sodium azide) before addition of antibodies. Nonspecific binding was blocked with anti-CD16/CD32 (BD Pharmingen) for 15 minutes at 4°C, before staining with anti-CD11c-fluorescein isothiocyanate (Serotec, Oxford, UK) and various phenotypic markers of DC maturation [anti-CD40-phycoerythrin (PE), anti-CD80-PE, anti-CD86-PE, and anti-Ia-PE appropriate for the mouse strain—all BD Pharmingen]. Cells were incubated for 45 minutes at 4°C before a further wash with buffer; cells were then resuspended and fixed in 300 μL 1% w/v paraformaldehyde solution before analysis with a FACSCalibur using CellQuest software (BD Biosciences).
Measurement of Cytokines
IL-12, IL-10, and IFN-γ were measured using standard enzyme-linked immunosorbent assay according to the manufacturer’s instructions. Briefly, flat-bottomed 96-well plates (Invitrogen) were coated with capture antibodies (BD Biosciences), incubated overnight with samples, and then developed with appropriate biotinylated secondary antibodies (BD Biosciences), followed by Extravidin (Sigma, Poole, UK) conjugate and substrate solution (p-nitrophenyl phosphate 1 mg/mL—Sigma) in 0.2-M Tris buffer (Sigma). Color change was measured with a plate reader (Titertek Multiskan MCC) at 405-nm wavelength.
Tumor necrosis factor (TNF)-α and IL-6 were measured using bead kits (Biosource, Nivelles, Belgium), which use spectrally encoded antibody-conjugated beads. The assay was performed in 96-well plate format and analyzed with a Luminex 100 instrument, which monitors the spectral properties of the capture bead and simultaneously measures the quantity of associated fluorophore.
Polymerase Chain Reaction
RNA was purified from samples using an RNeasy kit (Qiagen, Crawley, UK) and cDNA was produced using Superscript II reverse transcriptase (Invitrogen), both following manufacturer’s instructions. Primer pairs were manufactured by MWG (Milton Keynes, UK) according to previously published sequences.31 Polymerase chain reactions (PCRs) were performed over 30 cycles in a GeneAmp PCR System 9700 thermal cycler (PE Applied Biosystems, Warrington, UK). PCR buffer (10×; containing Mg and Taq polymerase (Roche, Welwyn Garden City, UK) with deoxyribonucleotide triphosphate (Invitrogen) were used for the reactions. Annealing temperatures were optimized and varied from 55°C to 60°C. Products were separated on 1.2% agarose gel containing ethidium bromide and visualized under ultraviolet light.
Optimization of HSP70 Production in B16 Cells
B16.hsp70.GFP cells respond to stressful stimuli by producing green fluorescent protein (GFP),28 which can be quantified by flow cytometry. Such cells were irradiated to 30 Gy in 50-mL tubes using a cesium-137 source (Gammacell 1000 Elite) or heat shocked for 25 minutes in a thermostatically controlled water bath (42°C). Controls received sham treatment. To determine the combination of these stimuli, which induced the maximum stress response, the timing of the insults was varied, as was the time of cell harvest for flow cytometry.
Western Blotting
Cells were treated as indicated, then washed in PBS (4°C) before lysis in buffer containing 50-mM N-2-hydroxyl piperazine-N′-2-ehane sulfonic acid (pH 7.5), 100-mM NaCl, 1-mM ethylene glycol-bis (b-aminoethyl ether), 1-mM DL-dithiothreitol (DTT), 1% (v/v) Triton X-100, 10-μg/mL aprotinin, 10-μg/mL leupeptin, 1-mM sodium orthovanadate, and 25-mM sodium fluoride. Protein concentration was assessed by the Bradford method (Bio-Rad, Hemel Hempstead, UK). Lysates (20 to 40 μg) were combined with loading buffer [100-mM Tris-HCl; pH 6.8, 4% sodium dodecyl sulfate (SDS), 0.2% (w/v) bromophenol blue, 20% glycerol, and 200-mM DTT] and separated by 10% SDS-polyacrylamide gel electrophoresis. Protein was transferred to Hybond-C Super membranes (Amersham, Bucks, UK). Membranes were incubated with anti-IκBα antibodies (Santa Cruz Biotechnology, CA) or anti-β actin antibodies (Abcam, Cambridge, UK) and visualized using horseradish peroxidase-labeled secondary antibodies (DakoCytomation, Ely, Cambs, UK) and chemiluminescent substrate (Pierce, Rockford, IL).
p38 Mitogen-activated Protein Kinase Activity Assay
The level of active p38 mitogen-activated protein kinase (MAPK) was analyzed using a nonradioactive assay (Cell Signaling Technology, Danvers, MA) according to the manufacturer’s instructions. Briefly, treated DCs were lysed in the lysis buffer provided [20-mM Tris-HCl (pH: 7.5), 150-mM NaCl, 1-mM ethylene diamine tetra acetic acid, 1-mM ethylene glycol-bis (b-aminoethyl ether), 1% Triton X-100, 2.5-mM sodium pyrophosphate, 1-mM β-glycerol phosphate, 1-mM sodium orthovanadate, and 1-μg/mL leupeptin] and supplemented with 1-mM phenylmethylsulfonyl fluoride. Phosphorylated p38 MAPK was immunoprecipitated from 200 μg of whole cell lysate protein, using an immobilized phospho-p38 MAPK (Thr-180/Tyr-182) monoclonal antibody, overnight at 4°C with constant agitation. The immunoprecipitates were washed twice with lysis buffer and twice with the kinase buffer [25-mM Tris (pH 7.5), 5-mM β-glycerol phosphate, 2-mM DTT, 0.1-mM sodium orthovanadate, and 10-mM MgCl2] provided with the kit. The immunoprecipitated p38 MAPK was then incubated for 30 minutes at 37°C in kinase buffer containing 200-μM adenosine triphosphate and 2 μg of activating transcription factor (ATF)-2 fusion protein. The reaction was terminated by adding 3 × sample buffer and heating the samples to 95°C for 5 minutes. The samples were then separated by 10% SDS-polyacrylamide gel electrophoresis and immunoblotted for phosphorylated ATF-2.
B3Z Assay
DCs or tumor cells were washed and resuspended at 1×106/mL. About 1×105 cells/well were then added to round-bottomed 96-well plates and incubated for 2 hours with SIINFEKL at varying concentrations. Cells were then washed with PBS and incubated overnight with B3Z cells (1:1 ratio) as previously described.29 Supernatants were then analyzed by enzyme-linked immunosorbent assay to measure IL-2.
Allogeneic Mixed Lymphocyte Reaction
C57BL/6 DCs were added to round-bottomed 96-well plates and serially diluted. After allowing the DCs to settle for 3 hours, LPS was added to each well. After an hour, allogeneic BALB/c splenocytes (100 μL) were then added to each well at 1×106/mL to give a range of effector to DC ratios (final LPS concentration 100 ng/mL). Coculture occurred over 96 hours. To assess proliferation, tritium (Amersham Biotech, Little Chalfont, Bucks, UK; 0.5 μCi/well) was added for the last 16 hours. Finally, the wells were aspirated and washed through a filter (Tomtec Harvester 96 Mach IIIM) to trap cells on a membrane. This was then exposed to substrate (Packard Ultima Gold) and activity detected by a scintillation counter (1450 Microbeta-Wallac Jet). Alternatively, supernatants were harvested at 96 hours for cytokine assay.
In Vivos
Animal experiments were approved and performed at the CRUK Clare Hall Animal Facility (Potters Bar, London). C57BL/6 mice were age-matched and sex-matched for individual experiments. To establish SC tumors, the indicated number of tumor cells were injected SC (100 μL) into the flank region. Subsequent assessments of growth were made as previously described.32
Statistics
The indicated analyses were performed using SPSS software (v12.0.1).
RESULTS
Effect of Freeze-thaw Necrotic Cell Lysates on DC Maturation
Previous investigators have shown that necrotic murine fibroblast cell lysates18 and human tumor cell lysates33 can activate immature DCs. We wished to confirm these findings in murine tumor models and see if lysates affected DC activation in response to a further maturation signal such as LPS. C57BL/6 and BALB/c DCs were pretreated with autologous tumor lysates (C57BL/6-B16.OVA and MC38; BALB/c-CT26) and matured overnight with LPS. Freeze-thaw lysates alone on immature DCs caused limited up-regulation of CD80 and, to a lesser extent, CD86 (Figs. 1A, B); CD40 and I-A were unaffected. More strikingly, however, the presence of lysates significantly reduced LPS-induced up-regulation of CD40, CD86, and I-A (Table 1), while leaving CD80 unchanged. Hence, lysate-pulsed, LPS-activated DCs showed a more immature phenotype with a greater ratio of CD80 to CD86 than unpulsed controls. These effects were seen in all 3 tumor models tested across 2 mouse strains. Direct toxicity of the lysates to the DCs was excluded, as cell viability (measured by trypan blue exclusion or annexin/propidium iodide assay) was unaffected at the end of the incubation (data not shown).
FIGURE 1.

Prevention of TLR-induced DC maturation by lysates. A, C57BL/6 and (B) BALB/c DCs (1 × 106 cells/mL) were cultured with autologous lysates or PBS control (green: B16.OVA, red: MC38, purple: CT26, black: PBS (ie, no lysate), and shaded: isotype control). The phenotype of gated CD11c+ cells was then assessed by FACS with or without overnight maturation by LPS (100 ng/mL). C, The production of inflammatory cytokines (pg/mL) was then assessed in the supernatants of these cocultures. A–C are representative of at least 5 independent experiments. D, RT-PCR of RNA isolated from such cocultures (5 h post-LPS) was then performed to evaluate the transcription of IL-12 subunits. E and F, Equivalent experiments to A-C were performed using poly I:C (100 μg/mL) and CpG (2 μg/mL) in combination. Representative of 3 independent experiments (B indicates B16.OVA; C, CT26; M, MC38). Error bars denote SD of triplicate repeats within the individual experiment shown (* = P<0.05 compared with LPS alone). DC indicates dendritic cell; FACS, fluorescence-activated cell sorting; IL, interleukin; LPS, lipopolysaccharide; PBS, phosphate-buffered saline; RT-PCR, reverse transcriptase-polymerase chain reaction; TLR, toll-like receptor.
Table 1.
The Mean Fluorescence Intensity (MFI) of Day 7 DCs (C57BL/6 and BALB/c), Matured Overnight With LPS (100 ng/mL), was Compared With the MFI When DCs Were Matured by LPS After Preexposure to Autologous Lysates
| n | LPS Alone, Mean MFI (SD) |
LPS + Lysate, Mean MFI (SD) |
Mean Absolute Reduction in MFI (SD) |
Mean % Reduction in MFI With Lysate (SD) |
P | |
|---|---|---|---|---|---|---|
| (A) B16.OVA | ||||||
| CD40 | 15 | 69 (35) | 41 (21) | 28 (24) | 35 (20) | * |
| CD80 | 11 | 357 (262) | 320 (256) | 37 (56) | 12 (18) | n.s. |
| CD86 | 24 | 387 (202) | 255 (170) | 132 (68) | 38 (16) | * |
| I-A | 20 | 1476 (873) | 1015 (586) | 461 (356) | 30 (13) | * |
| (B) MC38 | ||||||
| CD40 | 11 | 69 (40) | 23 (8) | 46 (34) | 60 (16) | * |
| CD80 | 10 | 374 (269) | 356 (250) | 19 (65) | 1 (14) | n.s. |
| CD86 | 19 | 368 (207) | 230 (168) | 139 (76) | 42 (19) | * |
| I-A | 14 | 1357 (739) | 718 (343) | 639 (478) | 43 (15) | * |
| (C) CT26 | ||||||
| CD40 | 8 | 90 (56) | 38 (16) | 52 (43) | 52 (14) | * |
| CD80 | 8 | 431 (283) | 366 (199) | 65 (121) | 9 (18) | n.s. |
| CD86 | 11 | 481 (200) | 264 (102) | 218 (112) | 44 (9) | * |
| I-A | 5 | 1459 (522) | 600 (264) | 859 (429) | 58 (15) | * |
Mean results of the n experiments performed are quoted with SDs. Mean absolute reductions in MFI and percentage reductions of MFI, in the presence of lysate, are also given with SDs. Paired t tests were performed of the pairs of results over the n experiments. Significance relates to the reduced expression in lysate-treated DCs compared with the fully matured controls (*P < 0.05).
DC indicates dendritic cell; LPS, lipopolysaccharide; MFI, mean fluorescence intensity; n.s., not significant.
Another important feature of effective DC maturation and activation is the production of inflammatory cytokines. We, therefore, tested whether freeze-thaw lysates influence LPS-induced cytokine release by DCs. The addition of lysates to immature DCs produced no significant release of IL-12 p70, IL-6, or TNF-α (Fig. 1C). In contrast, addition of LPS alone was a potent stimulus for secretion of all 3 inflammatory cytokines, as expected. Significantly, however, in accordance with the phenotypic data, the presence of lysates greatly altered the cytokine profile produced by LPS-activated DCs. In particular, there was a dramatic, and statistically significant, reduction in IL-12 p70 and TNF-α. IL-6 was also reduced significantly, but to a lesser extent. This was not a nonspecific effect of protein loading the DCs, as equivalent levels of recombinant protein (OVA or bovine serum albumin) did not produce the same effect (data not shown).
The most profound effect seen was the reduction of IL-12 p70, which is formed by 2 subunits (p35 and p40). Expression of these is usually controlled at the level of transcription.34 We have previously used semiquantitative real time-PCR to evaluate samples for cytokine mRNA expression.35 Therefore, to determine if tumor lysates were altering IL-12 release by influencing gene transcription, we used this technique to assess levels of mRNA after LPS stimulation (Fig. 1D). Relative to β-actin control, all 3 lysates markedly reduced the expression of both IL-12 subunits (p35 and p40). No cytokine mRNA was detected in extracts of the tumor cell lines themselves in the absence of DCs (data not shown).
Recent studies have shown that combining TLR ligands can enhance cytokine release by DCs,36 with the combination of poly I:C (a TLR3 agonist) and CpG (a TLR9 agonist), particularly effective for IL-12 p70 production.37 To test whether this potent combination could overcome the inhibition of phenotypic maturation and IL-12 p70 release caused by lysates, further experiments were conducted using poly I:C and CpG in place of LPS. As with LPS, up-regulation of CD40, CD86, and I-A was reduced by lysates, whereas CD80 remained unaffected (Fig. 1E); production of IL-12 p70 was again inhibited to a similar degree (Fig. 1F).
There is some evidence that adding IL-4 to culture during the preparation of DCs increases their immunostimulatory properties.38 However, repeats of these experiments, after preparing DCs with granulocyte macrophage colony-stimulating factor/IL-4 showed equivalent levels of suppression (data not shown). Uncentrifuged lysate (rather than the supernatant) and different ratios of lysate cell equivalents per DC (range, 1:1 to 10:1) also inhibited TLR-induced DC activation (data not shown).
Further experiments were also performed using OK-432 to mature DCs (OK-432 is a lyophilized preparation of whole, heat-killed streptococci that has been used as an immune adjuvant39 in cancer immunotherapy and is also a TLR4 ligand). Similar inhibition of DCs by lysate was again seen (data not shown).
Therefore, tumor cells freeze-thawed in vitro to generate ostensibly necrotic and hence, potentially immunogenic lysates, inhibit full DC phenotypic maturation and inflammatory cytokine release. These effects are broad, extending over a range of tumor models, mouse strains, and maturation stimuli.
Effect of Freeze-thaw Lysates on DC Interaction With T Cells in Vitro
To assess the effect of lysate pulsing on the ability of DCs to stimulate T cell responses, we first performed allogeneic mixed lymphocyte reactions between stimulator C57BL/6 DC (either lysate loaded or not, then matured with LPS) and BALB/c splenocytes as responders. As shown in Figure 2A, treatment of DCs with either B16.OVA or MC38 lysate did not affect the proliferation of responding splenocytes. However, the release of IFN-γ was significantly reduced in cocultures of lysate-loaded DCs and allogeneic splenocytes (P<0.05 over 4 experiments at the 20:1 ratio—indicated by asterisk in Fig. 2B). As IFN-γ is a key T helper (TH) 1 cytokine, this result suggests that lysate-loaded DCs may skew more toward a TH2 helper response on interaction with CD4 cells, although, in these cocultures, levels of IL-4 remained very low and were not significantly different between all conditions (data not shown).
FIGURE 2.

Effect of lysate pretreatment on DC interactions with T cells in vitro. C57BL/6 DCs, preincubated with lysates or PBS control, were stimulated with LPS (100 ng/mL) and then (A) cocultured at the indicated ratios with allogeneic BALB/c splenocytes. Proliferation was assessed by the addition of tritium (0.5 μCi/well) at 72 hours with the incorporated activity measured the next day using a scintillation counter (dashed line indicates B16.OVA; dotted, MC38; solid, PBS). B, The production of IFN-γ in these supernatants was measured by ELISA at 96 hours (A and B, representative of at least 3 independent experiments). C, Alternatively, the DCs were washed, exposed to a range of concentrations of SIINFEKL for 2 hours, washed again, and then cocultured overnight with B3Z cells at a 1:1 ratio. Supernatants were harvested and assayed by ELISA for the production of IL-2. Representative plot from 5 independent experiments (* = P<0.05 paired t test compared with relevant PBS control). DC indicates dendritic cell; ELISA, enzyme-linked immunosorbent assay; IFN, interferon; IL, interleukin; LPS, lipopolysaccharide; PBS, phosphate-buffered saline.
We also examined the presentation of specific antigens to CD8 cells by lysate-pulsed DCs. The B3Z T-cell hybridoma is specific for the OVA peptide SIINFEKL presented by the H-2-Kb MHC class I molecule (expressed in C57BL/6 mice) and upon recognition releases IL-2. However, repeated attempts to stimulate B3Z via cross-presentation using B16.OVA freeze-thaw lysate-loaded DCs were unsuccessful (data not shown). Similar results have also been reported by other groups using OVA-expressing murine cell lines,40-42 possibly reflecting relatively poor cross-presentation of soluble OVA, as opposed to cell-associated OVA as described by Li et al.43
In the light of the global suppression of DC maturation and function described above, we further speculated that lysate-pulsed DCs may be dysfunctional in CD8 activation even when not required to process and cross-present TAA. To test this hypothesis, we cocultured SIINFEKL-pulsed DCs (ie, exogenously loaded with the defined epitope for MHC I binding) with B3Z cells, with or without prior lysate loading of DCs. As shown in Figure 2C, peptide presentation by lysate-pulsed DCs was significantly impaired (indicated by asterisks), although this difference was not apparent at higher doses of SIINFEKL when peptide was in great excess. This effect was not owing to differences in MHC I surface expression between control and lysate-pulsed DCs (data not shown), and applied to MC38 and B16.OVA lysates.
These data show that DCs pulsed with freeze-thaw lysates are likely to skew CD4 responses away from an optimal TH1 profile, are unable to cross-present lysate-derived TAA, and are less effective at activating specific CD8 cells even when favorably loaded exogenously with specific peptide epitopes.
IL-10 Secretion by DCs is Increased by Lysates, but is not Responsible for Suppression of IL-12 p70
IL-10 has been shown to inhibit the release of IL-12 p70 in murine DCs,44 and suppressed IL-12 production by DCs has previously been attributed to increased IL-10 secretion.45 To address whether the reduction in IL-12 p70 from lysate-pulsed DCs seen in Figure 1C might be causally linked to increased IL-10, we next assayed IL-10 in these supernatants. As shown in Figure 3A, lysate pretreatment increased DC production of IL-10 after LPS (statistically significant over more than 10 independent experiments), although the absolute levels remained low, especially for BALB/c DC. A detectable increase in IL-10 mRNA transcription was also seen in lysate-pulsed C57BL/6 DCs (Fig. 3B), though this was not apparent for BALB/c DCs. To determine whether these increased levels of IL-10 were responsible for suppression of IL-12 p70 by lysates, DCs were prepared from IL-10−/− mice (Fig. 3C). When stimulated by LPS, the total IL-12 p70 release was slightly increased from IL-10−/− DCs compared with controls; however, the relative inhibition produced by freeze-thaw lysates was unchanged. Hence, although enhanced IL-10 production by lysate-pulsed DCs may account for other immunosuppressive effects, it is not responsible for the observed inhibition of IL-12 p70. Furthermore, the production of transforming growth factor (TGF)-β by DCs was not increased after lysate exposure, suggesting that it too is unrelated to IL-12 suppression (data not shown).
FIGURE 3.

Lysate preexposure increases the production of IL-10 from DCs in response to LPS, but this does not suppress IL-12 p70. A, DCs, preincubated with autologous lysates (B indicates B16.OVA; C, CT26; M, MC38) or PBS control, were stimulated overnight with LPS (100 ng/mL) or PBS control and the supernatants assayed by ELISA for IL-10. Representative of more than 10 independent experiments (* = P<0.001 compared with no lysate control). B, RT-PCR to assess transcription of the IL-10 gene was performed on similarly treated DCs (5 h post-LPS). C, Similar experiments using DCs from IL-10−/− mice failed to show any significant increase in IL-12 p70 production after lysate preexposure and LPS-induced maturation. Representative of 2 independent experiments. Error bars denote SD of triplicate repeats within the individual experiment shown. DC indicates dendritic cell; ELISA, enzyme-linked immunosorbent assay; IL, interleukin; LPS, lipopolysaccharide; PBS, phosphate-buffered saline; RT-PCR, reverse transcriptase-polymerase chain reaction.
Reduced IL-12 p70 Release by Lysate-pulsed DCs is Associated With Impaired p38 MAPK Signaling
As IL-12 is central to the effective priming of a TH1 antitumor response34 and is profoundly suppressed (in terms of both transcription and secretion) in lysate-pulsed DCs (Figs. 1C, F), we continued to focus on potential mechanisms underlying IL-12 inhibition. Given that enhanced IL-10 secretion was not responsible, we next examined 2 key intracellular pathways involved in DC IL-12 production, nuclear factor (NF)-κB, and p38 MAPK.46 Double knockout mice for the NF-κB subunits p50 and cRel are unable to make IL-12 p70 in response to LPS,47 whereas MKK3−/− mice with impaired p38 MAPK activity display similar inhibition of IL-12 p70 release from both DCs and macrophages.48 Using IκB-α degradation as a surrogate marker of NF-κB pathway activation in LPS-activated DCs,49 we found no evidence of differences between lysate-pulsed and control DCs (Fig. 4A). This lack of effect on NF-κB activation was also shown by similar experiments looking at the migration of the p50 subunit of NF-κB into the nucleus, after activation by LPS+/− lysate, which showed no difference between lysate pulsed and unpulsed DCs (data not shown). To investigate the p38 MAPK pathway, a kinase activity assay was used that immunoprecipitates phospho-p38 MAPK and determines its ability to phosphorylate a substrate, ATF-2. In contrast to the NF-κB pathway, there was clear evidence that the activity of p38 MAPK was inhibited after lysate exposure in all the 3 tumor models (Fig. 4B). These data suggest that freeze-thaw lysates have selective effects on specific intracellular signaling pathways involved in the release of inflammatory mediators by DCs.
FIGURE 4.
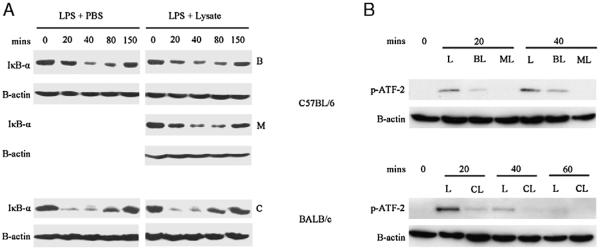
Effect of lysates on LPS-induced activation of NF-κB pathways and p38 MAPK activity. DCs were exposed to lysate or PBS control, then aliquots were stimulated with LPS (100 ng/mL) at timepoint 0. At the indicated timepoints, the DCs were centrifuged and prepared for immunoblotting. A, The amount of IκB-α was determined in comparison with β-actin control. B, DCs treated in a similar way were harvested and phospho-p38 MAPK immunoprecipitated overnight from 200-mg protein derived from each sample. This was then incubated with ATF-2 substrate and the quantity of phospho–ATF-2 determined by immunoblotting (as an indicator of active phospho-p38 MAPK). β-actin in the original samples was used as a loading control (B indicates B16.OVA; C, CT26; L, LPS; M, MC38). Each figure representative of 3 independent experiments. ATF indicates activating transcription factor; DC, dendritic cell; MAPK, mitogen-activated protein kinase; NF, nuclear factor; LPS, lipopolysaccharide; PBS, phosphate-buffered saline.
Induction of Tumor Cell Stress to Reverse Freeze-thaw Lysate-mediated DC Inhibition
The data presented above suggested that freeze-thaw treatment of tumor cells in vitro, to generate a lysate for antigen loading of DCs, may not be optimal for vaccine preparation. Although such lysates are a clinically practical and attractive source of TAA, they seem to be a poor representation of immunogenic in vivo necrotic cell death. Recent data have shown that heat shocking cells before lysis can enhance immunogenicity.50,51 Furthermore, irradiating cells can also have a variety of beneficial immune effects.52 As HSP70 is a well established danger signal, expressed in vivo by stressed cells, we sought to maximize its production in B16 cells using a combination of heat shock and radiotherapy. This approach effectively uses HSP70 as a surrogate marker for the many inducible changes associated with cellular “stress.” Using a B16 melanoma cell line (B16.hsp70.GFP) in which GFP is driven by the rat HSP70 promoter,28 we analyzed HSP70 induction in vitro after treatment of cells with radiation and/or heat shock. In these experiments, GFP expression, quantified by cell fluorescence, acted as a marker of HSP70 induction and hence the cell’s stress response. Optimal doses of radiation and heat shock alone were determined in preliminary experiments (data not shown). Heat shock led to a substantial, transient rise in B16.hsp70.GFP cell fluorescence, whereas radiation alone induced a smaller, more gradual increase (Fig. 5A). By testing different sequencing, we found that tumor cells harvested 48 hours after heat shock delivered 48 hours after radiation, expressed maximal levels of GFP. Effective induction of HSP70 protein after this treatment was confirmed by Western blot (Fig. 5B).
FIGURE 5.

Optimally stressed B16.OVA cell lysates are less inhibitory to LPS-induced DC maturation. A, Various sequences of irradiation (30 Gy, given first) and heat shock (42°C, 25 min, given immediately or deferred by periods of 24 h) were used to optimize stress in B16.hsp70.GFP cells. Production of GFP acted as a surrogate for HSP70 production. Representative of duplicate experiments. B, Production of high levels of HSP70 was confirmed in such cells by immunoblotting. C, Lysates of B16.OVA cells treated in this way were compared with control lysates in their ability to affect LPS-induced phenotypic maturation (expressed as % of expression relative to LPS alone controls) over n experiments (* = P<0.05 paired t test) where each line represents a separate experiment. D, Similar results were obtained for the production of IL-12 p70 (HX = lysate produced from cells optimally heated and irradiated). DC indicates dendritic cell; GFP, green fluorescent protein; HSP, heat shock protein; IL, interleukin; LPS, lipopolysaccharide.
Next, DCs were pulsed with freeze-thaw lysates prepared from tumor cells treated with this sequence of radiation/heat shock to assess whether this influenced their interaction with DCs compared with control lysates. Induction of cell stress significantly, though incompletely, reversed lysate-mediated inhibition of both phenotypic activation (Fig. 5C) and IL-12 production (Fig. 5D) by LPS-treated DCs. Hence, lysates prepared from cells after activation of a tumor cell stress response, associated with maximal HSP70 induction, are less suppressive to DC function.
To address whether these effects were the result of increased HSP70 alone, experiments were conducted adding recombinant human HSP70 to control lysates (the murine form is not commercially available). HSP70 addition was unable to reproduce the reversal of lysate-induced DC inhibition shown in Figure 5 (data not shown), either reflecting the importance of species-specific HSP70 or, more likely, the need for other/multiple danger signals induced by the stress response to reverse DC suppression.
DCs Pulsed With Freeze-thaw Lysates Derived From Stressed, but not Control, Tumor Cells Delay Tumor Growth in Vivo
To investigate the in vivo significance of stressing cells before lysate production, experiments were performed to determine if animals could be protected against a tumor challenge by 2 prior DC vaccinations. Experiments were conducted using LPS to mature DCs after lysate loading or OK-432 (which, as described above, is another TLR4 ligand that has the advantage of being suitable for clinical application53). In our hands, LPS-matured or OK-432–matured DCs, loaded with unmanipulated freeze-thaw B16.OVA lysates, were unable to protect mice against a live tumor challenge. In contrast, DCs pulsed with maximally stressed lysates significantly delayed tumor progression whether they were matured with LPS or OK-432 (Fig. 6). However, owing to the aggressiveness of the model, the number of animals remaining disease-free remained low, even with stressed lysate. Hence, induction of a stress response in tumor cells in vitro, with its associated reversal of lysate-induced DC suppression, uncovers a therapeutic benefit for lysate-pulsed DC vaccines in vivo.
FIGURE 6.

DCs pulsed with lysates of stressed, but not control, cells protect against tumor challenge in vivo. Mice were vaccinated twice (d1 and d8) with DC (1 × 105 cells/mouse/injection) that were either loaded with control (◆) or stressed (▲) B16.OVA lysates and matured with either (A) LPS (100 ng/mL) or (B) OK-432 (10 μg/mL). Progression-free survival was then recorded after live B16.OVA challenge (1×105 cells/mouse on d15) and compared with PBS control (■) (* indicates P<0.05 using log rank test). DC indicates dendritic cell; LPS, lipopolysaccharide; PBS, phosphate-buffered saline.
DISCUSSION
Pulsing DCs with TAA derived from freeze-thaw autologous tumor cell lysates is an attractive and practical approach to cancer immunotherapy, and has been widely applied in early clinical trials. Although it has been argued that freeze-thaw disruption of cells is nonapoptotic and, therefore, inevitably necrotic, we have argued that clear distinctions must be drawn between different modes of tumor cell death in vitro and in vivo.24 The context of in vivo necrosis is critical, in particular its association with the release and action of potential danger signals such as HSP. Such signals determine the response of sentinel immune effector cells, including DCs, which in turn control the eventual balance between priming of a tolerogenic versus an immunogenic response.
Although a large number of studies have examined the effect of different sorts of tumor cell death on DC activation, very few have considered the effect of dying cells on TLR-mediated DC activation. Stuart et al,17 in 2002, showed that murine DCs, which had phagocytosed apoptotic neutrophils, failed to mature fully with LPS. In particular, CD86 expression and IL-12 p70 release were inhibited. More recently, Idoyaga et al,54 using an almost identical method of DC preparation to our own, demonstrated that irradiated tumor cells (of which approximately 30% to 35% were apoptotic at 24 h) inhibited DC maturation triggered by LPS or poly I:C. In particular, they showed less induction of CD40, CD86, and MHC II expression, with impaired production of IL-12, TNF-α, and IL-6.
Here we have shown for the first time that freeze-thaw cell lysates also possess this property (Fig. 1), with inhibition extending to other TLR ligands such as OK-432 and the combination of poly I:C/CpG. Despite using different model systems, the effects are consistent with the published data cited above, with similar effects on the phenotype and cytokine profile. Interestingly, these comparative effects included maintenance of CD80 expression, leading to an increased ratio of CD80 to CD86 on DCs. Recently, CD86 and CD80 have been shown to differentially affect the function of regulatory T cells (Treg), with CD86 suppressing their activity and CD80 enhancing it.55 Hence, this altered ratio on lysate-pulsed DCs may impair stimulation of an antitumor immune response by supporting Treg function (we are currently investigating this possibility in our laboratory). Further to the work by Idoyaga et al, we also show that lysate loading affects DC interaction with T cells, both nonspecifically (impaired IFN-γ production in an allogeneic mixed lymphocyte reaction—Fig. 2A) and in the context of a specific TAA (diminished activation of B3Z cells by SIINFEKL-pulsed DCs—Fig. 2C).
Various mechanisms have been described by which intact tumor cells suppress DC function, including cytokines (eg, vascular endothelial growth factor, IL-10, TGF-β), ceramide,56 and other tumor-derived lipids.57 However, the inhibition of TLR-mediated DC activation by dead or dying tumor cells is a relatively recent finding, and the complete mechanism remains unclear. In our experiments, we found that (i) lysate was not toxic to DCs (as did Idoyaga et al), (ii) no IL-10 or TGF-β was detected in the lysates themselves, and (iii) TGF-β production by DCs was unaltered by lysate. Although LPS-induced IL-10 production was significantly increased after lysate pulsing (itself a characteristic of tolerogenic DCs58), this was not responsible for IL-12 suppression, as the same IL-12 inhibition was seen when DCs were cultured from IL-10 −/− mice (Fig. 3C).
Focusing further on specific signaling pathways, we have shown for the first time that p38 MAPK activity was suppressed in lysate-pulsed DC, whereas activation of the NF-κB pathway was unaffected (Fig. 4). Along with its importance in the release of IL-12 p70, the p38 MAPK pathway is also known to affect the production of TNF-α and IL-6. This is caused, at least in part, by disruption of a downstream kinase in the p38 pathway (mitogen-activated protein kinase 2, also known as MK2),59,60 which may explain the reduction seen in the release of all 3 of these cytokines in our system. Overall, these changes in p38 signaling, taken together with the differential effect of lysate on IL-10 and IL-12 p70 production, suggest important consequences of using tumor lysates for antigen loading that may interfere with an effective immune response.
If freeze-thaw lysates are suppressive to DC function, it is clinically important to conceive ways in which this suppression may be lifted to allow optimal priming of antitumor immunity on vaccinating patients. A significant body of evidence, from ourselves and other laboratories, has shown that stimulating a stress response in tumor cells, with induction of danger signals such as HSP, can enhance the efficacy of vaccine strategies.50,51,61 We found that activation of a maximal stress response in B16 cells, using HSP70 promoter activity as a surrogate marker, generated lysates that were less inhibitory to DC phenotype and cytokine production in vitro (Figs. 5C, D). We do not claim that HSP70 is the sole cause of this effect; indeed attempts to reproduce reversal of DC inhibition by addition of recombinant human HSP70 to unmanipulated lysates was unsuccessful. However, heat shock/radiation treatment of cells will induce multiple stress-associated proteins that are likely to be involved in enhancing an immune response, including other HSP family members.
Most importantly, although control lysate-pulsed DCs were ineffective as in vivo vaccines in the B16.OVA model, loading with stressed lysates enhanced vaccine immunogenicity using either LPS or OK-432 as a maturation agent (Fig. 6). Hence, in vitro imitation of immunogenic in vivo necrosis is a promising and practical strategy for improving the efficacy of cell lysate-based DC vaccines. Consistent with these data, stressing whole cells with heat shock and irradiation before loading onto DCs produced similar benefits in an in vivo animal model,62 and a highly promising lysate-based clinical cancer vaccine study (which did not include DC loading ex vivo) used tumor cells in which a stress response may have been triggered by IFN-γ before lysis.15
It is notable that there were some quantitative differences observed between the various lysates used in this study. Generally, B16.OVA lysate was less inhibitory than MC38 or CT26 preparations when loaded onto autologous DCs in vitro. However, it is not unusual for different models to vary, and the suppressive effects of tumor on DCs were remarkably consistent both within our own data and compared with the intact tumor cell system employed by Idoyaga et al. It could also be argued that our data uses a relatively high tumor cell:DC ratio of 3 tumor cells to 1 DC. However, this ratio has been commonly used in previous studies,9,10,63-66 and the suppression of DC activation by lysates was also apparent at loading ratios down to 1:1 (data not shown).
There are several questions this study has not yet addressed, which are the focus of ongoing work in our laboratory; the inhibitory factor(s) within the lysate responsible for DC inhibition; the precise mechanism(s) by which the induction of tumor cell stress reverses DC inhibition; the nature/range of stress-induced danger signals involved; and whether induction of cell stress reverses the effects of lysate on the p38 signaling pathway. The mechanism(s) by which stressed lysate-pulsed DCs provide tumor protection in vivo are also currently unclear. B16.OVA was chosen as 1 model in this work to allow tracking of TAA-specific responses. Interestingly, experiments to date have not detected OVA-specific immune priming in mice successfully vaccinated with stressed lysate-pulsed DCs. Moreover, we have not detected cross-presentation of SIINFEKL to B3Z in vitro even when stressed lysates are used to pulse DCs (data not shown). This suggests that protection by stressed lysate-pulsed DCs may be via mechanisms other than priming of specific anti-TAA cytotoxic T lymphocytes (potentially innate activation of eg, natural killer cells). The dependence of therapeutic vaccines on specific immune effector subsets and the consequences of vaccination on Treg populations are also the subject of continuing investigation.
In summary, we have shown that in vitro freeze-thaw of tumor cells generates a lysate which is profoundly suppressive to TLR-mediated DC maturation and function in vitro, and is ineffective as a loading strategy for DC therapy in vivo. However, induction of cell stress before lysate preparation, optimized to replicate the HSP70 production seen in pathologic necrosis, partially reverses DC suppression and enhances immunogenicity. This study highlights potential problems in the use of freeze-thaw lysates as a source on TAAs, suggests novel pathways by which tumor cell lysates may inhibit DC function, and defines how clinically practical tumor cell lysate-based vaccines might be improved for the benefit of patients.
ACKNOWLEDGMENT
The authors thank Dr Sandra Diebold of King’s College, London, for her helpful comments during the preparation of this manuscript.
Supported by Cancer Research UK, Mayo Foundation, and NIH Grants RO1 CA85931 and RO1 CA94180.
Footnotes
Financial Disclosure: The authors have declared there are no financial conflicts of interest in regards to this work.
REFERENCES
- 1.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 2.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 3.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 4.Scheffer SR, Nave H, Korangy F, et al. Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int J Cancer. 2003;103:205–211. doi: 10.1002/ijc.10777. [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann TK, Meidenbauer N, Dworacki G, et al. Generation of tumor-specific T lymphocytes by cross-priming with human dendritic cells ingesting apoptotic tumor cells. Cancer Res. 2000;60:3542–3549. [PubMed] [Google Scholar]
- 6.Goldszmid RS, Idoyaga J, Bravo AI, et al. Dendritic cells charged with apoptotic tumor cells induce long-lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J Immunol. 2003;171:5940–5947. doi: 10.4049/jimmunol.171.11.5940. [DOI] [PubMed] [Google Scholar]
- 7.Parkhurst MR, DePan C, Riley JP, et al. Hybrids of dendritic cells and tumor cells generated by electrofusion simultaneously present immunodominant epitopes from multiple human tumor-associated antigens in the context of MHC class I and class II molecules. J Immunol. 2003;170:5317–5325. doi: 10.4049/jimmunol.170.10.5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dai S, Wan T, Wang B, et al. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin Cancer Res. 2005;11:7554–7563. doi: 10.1158/1078-0432.CCR-05-0810. [DOI] [PubMed] [Google Scholar]
- 9.Fields RC, Shimizu K, Mule JJ. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc Natl Acad Sci USA. 1998;95:9482–9487. doi: 10.1073/pnas.95.16.9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jouanneau E, Poujol D, Gulia S, et al. Dendritic cells are essential for priming but inefficient for boosting antitumor immune response in an orthotopic murine glioma model. Cancer Immunol Immunother. 2005;55:254–267. doi: 10.1007/s00262-005-0040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Yoneyama H, Wang Y, et al. Mobilization of dendritic cell precursors into the circulation by administration of MIP-1alpha in mice. J Natl Cancer Inst. 2004;96:201–209. doi: 10.1093/jnci/djh024. [DOI] [PubMed] [Google Scholar]
- 12.Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4:328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- 13.Holtl L, Zelle-Rieser C, Gander H, et al. Immunotherapy of metastatic renal cell carcinoma with tumor lysate-pulsed autologous dendritic cells. Clin Cancer Res. 2002;8:3369–3376. [PubMed] [Google Scholar]
- 14.Hersey P, Menzies SW, Halliday GM, et al. Phase I/II study of treatment with dendritic cell vaccines in patients with disseminated melanoma. Cancer Immunol Immunother. 2004;53:125–134. doi: 10.1007/s00262-003-0429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jocham D, Richter A, Hoffmann L, et al. Adjuvant autologous renal tumor cell vaccine and risk of tumor progression in patients with renal-cell carcinoma after radical nephrectomy: phase III, randomised controlled trial. Lancet. 2004;363:594–599. doi: 10.1016/S0140-6736(04)15590-6. [DOI] [PubMed] [Google Scholar]
- 16.Voll RE, Herrmann M, Roth EA, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 17.Stuart LM, Lucas M, Simpson C, et al. Inhibitory effects of apoptotic cell ingestion upon endotoxin-driven myeloid dendritic cell maturation. J Immunol. 2002;168:1627–1635. doi: 10.4049/jimmunol.168.4.1627. [DOI] [PubMed] [Google Scholar]
- 18.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 19.Sauter B, Albert ML, Francisco L, et al. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J Exp Med. 2000;191:423–434. doi: 10.1084/jem.191.3.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rovere P, Vallinoto C, Bondanza A, et al. Cutting edge: bystander apoptosis triggers dendritic cell maturation and antigen-presenting function. J Immunol. 1998;161:4467–4471. [PubMed] [Google Scholar]
- 21.Buttiglieri S, Galetto A, Forno S, et al. Influence of drug-induced apoptotic death on processing and presentation of tumor antigens by dendritic cells. Int J Cancer. 2003;106:516–520. doi: 10.1002/ijc.11243. [DOI] [PubMed] [Google Scholar]
- 22.Kotera Y, Shimizu K, Mule JJ. Comparative analysis of necrotic and apoptotic tumor cells as a source of antigen(s) in dendritic cell-based immunization. Cancer Res. 2001;61:8105–8109. [PubMed] [Google Scholar]
- 23.Larmonier N, Merino D, Nicolas A, et al. Apoptotic, necrotic, or fused tumor cells: an equivalent source of antigen for dendritic cell loading. Apoptosis. 2006;11:1513–1524. doi: 10.1007/s10495-006-8765-0. [DOI] [PubMed] [Google Scholar]
- 24.Melcher A, Gough M, Todryk S, et al. Apoptosis or necrosis for tumor immunotherapy: what’s in a name? J Mol Med. 1999;77:824–833. doi: 10.1007/s001099900066. [DOI] [PubMed] [Google Scholar]
- 25.Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. 2002;2:185–194. doi: 10.1038/nri749. [DOI] [PubMed] [Google Scholar]
- 26.Feng H, Zeng Y, Whitesell L, et al. Stressed apoptotic tumor cells express heat shock proteins and elicit tumor-specific immunity. Blood. 2001;97:3505–3512. doi: 10.1182/blood.v97.11.3505. [DOI] [PubMed] [Google Scholar]
- 27.Feng H, Zeng Y, Graner MW, et al. Stressed apoptotic tumor cells stimulate dendritic cells and induce specific cytotoxic T cells. Blood. 2002;100:4108–4115. doi: 10.1182/blood-2002-05-1389. [DOI] [PubMed] [Google Scholar]
- 28.Wysocka A, Krawczyk Z. Green fluorescent protein as a marker for monitoring activity of stress-inducible hsp70 rat gene promoter. Mol Cell Biochem. 2000;215:153–156. doi: 10.1023/a:1026523305294. [DOI] [PubMed] [Google Scholar]
- 29.Cheadle EJ, O’Donnell D, Selby PJ, et al. closely related mycobacterial strains demonstrate contrasting levels of efficacy as antitumor vaccines and are processed for major histocompatibility complex class I presentation by multiple routes in dendritic cells. Infect Immun. 2005;73:784–794. doi: 10.1128/IAI.73.2.784-794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Melcher A, Todryk S, Bateman A, et al. Adoptive transfer of immature dendritic cells with autologous or allogeneic tumor cells generates systemic antitumor immunity. Cancer Res. 1999;59:2802–2805. [PubMed] [Google Scholar]
- 31.Maffei CM, Mirels LF, Sobel RA, et al. Cytokine and inducible nitric oxide synthase mRNA expression during experimental murine cryptococcal meningoencephalitis. Infect Immun. 2004;72:2338–2349. doi: 10.1128/IAI.72.4.2338-2349.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Errington F, Bateman A, Kottke T, et al. Allogeneic tumor cells expressing fusogenic membrane glycoproteins as a platform for clinical cancer immunotherapy. Clin Cancer Res. 2006;12:1333–1341. doi: 10.1158/1078-0432.CCR-05-1113. [DOI] [PubMed] [Google Scholar]
- 33.Somersan S, Larsson M, Fonteneau JF, et al. Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J Immunol. 2001;167:4844–4852. doi: 10.4049/jimmunol.167.9.4844. [DOI] [PubMed] [Google Scholar]
- 34.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 35.Linardakis E, Bateman A, Phan V, et al. Enhancing the efficacy of a weak allogeneic melanoma vaccine by viral fusogenic membrane glycoprotein-mediated tumor cell-tumor cell fusion. Cancer Res. 2002;62:5495–5504. [PubMed] [Google Scholar]
- 36.Napolitani G, Rinaldi A, Bertoni F, et al. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hokey DA, Larregina AT, Erdos G, et al. Tumor cell loaded type-1 polarized dendritic cells induce TH1-mediated tumor immunity. Cancer Res. 2005;65:10059–10067. doi: 10.1158/0008-5472.CAN-05-1692. [DOI] [PubMed] [Google Scholar]
- 38.Wells JW, Darling D, Farzaneh F, et al. Influence of interleukin-4 on the phenotype and function of bone marrow-derived murine dendritic cells generated under serum-free conditions. Scand J Immunol. 2005;61:251–259. doi: 10.1111/j.1365-3083.2005.01556.x. [DOI] [PubMed] [Google Scholar]
- 39.Yamanaka R, Homma J, Yajima N, et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res. 2005;11:4160–4167. doi: 10.1158/1078-0432.CCR-05-0120. [DOI] [PubMed] [Google Scholar]
- 40.Strome SE, Voss S, Wilcox R, et al. Strategies for antigen loading of dendritic cells to enhance the antitumor immune response. Cancer Res. 2002;62:1884–1889. [PubMed] [Google Scholar]
- 41.Galea-Lauri J, Wells JW, Darling D, et al. Strategies for antigen choice and priming of dendritic cells influence the polarization and efficacy of antitumor T-cell responses in dendritic cell-based cancer vaccination. Cancer Immunol Immunother. 2004;53:963–977. doi: 10.1007/s00262-004-0542-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casey DG, Lysaght J, James T, et al. Heat shock protein derived from a non-autologous tumor can be used as an anti-tumor vaccine. Immunology. 2003;110:105–111. doi: 10.1046/j.1365-2567.2003.01726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li M, Davey GM, Sutherland RM, et al. Cell-associated ovalbumin is cross-presented much more efficiently than soluble ovalbumin in vivo. J Immunol. 2001;166:6099–6103. doi: 10.4049/jimmunol.166.10.6099. [DOI] [PubMed] [Google Scholar]
- 44.Koch F, Stanzl U, Jennewein P, et al. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J Exp Med. 1996;184:741–746. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loscher CE, Draper E, Leavy O, et al. Conjugated linoleic acid suppresses NF-kappaB activation and IL-12 production in dendritic cells through ERK-mediated IL-10 induction. J Immunol. 2005;175:4990–4998. doi: 10.4049/jimmunol.175.8.4990. [DOI] [PubMed] [Google Scholar]
- 46.Ma X, Trinchieri G. Regulation of interleukin-12 production in antigen-presenting cells. Adv Immunol. 2001;79:55–92. doi: 10.1016/s0065-2776(01)79002-5. [DOI] [PubMed] [Google Scholar]
- 47.Ouaaz F, Arron J, Zheng Y, et al. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002;16:257–270. doi: 10.1016/s1074-7613(02)00272-8. [DOI] [PubMed] [Google Scholar]
- 48.Lu HT, Yang DD, Wysk M, et al. Defective IL-12 production in mitogen-activated protein (MAP) kinase 3 (Mkk3)-deficient mice. Embo J. 1999;18:1845–1857. doi: 10.1093/emboj/18.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang JS, Feng WG, Li CL, et al. NF-kappa B regulates the LPS-induced expression of interleukin 12 p40 in murine peritoneal macrophages: roles of PKC, PKA, ERK, p38 MAPK, and proteasome. Cell Immunol. 2000;204:38–45. doi: 10.1006/cimm.2000.1690. [DOI] [PubMed] [Google Scholar]
- 50.Qiu J, Li GW, Sui YF, et al. Heat-shocked tumor cell lysate-pulsed dendritic cells induce effective anti-tumor immune response in vivo. World J Gastroenterol. 2006;12:473–478. doi: 10.3748/wjg.v12.i3.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim HS, Choo YS, Koo T, et al. Enhancement of antitumor immunity of dendritic cells pulsed with heat-treated tumor lysate in murine pancreatic cancer. Immunol Lett. 2006;103:142–148. doi: 10.1016/j.imlet.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 52.Hatfield P, Merrick A, Harrington K, et al. Radiation-induced cell death and dendritic cells: potential for cancer immunotherapy? Clin Oncol (R Coll Radiol) 2005;17:1–11. doi: 10.1016/j.clon.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 53.Okamoto M, Furuichi S, Nishioka Y, et al. Expression of toll-like receptor 4 on dendritic cells is significant for anticancer effect of dendritic cell-based immunotherapy in combination with an active component of OK-432, a streptococcal preparation. Cancer Res. 2004;64:5461–5470. doi: 10.1158/0008-5472.CAN-03-4005. [DOI] [PubMed] [Google Scholar]
- 54.Idoyaga J, Moreno J, Bonifaz L. Tumor cells prevent mouse dendritic cell maturation induced by TLR ligands. Cancer Immunol Immunother. 2007;56:1237–1250. doi: 10.1007/s00262-006-0275-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng Y, Manzotti CN, Liu M, et al. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J Immunol. 2004;172:2778–2784. doi: 10.4049/jimmunol.172.5.2778. [DOI] [PubMed] [Google Scholar]
- 56.Kanto T, Kalinski P, Hunter OC, et al. Ceramide mediates tumor-induced dendritic cell apoptosis. J Immunol. 2001;167:3773–3784. doi: 10.4049/jimmunol.167.7.3773. [DOI] [PubMed] [Google Scholar]
- 57.Shurin GV, Shurin MR, Bykovskaia S, et al. Neuroblastoma-derived gangliosides inhibit dendritic cell generation and function. Cancer Res. 2001;61:363–369. [PubMed] [Google Scholar]
- 58.Monti P, Leone BE, Zerbi A, et al. Tumor-derived MUC1 mucins interact with differentiating monocytes and induce IL-10 high IL-12 low regulatory dendritic cell. J Immunol. 2004;172:7341–7349. doi: 10.4049/jimmunol.172.12.7341. [DOI] [PubMed] [Google Scholar]
- 59.Neininger A, Kontoyiannis D, Kotlyarov A, et al. MK2 targets AU-rich elements and regulates biosynthesis of tumor necrosis factor and interleukin-6 independently at different post-transcriptional levels. J Biol Chem. 2002;277:3065–3068. doi: 10.1074/jbc.C100685200. [DOI] [PubMed] [Google Scholar]
- 60.Clark AR, Dean JL, Saklatvala J. Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett. 2003;546:37–44. doi: 10.1016/s0014-5793(03)00439-3. [DOI] [PubMed] [Google Scholar]
- 61.Melcher A, Todryk S, Hardwick N, et al. Tumor immunogenicity is determined by the mechanism of cell death via induction of heat shock protein expression. Nat Med. 1998;4:581–587. doi: 10.1038/nm0598-581. [DOI] [PubMed] [Google Scholar]
- 62.Prasad SJ, Farrand KJ, Matthews SA, et al. Dendritic cells loaded with stressed tumor cells elicit long-lasting protective tumor immunity in mice depleted of CD4+CD25+ regulatory T cells. J Immunol. 2005;174:90–98. doi: 10.4049/jimmunol.174.1.90. [DOI] [PubMed] [Google Scholar]
- 63.Shimizu K, Thomas EK, Giedlin M, et al. Enhancement of tumor lysate- and peptide-pulsed dendritic cell-based vaccines by the addition of foreign helper protein. Cancer Res. 2001;61:2618–2624. [PubMed] [Google Scholar]
- 64.Grolleau A, Misek DE, Kuick R, et al. Inducible expression of macrophage receptor Marco by dendritic cells following phagocytic uptake of dead cells uncovered by oligonucleotide arrays. J Immunol. 2003;171:2879–2888. doi: 10.4049/jimmunol.171.6.2879. [DOI] [PubMed] [Google Scholar]
- 65.Teitz-Tennenbaum S, Li Q, Rynkiewicz S, et al. Radiotherapy potentiates the therapeutic efficacy of intratumoral dendritic cell administration. Cancer Res. 2003;63:8466–8475. [PubMed] [Google Scholar]
- 66.Ito F, Li Q, Shreiner AB, et al. Anti-CD137 monoclonal antibody administration augments the antitumor efficacy of dendritic cell-based vaccines. Cancer Res. 2004;64:8411–8419. doi: 10.1158/0008-5472.CAN-04-0590. [DOI] [PubMed] [Google Scholar]