

Abstract
Background
Common genetic variation is associated with increased risk of common metabolic diseases such as type 1 and type 2 diabetes, and obesity. Increasing experience with genetic association studies has led to an understanding of the large sample sizes required to detect a weak to moderate genetic predisposition to disease, the need to reproduce such associations in independent cohorts, and the statistical criteria required to detect a true association. This approach has been used successfully to identify disease- associated gene variation usually in representative populations of large numbers.
Objective
To review the current understanding of how common genetic variation influences predisposition to, and treatment of, metabolic disease.
Methodology
Review of scientific literature.
Results
While there has been progress in understanding how genetic variation predisposes to diabetes and obesity, and how candidate genes may alter drug response, several caveats limit the interpretation and significance of pharmacogenetic studies published to date: those caveats typically include relatively small numbers of participants and choice of endpoints in determining gene- associated differences in response, which may not be clinically significant or relevant as a biomarker or predictor of a desired clinical effect. The genetic variants studied at a given locus are often limited in number and may not represent a comprehensive map of the region under study.
Conclusions
The pharmacogenetic associations in diabetes and obesity that have been reported to date have had limited impact on the choice of individual treatments. We perceive, however, that this field is in its infancy in these multifactorial metabolic diseases, and with further advances and future drug intervention trials designed in a way that allows a more clear interpretation of the impact of genetic variation on differences in drug response in obesity and diabetes, it is anticipated that pharmacogenetics will have a significant impact on individualizing medical care.
Keywords: common genetic variation, glucagon-like peptide-1, obesity, pharmacogenetics, sulfonylureas, thiazolidinediones, type 1 diabetes, type 2 diabetes
1. Introduction
The characterization and annotation of the human genome, coupled with the development of resequencing and genotyping technologies, have led to an exponential increase in the number of published genetic association studies, reflecting in part the relative ease with which such studies can be performed [1]. Such studies contribute to an understanding of the genetic versus environmental contribution to disease, and the identification and characterization of pathways essential to the pathophysiology of the disease of interest [2]. In an ideal situation, when the gene of interest is pivotal to the development of the disease, identification of the genes (and the proteins they encode) that lead to disease would allow the rational development of pharmacotherapy directed at disease pathogenesis, and potentially lead to disease prevention strategies.
Genetic association studies have proven to be a powerful tool in understanding the pathophysiology of type 1 diabetes, an autoimmune disorder characterized by the breakdown of self-tolerance and immune-mediated destruction of the pancreatic β-cells. The events leading to disease development occur in a variable and unknown period of time prior to disease presentation, and in sites, such as the thymus, peri-pancreatic lymph nodes and within the pancreatic islets; such sites are inaccessible for study in humans. Furthermore, the environmental events that may precipitate, or perhaps accelerate, the disease process are unknown and may differ from person to person. Given these challenges, genetic association studies have contributed significantly, as they have provided strong evidence that this is a disorder of (T-cell) immune regulation [3] and that it shares predisposition with other immune disorders [4].
Obesity strongly predisposes to type 2 diabetes. The recent characterization of a common variant associated with type 2 diabetes that confers diabetes risk by increasing bodyweight supports this notion [5]. Thus, multiple loci conferring predisposition to type 2 diabetes have recently been discovered or confirmed by several independent genome-wide association studies (GWASs) [5–10]. To date, none of these loci has been associated with significant changes in the response to pharmacotherapy.
Our goal is to provide a brief overview of the current treatments for diabetes and obesity, to review what is currently known about the genetic predisposition to diabetes and obesity and how this might potentially influence the response to pharmacotherapy, and to explore evidence of the potential candidate genes that may alter drug responses in diabetes and obesity, independent of their effects on disease predisposition.
Before discussing the data on pharmacogenetics, it is relevant to briefly discuss current treatments, genetic predisposition to these diseases with particular focus on specific genes associated with diabetes and obesity, and how genetic predisposition to disease affects response to pharmacotherapy. For pharmacogenetics to be relevant to the treatment of metabolic disease, its application needs to demonstrably improve on the standard of care. The pharmacogenetic associations in diabetes and obesity that have been reported to date have had limited impact on the choice of individual treatments. We perceive, however, that this field is in its infancy as it relates to multifactorial metabolic disease, and with further advances and with future drug intervention trials designed in a way that allows a more clear interpretation of the impact of genetic variation on differences in drug response in obesity and diabetes, it is anticipated that pharmacogenetics will have a significant impact on individualizing medical care.
The paper will cover the following topics:
Current treatment of metabolic disease. A review of the goals of therapy and current treatment modalities for diabetes and obesity.
Common genetic variation and predisposition to disease. An overview of recent data from GWASs and the association of common genetic variation with metabolic disease.
Monogenic predisposition to metabolic disease and effect on pharmacotherapy. Monogenic metabolic disorders are associated with differential response to pharmacotherapy.
Genetic predisposition to disease affecting response to pharmacotherapy. Disease-associated variation may alter response to pharmacotherapy.
Role of other genetic variations affecting response to pharmacotherapy. Genetic variation may alter drug metabolism/action independent of effects on disease predisposition.
Conclusions.
2. Current treatment of obesity, diabetes and associated metabolic conditions
2.1 Goals of therapy
The main goal of therapy for diabetes is the prevention of complications of the disease: these include microvascular (retinopathy, nephropathy, and neuropathy) and macrovascular sequelae (including ischemic heart disease). The Diabetes Control and Complications Trial (DCCT) demonstrated that ‘tight’ glucose control prevented or delayed retinopathy and nephropathy in people with type 1 diabetes [11]. Similar findings have been reported by the UK Prospective Diabetes Study group (UKPDS) for type 2 diabetes [12,13].
In addition to treatment of hyperglycemia, aggressive therapy of cardiovascular risk factors such as hypertension or hyperlipidemia is usually undertaken in patients with diabetes. Obesity adversely affects cardiovascular risk and also predisposes to type 2 diabetes. Exercise, weight loss and an appropriate diet are important foundations for treatments of diabetes and may allow simpler regimens to be used or enhance glycemic control.
2.2 Insulin secretagogues: sulfonylureas and meglitinides
The sulfonylurea receptor on pancreatic β cells is part of a potassium channel complex that regulates potassium influx into the cell. Binding of a sulfonylurea to this complex leads to exocytosis of insulin [14]. The consequence of this insulin secretion is a suppression of endogenous glucose production and stimulation of glucose disappearance. This leads to lower fasting and postprandial blood glucose concentrations.
Repaglinide and nateglinide are short-acting non-sulfonylurea insulin secretagogues collectively known as meglitinides. Both compounds are rapidly absorbed and rapidly cleared from the circulation so that administration results in a rapid but brief rise in insulin secretion. Like sulfonylureas, meglitinides inhibit ATP-sensitive K+ channels, thereby leading to insulin release; however, they bind to a site distinct from that of conventional sulfonylureas [15].
The ability of islets to secrete insulin determines, at least in part, the response to sulfonylureas. Thus, patients with evidence of severely impaired secretion (fasting glucose > 15 mmol/l, low fasting C-peptide concentrations) or evidence of islet autoimmunity are unlikely to respond adequately to such secretagogue therapy. It has been commonly believed that sulfonylureas may predispose to or directly contribute to β-cell failure; however, a similar rate of secondary β-cell failure with metformin has been observed in the UKPDS trial [16].
2.3 Glucagon-like peptide-1 based therapy
Glucagon-like peptide-1 (GLP-1) is a gut hormone secreted by intestinal L cells in response to meal ingestion. It is a powerful insulin secretagogue [17]. In addition, it inhibits glucagon secretion and delays gastric emptying [18]. The short half-life of active GLP-1 in the circulation (2 – 5 min) reduced its therapeutic potential. Longer-acting GLP-1 receptor agonists, or drugs which raise concentrations of endogenous GLP-1, have been developed for clinical use.
Exenatide is a naturally occurring analog of GLP-1 that acts as a GLP-1 receptor agonist and has a half-life of ~ 2 h. In humans, it is a powerful insulin secretagogue, suppresses glucagon secretion, delays gastric emptying and is often accompanied by nausea and weight loss [19].
Native GLP-1 is cleaved at the N-terminus, and is inactivated by a widely distributed peptidase, dipeptidyl-peptidase-4 (DPP-4). Several inhibitors of DPP-4 are being developed and sitagliptin has been approved for clinical use. This class of agents inhibits DPP-4 and the breakdown of endogenous GLP-1, and therefore raises GLP-1 concentrations after meal ingestion. In clinical trials, these compounds have no effect on weight but they have not been associated with nausea, vomiting or other gastrointestinal symptoms [20] or delayed gastric emptying [21].
2.4 Insulin sensitizers: metformin and thiazolidinediones
Metformin improves insulin action in hepatic and extra- hepatic tissues. It may decrease endogenous glucose production by inhibiting glycogenolysis and gluconeogenesis. In muscle, it increases insulin receptor tyrosine kinase activity and increases the number and activity of GLUT-4 transporters. All of these actions enhance glucose disposal [22]. The receptor(s) through which metformin mediates these actions, however, is as yet unknown.
The peroxisome proliferator activated receptor (PPAR) is activated by fatty acids and fatty acid-derived eicosanoids. PPAR exists in three isoforms with different tissue specificity. Activation of PPAR leads to the formation of heterodimers with retinoid-X receptors (RXRs). These bind to genomic PPAR response elements that regulate the transcription of multiple genes. The PPAR-γ isoform is mainly expressed in white and brown adipose tissue, although it is also expressed in cardiac and skeletal muscle and blood vessels.
Thiazolidinediones bind to PPAR-γ. This isoform is important in adipocyte differentiation and also mediates the expression of the glucose transporters GLUT-1 and GLUT-4, thereby increasing glucose uptake by the liver and skeletal muscle [23]. At present two thiazolidinediones are available for clinical use: pioglitazone and rosiglitazone.
2.5 Insulin
Insulin is the mainstay of therapy for people with type 1 diabetes and some patients with type 2 diabetes. Insulin regimens have evolved over the years in attempts to mimic normal endogenous insulin secretion. Different formulations of insulin alter the duration and onset of action by altering the rate of absorption of insulin from the subcutaneous injection site.
2.6 Pharmacotherapy of obesity
2.6.1 Orlistat
Orlistat is a lipase inhibitor that works within the gut to inhibit the digestion and ultimately the absorption of fat. Its use (and efficacy) is limited by its gastrointestinal side effects.
2.6.2 Sibutramine
Sibutramine is a potent inhibitor of serotonin and noradrenaline re-uptake. Its use leads to a centrally mediated decrease in food intake. Several double-blind, placebo- controlled studies have established that the compound produces modest decreases in waist circumference and body fat during treatment [24], and its efficacy is greater when combined with behavioral therapy.
2.6.3 Rimonabant
Rimonabant is a cannabinoid type 1 receptor antagonist which leads to weight loss in obese individuals. It has also been associated with improved glycemic control and improved lipid profiles in obese patients with type 2 diabetes. An increased incidence of depression while on treatment has limited its widespread approval. Like sibutramine, rimonabant is metabolized in the liver [25].
3. Genetic predisposition to metabolic disease
Type 1 diabetes, obesity and type 2 diabetes arise out of complex interactions between genes and the environment. Until less than a century ago, the greatest selective pressures on human survival arose from infection and famine. It is conceivable that genetic variation conferring a survival advantage in responding to (and surviving) infection may predispose individuals to developing autoimmune disease. Similarly, variation that allowed more efficient conservation of energy in situations of food scarcity may predispose individuals to obesity and type 2 diabetes when calories are abundant and physical exertion is optional. In the past 2 years, several studies have enhanced our understanding of the genetic predisposition to these common metabolic diseases.
3.1 Type 2 diabetes
A few, commonly encountered polymorphisms have been associated with an increased risk of type 2 diabetes. Variation in three genes (TCF7L2[26], KCNJ11[27] and PPARG[28]) independently and interactively increase risk of progression from impaired fasting glucose and impaired glucose tolerance to overt diabetes [29].
While variation in KCNJ11 may alter insulin secretion and variation in PPARG may alter insulin action [30], the mechanisms by which the disease-associated variation in TCF7L2 causes diabetes is unknown. Florez et al.[31] reported that two single nucleotide polymorphisms (SNPs) in TCF7L2 (rs7903146 and rs12255372) predicted the progression to diabetes of persons with impaired glucose tolerance who were enrolled in the Diabetes Prevention Program. Over the 3-year period of observation, participants homozygous for the risk-conferring allele were more likely to develop diabetes.
Numerous studies have since shown that greater diabetes risk is conferred by the T allele of rs7903146 than the T allele of rs12255372 [31–34]. Helgason et al.[35] demonstrated rs7903146 to be the variant most strongly associated with disease. More importantly, in a West African cohort, the authors replicated their results, with the T allele of rs7903146 being most strongly associated with disease. Resequencing of the entire linkage disequilibrium block containing rs7903146 did not uncover any additional variants with a stronger association to type 2 diabetes.
The recent publication of several independent GWASs has helped identify several other loci conferring risk of type 2 diabetes. These loci [6–10,36] include the following:
CDKN2A/B, a cyclin-dependent kinase inhibitor.
IGF2BP2, which encodes a binding protein that alters translation of insulin-like growth factor 2 (IGF2).
CDKALI and HHEX.
SLC30A8, which is responsible for the integration of zinc into the insulin granule.
More recently, WFS1 has been associated with type 2 diabetes [37]. This finding has now been independently verified [38]. In addition, TCF2 has been associated with prostate cancer and type 2 diabetes [39,40].
3.2 Diabetes and obesity
One of the loci identified by these studies encodes a gene of unknown function, FTO, whose effect on diabetes predisposition disappears when affected cases and controls are matched for weight. This implies that variation in this gene predisposes individuals to diabetes by increasing weight. An additive association of the variant(s) in FTO with body mass index was demonstrated in 38,759 participants from 13 independent cohorts [5]. Adults homozygous for the risk allele (A – rs9939609) weighed about 3 kg more and had a 1.67-fold increase in obesity risk when compared with those not inheriting a risk allele (homozygous for the T allele). Dina et al.[41] confirmed the association of FTO with obesity when they reported that in 2900 obese children and adults (compared with 5100 controls), a haplotype in FTO was strongly associated with obesity (p = 1.67 × 10−26). At present, little is known about how FTO alters energy balance and predisposition to obesity. A similar association of FTO with obesity was reported by Scuteri et al.[42].
3.3 Type I diabetes
Some loci associated with type 1 diabetes have been shown to predispose individuals to other autoimmune diseases, such as Graves’ disease and multiple sclerosis [43]. Variation in the HLA class II genes is strongly associated with type 1 diabetes predisposition [odds ratio (OR) ~ 7.0]. Variation in the insulin gene, INS, is also associated with type 1 diabetes. Both HLA class II antigens, which play an important part in antigen presentation, and a change in the degree of expression of insulin likely lead to a breakdown in self-tolerance. PTPN22[44] encodes a tyrosine kinase that is an important part of T-cell receptor signaling. CTLA4[43] and IL2RA[45,46] both encode proteins that are modulators of T-cell activation. Diabetes-associated variation in CTLA4 alters the ratio between soluble and membrane-bound forms of CTLA-4. The variants in IL2RA most strongly associated with type 1 diabetes may alter concentrations of soluble CD25 (the protein product of IL2RA), a marker of T-cell activation, but the mechanism by which these variants predispose individuals to breakdown of self-tolerance and type 1 diabetes is unclear.
Other loci identified by GWASs in association with type I diabetes include IFN-1 and loci on chromosomes 12q24, 12q13, 18p11 and 16p13. These loci all have functions related to immune activation and regulation [3]. The risk conferred by all of these genes other than HLA II is relatively small (OR 1.5 – 3 compared with HLA II, OR ~ 7).
4. Monogenic predisposition to metabolic disease and effect on pharmacotherapy
4.1 Monogenic forms of diabetes
Maturity onset diabetes of the young (MODY) is a heterogenous group of disorders where diabetes is inherited as an autosomal dominant trait. MODY manifests in young individuals (typically before age 25) but absolute insulin dependence is not usually a feature initially [47]. Patients with MODY who have mutations in glucokinase or HNF1α respond to insulin secretagogues but not to insulin sensitizers [48]. In contrast, patients with mutations in HNF1β do not respond to sulfonylureas and rapidly require treatment with exogenous insulin [49].
Such observations provide important insights into how genetics may influence response to pharmacotherapy, although it is unknown whether common genetic variation in MODY genes also influences response to pharmacotherapy in patients with polygenic type 2 diabetes.
4.2 Lipoatrophy or lipodystrophy
These are associated with severe defects in insulin action and hypertriglyceridemia. There is a high incidence of diabetes in affected patients. Some forms of lipodystrophy arise from mutations in genes such as PPARG, LMNA and BSCL2 that are important in adipocyte differentiation and metabolism [50]. There is some evidence that these patients experience significant benefit in glycemic control after addition of thiazolidinediones to their treatment regimen [51].
4.3 Monogenic forms of obesity
The discovery of a patient with genetic leptin deficiency led to the use of recombinant leptin to treat this severe form of monogenic obesity [52]. Unfortunately the use of this hormone to treat polygenic obesity has been less successful [53,54]. Although other monogenic forms of obesity have been identified, these monogenic forms do not appear to confer any alteration on response to treatment, and to date, they appear to have no clinical impact on treatment choice [55].
5. Genetic predisposition to disease affecting response to pharmacotherapy
The metabolic effects of disease-associated polymorphisms in type 2 diabetes have been partly characterized for KCNJ11, PPARG and TCF7L2. The demonstration of a reproducible association of these loci with disease led to studies of their effects on glucose metabolism [56]. Intriguingly, PPARG and KCNJ11 are the sites of action for thiazolidinediones and sulfonylureas, respectively; this has aroused interest in the effect of these variants on response to oral therapy for type 2 diabetes.
A genetic defect producing a global impairment in insulin secretion, in addition to a predisposition to diabetes, will likely alter the response to insulin secretagogues. Since variation in KCNJ11 alters glucose-induced insulin secretion, it follows that the secretory response to sulfonylureas is also impaired, leading to failure of sulfonylurea therapy [57]; however, this has not been true in all studies [58]. In the Diabetes Prevention Program, the same polymorphism also altered response to metformin monotherapy [59], and the mechanism of this alteration in response is unclear given the current understanding of the mechanism of action of metformin.
The product of TCF7L2 is a member of the TCF (transcription factor) family and is therefore an important constituent of the Wnt signaling pathway. This pathway regulates gene expression, cell–cell adhesion and cell cycle control and is initiated when Wnt binds the transmembrane, cysteine-rich Frizzled family of receptors. The resulting receptor kinase action ultimately allows β-catenin, the main effector of the signaling pathway, to accumulate in the cytoplasm and nucleoplasm.
Within the nucleus, β-catenin heterodimerizes with one of four TCFs, including TCF7L2 (formerly known as TCF-4) to mediate its regulatory role [60]. The incretin hormone GLP-1 results from post-translational processing of the glucagon gene (GCG) product. In the pancreatic α cells, prohormone Convertase-1 (PC-1) calalyzes the synthesis of glucagon, whereas in intestinal L cells and in the brain, PC-2 leads to the synthesis of GLP-1 and GLP-2. Yi et al.[61] have shown that the β-catenin–TCF7L2 heterodimer regulates GCG expression in the intestinal L cells but not in pancreatic α cells. This led to the suggestion that the disease-associated alleles lead to defects in the incretin system and subsequently diabetes [26]. As expected, the disease-associated decrease in insulin secretion impairs the response to glucose, sulfonylureas and other secretagogues [62]. Polymorphisms in TCF7L2 do not alter response to interventions that improve insulin action in glucose-intolerant, obese patients [31].
A polymorphism in the GLP-1 receptor has been shown to alter response to GLP-1 in vitro[63]. To date, variation in GLP1R has not been associated with predisposition to type 2 diabetes. With the availability of GLP-1-based treatment of diabetes, it is relevant to find out whether polymorphisms in GLP1R alter response to GLP-1 receptor agonists or DPP-4 inhibitors, and such studies are keenly awaited.
The common non-synonymous SNP that leads to substitution of a proline for an alanine at position 12 in PPARG (P12A) is associated with type 2 diabetes and alters insulin action. When 131 patients were treated with pioglitazone for 26 weeks, the proportion of patients experiencing a > 15% decrease in glycosylated hemoglobin did not differ between patients homozygous for the disease allele and those carrying the disease-protective allele [64]. The Troglitazone in the Prevention of Diabetes (TRIPOD) study [65] also examined the effect of PPARG P12A on insulin action in response to troglitazone in the study cohort and found no association with response to treatment. A subsequent reanalysis using a dense SNP map of the locus suggested that other variants in PPARG might alter response to troglitazone [66]. Florez et al.[67] examined the effect of these variants together with P12A on the response to troglitazone in the 554 patients originally randomized to this intervention and were unable to reproduce the association(s) reported by the TRIPOD investigators or with the non-synonymous SNP in PPARG.
6. Role of other genetic variations affecting response to pharmacotherapy
Variation in the genes encoding proteins that are responsible for the transport of a drug to its active site, or proteins responsible for drug activation, metabolism and excretion may also influence drug response (Figure 1). Some interesting candidate genes that appear to affect drug responses in diabetes and obesity have emerged. The studies published so far are often small and usually focus on a few variants in a candidate gene rather than undertaking comprehensive genetic analysis of a candidate locus. A few selected examples are discussed below.
Figure 1.

Putative mechanisms of how genetic variation in candidate genes (shaded areas) alters drug action.
6.1 Diabetes
Metformin may mediate most of its (beneficial) actions on glucose metabolism by phosphorylating and therefore activating AMP-activated protein kinase (AMPK), which suppresses glucagon-stimulated glucose production in hepatic cells. In addition, AMPK stimulates glucose uptake by muscle cells [68]. Metformin is a substrate for organic cation transporters (OCTs) including OCT1, which is specifically expressed in the liver [69]. It has been shown that Oct−/− mice have reduced uptake of metformin by the liver [70], and decreased action of metformin on isolated hepatocytes and intact mice [71]. In humans, OCT1 (also known as SLC22A1) is a highly polymorphic gene and stable transfection of 12 non-synonymous OCT1 human variants in stable cell lines showed significant loss of metformin uptake in seven of the 12 cell lines despite similar levels of mRNA expression [71].
In an effort to determine how variation in OCT1 affects response to metformin in humans, Shu et al.[71] studied a 75 g oral glucose tolerance load in non-diabetic human subjects at baseline and after receiving two doses of metformin. Metformin lowered the glycemic excursion (expressed as area under the concentration/time curve, AUC) by 7.5% in individuals with reference alleles that did not alter OCT1 function. In contrast, metformin did not alter AUC in individuals with polymorphisms in OCT1 that decreased uptake of metformin [71]. More comprehensive studies are required to confirm if variation in OCT1 influences response to metformin in people with diabetes.
6.2 Obesity
Sibutramine raises hypothalamic serotonin and noradrenaline concentrations by inhibiting their reuptake; this decreases food intake [72]. There have been three candidate gene studies that explored the association with weight loss outcome in response to sibutramine (Figure 2). See [73] for a complete review.
Figure 2. Variation in the functions of the serotonin transporter protein and of the G protein translation of ligand receptor stimulation.

The literature provides evidence to support the potential for genetic variation to influence the weight loss response to sibutramine treatment and its active M1 and M2 metabolites.
Reproduced from Camilleri [73].
PNMT: Phenylethanolamine N-methyl transferase.
Genetic variation in PNMT, which encodes the enzyme phenylethanolamine N-methyltransferase, responsible for the conversion of noradrenaline to adrenaline, may influence response to this compound [74]. Similarly, variation (C825T) in postreceptor signaling (GNB3, encoding guanine nucleotide binding protein, β polypeptide 3) may also influence weight loss in response to sibutramine in a 1-year treatment trial [75]. Interestingly, the CC genotype was associated with greater weight loss in response to sibutramine, whereas the TC/TT genotype was associated with greater weight loss with placebo.
Variation in several other candidate genes may modulate serotonin reuptake and the function of the α2 adreno-receptors, which are involved in central control of appetite as well as lipolysis [76]. Recently a variant in SLC6A4, which encodes a protein that transports serotonin from the synaptic cleft to the presynaptic neurons, thereby terminating its effect, also influences weight loss in response to sibutramine (Figure 3)[77]. Weight loss was associated with 12 weeks of treatment of obesity in 24 overweight or obese individuals randomized to placebo or sibutramine 10 mg per day. Note the SLC6A4 gene variation influences response to treatment in those in whom the gene promoter includes a short (S) allele in contrast to the response in the presence of homozygosity for the long allele. Lesch et al.[78] demonstrated that the short allele results in functional impairment of the gene and the related transporter protein. Such observations require confirmation in larger studies. If confirmed, detailed resequencing of the locus to determine which variant might alter drug response will be required.
Figure 3. Weight loss associated with 12-week treatment of obesity in 24 overweight or obese individuals randomized to placebo or sibutramine 10 mg per day.
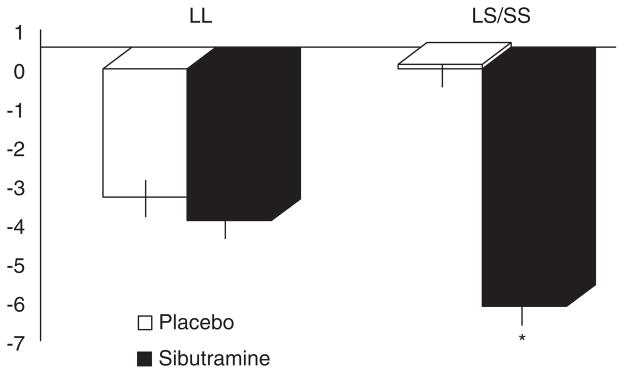
Note the SLC6A4 gene variation influences response to treatment in those in whom the gene promoter includes a short (S) allele in contrast to the response in the presence of homozygosity for the long (L) allele.
*p = 0.024.
Plotted from Vazquez Roque MI et al. [77].
6.3 Hyperlipidemia
Hydroxymethylglutaryl coenzyme A (HMG-CoA) reductase inhibitors are widely used for the treatment of hyperlipidemia and are effective at lowering cholesterol because of their strong inhibition of the rate-limiting enzyme (HGM-CoA reductase) in cholesterol biosynthesis. A significant proportion of patients taking these medications report one or more musculoskeletal side effects [79]: myalgia, myositis and rarely rhabdomyolysis. It is unknown if the side effects are influenced by circulating drug concentrations, conversion to active metabolites, delayed hepatic uptake or delayed clearance of these drugs.
A retrospective study of 68 cases and 69 controls suggested that variation in a candidate gene for drug metabolism (CYP3A5) may alter the degree of creatine kinase (a marker of muscle damage) in atorvastatin-induced myositis [80]. Other genes have been implicated in statin-induced myositis including COQ2, which is involved in the ubiquitylation pathway [81]. Data available to date correlating statin-induced myositis with common genetic variation have been reviewed previously [82]. The conclusions from such studies, however, may not be generalizable because of variable definitions of the clinical syndrome, small numbers and a lack of comprehensive genetic analysis of the candidate loci. A comprehensive genetic analysis of cholesteryl esterase transfer protein (CETP) in 98 patients suggested that some haplotypes may be predictive of response to statin therapy [83].
7. Conclusion
A better understanding of the genetic predisposition to disease may identify targets for future drug development and perhaps individualize prediction of disease risk beyond risk based on conventional criteria. It is theoretically possible that this may lead to intervention at an earlier, preclinical disease stage, and to prevent progression to clinically significant disease. In addition, common genetic variation may alter response to pharmacotherapy irrespective of whether it alters predisposition to disease itself. In the fields of diabetes and obesity, however, the application of pharmacogenetics to individualized therapy and prevention of disease has not yet reached clinical relevance. For the prevention of type 2 diabetes and obesity, lifestyle modification with decreased caloric intake and increased energy expenditure are still the most effective interventions that are generally applicable, and contrast with recommendations based on genetic variation which will be applicable to subpopulations of people with increased predisposition to disease or variation in response to treatment.
We suggest that pharmacogenetics has a potential role in individualizing the prevention and treatment of complex metabolic diseases such as diabetes and obesity. In addition to identifying targets for intervention of new therapeutic agents, the role of genetic variation in affecting clearance or action of a drug should be considered in the design of future studies involving pharmacological interventions. Similarly, the discovery of genetic determinants of heterogeneity in drug response will likely increase significantly in the coming years and could lead to changes in the design of drug studies for metabolic disease, ensuring greater homogeneity between studies and also ensuring that cases and controls have similar predisposition to disease/disease progression or complications of disease. By the same token, these considerations may also significantly impact the choices of therapy in clinical practice for individuals based on genetic variation. Clearly, this field is still in its infancy and well constructed and powered studies are needed to bring the promise of individualized therapy from the conceptual to the clinical and practical level.
8. Expert opinion
All interventions used in the treatment of metabolic disease, be they behavioral or pharmacologic, exhibit individual variation in the magnitude of their effect. Just as common genetic variation affects predisposition to disease, it is likely that variation in genes encoding the proteins responsible for determining drug uptake, metabolism and effect also influence response to these therapies; they may also alter the risk of toxicity associated with such treatment. The desire to provide the optimal and safest treatment to the individual patient, as well as identifying populations that respond better than others to a given intervention, has spurred the science of pharmacogenetics.
It is also theoretically possible that interventions may be applied at an earlier, preclinical disease stage, thus preventing progression to clinically significant disease. At the present time, however, lifestyle modification is a recommendation that can be universally applied to the prevention of type 2 diabetes with little associated risk. In contrast, the interventions for type 1 diabetes presenting prior to insulin dependency have such potential long-term toxicities compared with insulin that genetic prediction of individual disease predisposition, if it were possible at present, would have no clear clinical benefit.
Increasing experience with genetic association studies has led to an understanding of the large sample sizes required to detect a weak to moderate genetic effect on disease predisposition. Given the patterns of linkage disequilibrium in the human genome, multiple variants across a locus will need to be studied to find the true etiologic variant. This progress has not been evident in most pharmacogenetic studies published to date, which are hampered by variable endpoints, small sample sizes and one or very few genetic variants at a given locus tested for association with endpoints, drug effect or adverse events. A review of the literature, however, reveals some progress in understanding how disease-associated variation and candidate genes may alter drug response. For example, variation in KCNJ11 alters insulin secretion, predisposition to type 2 diabetes and may influence sulfonylurea efficacy. Variation in OCT1, however, is not known to influence predisposition to metabolic disease, but by altering delivery of metformin to its putative target, may dramatically influence metformin efficacy.
The discovery of genetic determinants of heterogeneity in drug response will likely increase significantly in the coming years and could alter the design of drug studies for metabolic disease, ensuring greater homogeneity between studies and also ensuring that cases and controls have similar predisposition to disease/disease progression or complications of disease. By the same token, these considerations may also significantly impact on the choices of therapy in clinical practice for individuals based on genetic variation.
Footnotes
Declaration of interest
Adrian Vella receives grant support from Amylin/Lilly and Merck.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (• •) to readers.
- 1.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 2.Todd JA. Statistical false positive or true disease pathway? Nat Genet. 2006;38:731–3. doi: 10.1038/ng0706-731. [DOI] [PubMed] [Google Scholar]
- 3•.Todd JA, Walker NM, Cooper JD, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39:857–64. doi: 10.1038/ng2068. GWAS of type 1 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matesanz F, Caro-Maldonado A, Fedetz M, et al. IL2RA/CD25 polymorphisms contribute to multiple sclerosis susceptibility. J Neurol. 2007;254:682–4. doi: 10.1007/s00415-006-0416-4. [DOI] [PubMed] [Google Scholar]
- 5•.Frayling TM, Timpson NJ, Weedon MN, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–94. doi: 10.1126/science.1141634. Identification of a common genetic variant that alters bodyweight. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6•.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–41. doi: 10.1126/science.1142364. GWAS of type 2 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7•.Sladek R, Rocheleau G, Rung J, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. 2007;445:881–5. doi: 10.1038/nature05616. GWAS of type 2 diabetes. [DOI] [PubMed] [Google Scholar]
- 8•.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–5. doi: 10.1126/science.1142382. GWAS of type 2 diabetes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9•.Saxena R, Voight BF, Lyssenko V, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–6. doi: 10.1126/science.1142358. GWAS of type 2 diabetes. [DOI] [PubMed] [Google Scholar]
- 10•.Steinthorsdottir V, Thorleifsson G, Reynisdottir I, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet. 2007;39:770–5. doi: 10.1038/ng2043. GWAS of type 2 diabetes. [DOI] [PubMed] [Google Scholar]
- 11.The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–86. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 12.Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:837–53. [PubMed] [Google Scholar]
- 13.Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34) UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352:854–65. [PubMed] [Google Scholar]
- 14.Siconolfi-Baez L, Banerji MA, Lebovitz HE. Characterization and significance of sulfonylurea receptors. Diabetes Care. 1990;13(Suppl 3):2–8. doi: 10.2337/diacare.13.3.2. [DOI] [PubMed] [Google Scholar]
- 15.Fuhlendorff J, Rorsman P, Kofod H, et al. Stimulation of insulin release by repaglinide and glibenclamide involves both common and distinct processes. Diabetes. 1998;47:345–51. doi: 10.2337/diabetes.47.3.345. [DOI] [PubMed] [Google Scholar]
- 16.United Kingdom Prospective Diabetes Study 24: a 6-year, randomized controlled trial comparing sulfonylurea, insulin, and metformin therapy in patients with newly diagnosed type 2 diabetes that could not be controlled with diet therapy. United Kingdom Prospective Diabetes Study Group. Ann Intern Med. 1998;128:165–75. doi: 10.7326/0003-4819-128-3-199802010-00001. [DOI] [PubMed] [Google Scholar]
- 17.Kieffer TJ, Habener JF. The glucagon-like peptides. Endocr Rev. 1999;20:876–913. doi: 10.1210/edrv.20.6.0385. [DOI] [PubMed] [Google Scholar]
- 18.Drucker DJ. Development of glucagon-like peptide-1-based pharmaceuticals as therapeutic agents for the treatment of diabetes. Curr Pharm Des. 2001;7:1399–412. doi: 10.2174/1381612013397401. [DOI] [PubMed] [Google Scholar]
- 19.DeFronzo RA, Ratner RE, Han J, et al. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1092–100. doi: 10.2337/diacare.28.5.1092. [DOI] [PubMed] [Google Scholar]
- 20.Ristic S, Byiers S, Foley J, et al. Improved glycaemic control with dipeptidyl peptidase-4 inhibition in patients with type 2 diabetes: vildagliptin (LAF237) dose response. Diabetes Obes Metab. 2005;7:692–8. doi: 10.1111/j.1463-1326.2005.00539.x. [DOI] [PubMed] [Google Scholar]
- 21.Vella A, Bock G, Giesler PD, et al. Effects of dipeptidyl peptidase-4 inhibition on gastrointestinal function, meal appearance, and glucose metabolism in type 2 diabetes. Diabetes. 2007;56:1475–80. doi: 10.2337/db07-0136. [DOI] [PubMed] [Google Scholar]
- 22.Klip A, Leiter LA. Cellular mechanism of action of metformin. Diabetes Care. 1990;13:696–704. doi: 10.2337/diacare.13.6.696. [DOI] [PubMed] [Google Scholar]
- 23.Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54:2460–70. doi: 10.2337/diabetes.54.8.2460. [DOI] [PubMed] [Google Scholar]
- 24.Arterburn DE, Crane PK, Veenstra DL. The efficacy and safety of sibutramine for weight loss: a systematic review. Arch Intern Med. 2004;164:994–1003. doi: 10.1001/archinte.164.9.994. [DOI] [PubMed] [Google Scholar]
- 25.Despres JP, Golay A, Sjostrom L. Effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N Engl J Med. 2005;353:2121–34. doi: 10.1056/NEJMoa044537. [DOI] [PubMed] [Google Scholar]
- 26.Grant SF, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–3. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- 27.Gloyn AL, Weedon MN, Owen KR, et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6. 2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52:568–72. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- 28.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. 2000;26:76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 29.Lyssenko V, Almgren P, Anevski D, et al. Genetic prediction of future type 2 diabetes. PLoS Med. 2005;2:e345. doi: 10.1371/journal.pmed.0020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30•.Hansen SK, Nielsen EM, Ek J, et al. Analysis of separate and combined effects of common variation in KCNJ11 and PPARG on risk of type 2 diabetes. J Clin Endocrinol Metab. 2005;90:3629–37. doi: 10.1210/jc.2004-1942. Characterization of metabolic effects of common variants associated with type 2 diabetes. [DOI] [PubMed] [Google Scholar]
- 31.Florez JC, Jablonski KA, Bayley N, et al. TCF7L2 polymorphisms and progression to diabetes in the Diabetes Prevention Program. N Engl J Med. 2006;355:241–50. doi: 10.1056/NEJMoa062418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Humphries SE, Gable D, Cooper JA, et al. Common variants in the TCF7L2 gene and predisposition to type 2 diabetes in UK European Whites, Indian Asians and Afro-Caribbean men and women. J Mol Med. 2006;84:1–10. doi: 10.1007/s00109-006-0108-7. [DOI] [PubMed] [Google Scholar]
- 33.Damcott CM, Pollin TI, Reinhart LJ, et al. Polymorphisms in the transcription factor 7-like 2 (TCF7L2) gene are associated with type 2 diabetes in the Amish: replication and evidence for a role in both insulin secretion and insulin resistance. Diabetes. 2006;55:2654–9. doi: 10.2337/db06-0338. [DOI] [PubMed] [Google Scholar]
- 34.Saxena R, Gianniny L, Burtt NP, et al. Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes. 2006;55:2890–5. doi: 10.2337/db06-0381. [DOI] [PubMed] [Google Scholar]
- 35.Helgason A, Palsson S, Thorleifsson G, et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet. 2007;39:218–25. doi: 10.1038/ng1960. [DOI] [PubMed] [Google Scholar]
- 36.Tremelling M, Parkes M. Genome-wide association scans identify multiple confirmed susceptibility loci for Crohn’s disease: lessons for study design. Inflamm Bowel Dis. 2007;13:1554–60. doi: 10.1002/ibd.20239. [DOI] [PubMed] [Google Scholar]
- 37.Sandhu MS, Weedon MN, Fawcett KA, et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet. 2007;39:951–3. doi: 10.1038/ng2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Florez JC, Jablonski KA, McAteer J, et al. Testing of diabetes-associated WFS1 polymorphisms in the Diabetes Prevention Program. Diabetologia. 2008;51:451–7. doi: 10.1007/s00125-007-0891-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gudmundsson J, Sulem P, Steinthorsdottir V, et al. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet. 2007;39:977–83. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 40.Winckler W, Weedon MN, Graham RR, et al. Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007;56:685–93. doi: 10.2337/db06-0202. [DOI] [PubMed] [Google Scholar]
- 41.Dina C, Meyre D, Gallina S, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39:724–6. doi: 10.1038/ng2048. [DOI] [PubMed] [Google Scholar]
- 42.Scuteri A, Sanna S, Chen WM, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3:e115. doi: 10.1371/journal.pgen.0030115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Ueda H, Howson JM, Esposito L, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–11. doi: 10.1038/nature01621. Fine mapping helps characterize the functional variant associated with disease. [DOI] [PubMed] [Google Scholar]
- 44.Bottini N, Musumeci L, Alonso A, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36:337–8. doi: 10.1038/ng1323. [DOI] [PubMed] [Google Scholar]
- 45.Vella A, Cooper JD, Lowe CE, et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am J Hum Genet. 2005;76:773–9. doi: 10.1086/429843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lowe CE, Cooper JD, Brusko T, et al. Large-scale genetic fine mapping and genotype–phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet. 2007;39:1074–82. doi: 10.1038/ng2102. [DOI] [PubMed] [Google Scholar]
- 47.Hattersley AT. Molecular genetics goes to the diabetes clinic. Clin Med. 2005;5:476–81. doi: 10.7861/clinmedicine.5-5-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pearson ER, Flechtner I, Njolstad PR, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6. 2 mutations. N Engl J Med. 2006;355:467–77. doi: 10.1056/NEJMoa061759. [DOI] [PubMed] [Google Scholar]
- 49•.Hattersley AT, Pearson ER. Minireview: pharmacogenetics and beyond: the interaction of therapeutic response, beta-cell physiology, and genetics in diabetes. Endocrinology. 2006;147:2657–63. doi: 10.1210/en.2006-0152. Mendelian forms of diabetes mellitus influence therapeutic response to pharmacotherapy. [DOI] [PubMed] [Google Scholar]
- 50.Hegele RA, Joy TR, Al-Attar SA, et al. Thematic review series: Adipocyte biology. Lipodystrophies: windows on adipose biology and metabolism. J Lipid Res. 2007;48:1433–44. doi: 10.1194/jlr.R700004-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Arioglu E, Duncan-Morin J, Sebring N, et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med. 2000;133:263–74. doi: 10.7326/0003-4819-133-4-200008150-00009. [DOI] [PubMed] [Google Scholar]
- 52.Farooqi IS, Jebb SA, Langmack G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–84. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 53.Hukshorn CJ, Saris WH, Westerterp-Plantenga MS, et al. Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB) administration in obese men. J Clin Endocrinol Metab. 2000;85:4003–9. doi: 10.1210/jcem.85.11.6955. [DOI] [PubMed] [Google Scholar]
- 54.Heymsfield SB, Greenberg AS, Fujioka K, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA. 1999;282:1568–75. doi: 10.1001/jama.282.16.1568. [DOI] [PubMed] [Google Scholar]
- 55.Farooqi S, O’Rahilly S. Genetics of obesity in humans. Endocr Rev. 2006;27:710–8. doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- 56.Lyssenko V, Almgren P, Anevski D, et al. Genetic prediction of future type 2 diabetes. PLoS Med. 2005;2:e345. doi: 10.1371/journal.pmed.0020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sesti G, Laratta E, Cardellini M, et al. The E23K variant of KCNJ11 encoding the pancreatic beta-cell adenosine 5′-triphosphate-sensitive potassium channel subunit Kir6. 2 is associated with an increased risk of secondary failure to sulfonylurea in patients with type 2 diabetes. J Clin Endocrinol Metab. 2006;91:2334–9. doi: 10.1210/jc.2005-2323. [DOI] [PubMed] [Google Scholar]
- 58.Gloyn AL, Hashim Y, Ashcroft SJ, et al. Association studies of variants in promoter and coding regions of beta-cell ATP-sensitive K-channel genes SUR1 and Kir6. 2 with type 2 diabetes mellitus (UKPDS 53) Diabet Med. 2001;18:206–12. doi: 10.1046/j.1464-5491.2001.00449.x. [DOI] [PubMed] [Google Scholar]
- 59.Florez JC, Jablonski KA, Kahn SE, et al. Type 2 diabetes-associated missense polymorphisms KCNJ11 E23K and ABCC8 A1369S influence progression to diabetes and response to interventions in the Diabetes Prevention Program. Diabetes. 2007;56:531–6. doi: 10.2337/db06-0966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mulholland DJ, Dedhar S, Coetzee GA, et al. Interaction of nuclear receptors with the Wnt/beta-catenin/Tcf signaling axis: Wnt you like to know? Endocr Rev. 2005;26:898–915. doi: 10.1210/er.2003-0034. [DOI] [PubMed] [Google Scholar]
- 61.Yi F, Brubaker PL, Jin T. TCF-4 mediates cell type-specific regulation of proglucagon gene expression by beta-catenin and glycogen synthase kinase-3beta. J Biol Chem. 2005;280:1457–64. doi: 10.1074/jbc.M411487200. [DOI] [PubMed] [Google Scholar]
- 62.Lyssenko V, Lupi R, Marchetti P, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. 2007;117:2155–63. doi: 10.1172/JCI30706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beinborn M, Worrall CI, McBride EW, et al. A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul Pept. 2005;130:1–6. doi: 10.1016/j.regpep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 64.Bluher M, Lubben G, Paschke R. Analysis of the relationship between the Pro12Ala variant in the PPAR-gamma2 gene and the response rate to therapy with pioglitazone in patients with type 2 diabetes. Diabetes Care. 2003;26:825–31. doi: 10.2337/diacare.26.3.825. [DOI] [PubMed] [Google Scholar]
- 65.Snitker S, Watanabe RM, Ani I, et al. Changes in insulin sensitivity in response to troglitazone do not differ between subjects with and without the common, functional Pro12Ala peroxisome proliferator-activated receptor-gamma2 gene variant: results from the Troglitazone in Prevention of Diabetes (TRIPOD) study. Diabetes Care. 2004;27:1365–8. doi: 10.2337/diacare.27.6.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wolford JK, Yeatts KA, Dhanjal SK, et al. Sequence variation in PPARG may underlie differential response to troglitazone. Diabetes. 2005;54:3319–25. doi: 10.2337/diabetes.54.11.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Florez JC, Jablonski KA, Sun MW, et al. Effects of the type 2 diabetes-associated PPARG P12A polymorphism on progression to diabetes and response to troglitazone. J Clin Endocrinol Metab. 2007;92:1502–9. doi: 10.1210/jc.2006-2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang DS, Jonker JW, Kato Y, et al. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002;302:510–5. doi: 10.1124/jpet.102.034140. [DOI] [PubMed] [Google Scholar]
- 70.Jonker JW, Wagenaar E, Mol CA, et al. Reduced hepatic uptake and intestinal excretion of organic cations in mice with a targeted disruption of the organic cation transporter 1 (Oct1 [Slc22a1]) gene. Mol Cell Biol. 2001;21:5471–7. doi: 10.1128/MCB.21.16.5471-5477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71•.Shu Y, Sheardown SA, Brown C, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117:1422–31. doi: 10.1172/JCI30558. Characterization of the potential effect of common genetic variation in OCT1 on metformin response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weintraub M. Long-term weight control study: conclusions. Clin Pharmacol Ther. 1992;51:642–6. doi: 10.1038/clpt.1992.76. [DOI] [PubMed] [Google Scholar]
- 73.Camilleri M. Pharmacogenomics and serotonergic agents: research observations and potential clinical practice implications. Neurogastroenterol Motil. 2007;19(Suppl 2):40–5. doi: 10.1111/j.1365-2982.2007.00961.x. [DOI] [PubMed] [Google Scholar]
- 74.Peters WR, MacMurry JP, Walker J, et al. Phenylethanolamine N-methyltransferase G-148A genetic variant and weight loss in obese women. Obes Res. 2003;11:415–9. doi: 10.1038/oby.2003.56. [DOI] [PubMed] [Google Scholar]
- 75.Hauner H, Meier M, Jockel KH, et al. Prediction of successful weight reduction under sibutramine therapy through genotyping of the G-protein beta3 subunit gene (GNB3) C825T polymorphism. Pharmacogenetics. 2003;13:453–9. doi: 10.1097/00008571-200308000-00003. [DOI] [PubMed] [Google Scholar]
- 76.Kim HJ, Camilleri M, Carlson PJ, et al. Association of distinct alpha(2) adrenoceptor and serotonin transporter polymorphisms with constipation and somatic symptoms in functional gastrointestinal disorders. Gut. 2004;53:829–37. doi: 10.1136/gut.2003.030882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vazquez Roque MI, Camilleri M, Clark MM, et al. Alteration of gastric functions and candidate genes associated with weight reduction in response to sibutramine. Clin Gastroenterol Hepatol. 2007;5:829–37. doi: 10.1016/j.cgh.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 78.Lesch KP, Bengel D, Heils A, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–31. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]
- 79.McClure DL, Valuck RJ, Glanz M, et al. Systematic review and meta-analysis of clinically relevant adverse events from HMG CoA reductase inhibitor trials worldwide from 1982 to present. Pharmacoepidemiol Drug Saf. 2007;16:132–43. doi: 10.1002/pds.1341. [DOI] [PubMed] [Google Scholar]
- 80•.Wilke RA, Berg RL, Vidaillet HJ, et al. Impact of age, CYP2C9 genotype and concomitant medication on the rate of rise for prothrombin time during the first 30 days of warfarin therapy. Clin Med Res. 2005;3:207–13. doi: 10.3121/cmr.3.4.207. Review of the reported associations between common genetic variants and adverse drug reactions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Oh J, Ban MR, Miskie BA, et al. Genetic determinants of statin intolerance. Lipids Health Dis. 2007;6:7. doi: 10.1186/1476-511X-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wilke RA, Lin DW, Roden DM, et al. Identifying genetic risk factors for serious adverse drug reactions: current progress and challenges. Nat Rev Drug Discov. 2007;6:904–16. doi: 10.1038/nrd2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Winkelmann BR, Hoffmann MM, Nauck M, et al. Haplotypes of the cholesteryl ester transfer protein gene predict lipid-modifying response to statin therapy. Pharmacogenomics J. 2003;3:284–96. doi: 10.1038/sj.tpj.6500195. [DOI] [PubMed] [Google Scholar]