

Abstract
It has been shown that ROS (reactive oxygen species, superoxide and hydrogen peroxide) regulate major epigenetic processes, DNA methylation and histone acetylation, although the mechanism of ROS action (ROS signaling) is still unknown. Both DNA methylation and histone acetylation are nucleophilic processes and therefore ROS signaling through typical free radical processes, for example hydrogen atom abstraction is impossible. However, being “super-nucleophile” superoxide can participate in these reactions. Now we propose new nucleophilic mechanisms of DNA methylation and histone modification. During DNA methylation superoxide can deprotonate the cytosine molecule at C-5 position and by this accelerate the reaction of DNA with the positive-charged intermediate S-adenosyl-L-methionine (SAM). Superoxide can also deprotonate histone N-terminal tail lysines and accelerate the formation of their complexes with acetyl-coenzyme A (AcCoA), the supplier of acetyl groups. In cancer cells ROS enhance DNA methylation causing the silencing of tumor suppressor and antioxidant genes and enhancing the proliferation of cancer cells under condition of oxidative stress. ROS signaling in senescent cells probably causes DNA hypomethylation although there are insufficient data for such proposal.
Keywords: ROS, DNA Methylation, Histone Modification, Nucleophilic Mechanisms
Important role of reactive oxygen species (ROS) signaling in genetic and epigenetic processes is well documented although a mode of their action still remains enigmatic apart from the mechanisms of ROS-induced DNA damage. We and others have already considered the mechanisms of ROS damaging effects on DNA. Although there is uncertainty in the nature of reactive oxygen species involved in DNA damage, only possible candidate is the hydroxyl radical formed by the ferrous ion-catalyzed decomposition of hydrogen peroxide (for example on the “iron fingers” of DNA [1]). (Free hydroxyl radicals are frequently considered to be such culprits but they are not able to achieve a DNA molecule due to their extraordinary reactivity).
However it has now been shown that in addition to DNA damage through the direct attack of free radicals on DNA molecules ROS signaling is the important factor of epigenetic processes such as DNA methylation and histone modification. It makes the role of free radicals and other reactive oxygen species significantly more important in epigenetic and genetic processes. Therefore in this work we are going to consider the possible mechanisms of ROS signaling in epigenetic processes under physiological and pathological (cancer and aging) conditions.
Nucleophilic mechanisms of ros signaling
Typical reactions of most free radicals (paramagnetic species with unpaired electrons) are the reactions of hydrogen atom abstraction. However we have earlier already demonstrated that superoxide anion radical (O2•−) possesses strong nucleophilic properties (being probably the known strongest “supernucleophile”) [2–5]. It will be shown below that the epigenetic processes of DNA methylation and histone modification proceed by nucleophilic mechanisms. Therefore together with diamagnetic molecule hydrogen peroxide (H2O2) superoxide might be the most important signaling species in these nucleophilic reactions.
Important superoxide-induced reactions of nucleophilic substitution are deetherification and deprotonation (Fig. 1). At the time the role of hydrogen peroxide in nucleophilic substitution is not clear. Likewise hydrogen peroxide is able to activate numerous enzymatic processes, but the mechanisms of such processes are also uncertain. It is usually assumed that the principal reaction of hydrogen peroxide is the Fenton reaction – the ferrous ion catalyzed decomposition to hydroxyl radicals, however hydrogen peroxide cannot participate in signaling processes by such a way. We earlier proposed [4] that hydrogen peroxide can be converted into superoxide by a reversible reaction catalyzed by ubiquitous enzyme superoxide dismutase:
Figure 1.
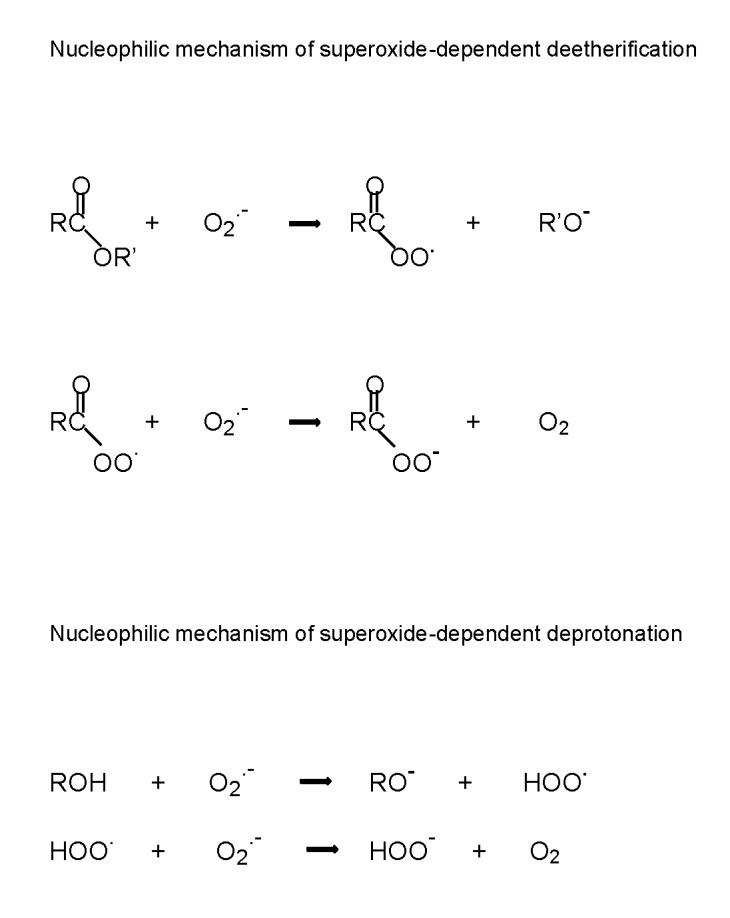
Nucleophilic mechanism of superoxide-dependent deetherification
Therefore it is quite possible that superoxide is also responsible for nucleophilic processes with participation of hydrogen peroxide.
Ros signaling in DNA methylation
ROS signaling has been demonstrated in many studies of DNA methylation under both physiologic and pathologic conditions. (See also recent review by Hayes and Knaus [6]). It is known that DNA methylation is catalyzed by enzymes methyltransferases (DNMT) through the positive-charged intermediate S-adenosyl-L-methionine (SAM). Therefore it is possible that ROS (superoxide) participates in this process as a nucleophile. We will now quote some examples of ROS signaling in DNA methylation.
ROS usually increase DNA methylation. Thus it has been found that the norepinephrine-induced ROS production increased the protein kinase C epsilon (PKCɛ) promoter methylation at Egr-1 and Sp-1 binding sites, initiating PKCɛ gene repression in fetal rat hearts [7]. ROS stimulated CpG methylation of the SP1-binding sites at PKCɛ promoter under hypoxic conditions [8]. DNA hypermethylation of a single CpG dinucleotide close to the transcription start site was found at the early onset of autonomic dysfunction in adult individuals born preterm which associated with enhanced oxidative stress and decreased expression of the SOD2 gene [9].
ROS-induced DNA Methylation in Cancer
However ROS signaling produces probably the greatest effects on DNA methylation in cancer. It may be explained by the fact that cancer cells are characterized by enhanced oxidative stress. (Numerous examples of enhanced oxidative stress in cancer cells are cited in Ref. [10]). It has been shown that ROS-dependent DNA methylation silences some tumor suppressor genes and initiated subsequent tumor progression. For example, prolonged exposure to ROS in hepatocellular carcinoma induced CpG island II methylation on the cadherin promoter [11]. Hypermethylation of the p16 gene promoter leads to the inactivation of tumor suppressor gene p16 and progression from Barrett’s esophagus (BE) to esophageal adenocarcinoma (EA). It was proposed that acid reflux presented in BE patients activated NADPH oxidase NOX5-S and increased ROS production which in turn increased p16 promoter methylation enhancing the progression from BE to EA [12].
It has been shown that catalase levels were reduced in hepatocellular carcinoma (HCC). ROS induced the methylation of CpG island II on the catalase promoter and downregulated catalase expression at HCC transcriptional level. Therefore it was proposed that the development of HCC was caused by ROS downregulation of catalase gene through the methylation of catalase promoter [13]. ROS (hydrogen peroxide) silenced the runt domain transcription factor 3 (RUNX3) through hypermethylation of its promoter leading to the association of RUNX3 with the progression of colorectal cancer [14].
Overexpression of epidermal growth factors ERBB2 and ERBB3 is linked with cancer development, poor prognosis, and drug resistance. Treatment of cancer cells with hydrogen peroxide induced hypermethylation and inhibited miRNAs through DNMT1-mediated DNA methylation. Repression of miR-199a and miR-125b promoters led to the activation of their direct targets ERBB2 and ERBB3 and the induction of tumor growth [15]. Hydrogen peroxide initiated the formation and relocalization of a silencing complex containing DNA methyltransferases, Sirt1, and polycomb members and stimulated the cancer-specific aberrant DNA methylation and transcriptional silencing [16].
Exposure to the particulate matter particles 2.5 μm in diameter (PM (2.5)) increased ROS production and enhanced the expression of methyltransferase 1 (DNMT1). It induced methylation of the p16 promoter in murine alveolar epithelial cells and caused lung cancer development [17]. Examination of the promoter of antioxidant enzyme glutathione peroxide 3 (GPX3) suggested that DNA hypermethylation in gastric cancer lines was induced by deregulation of ROS generation [18]. Hydrogen peroxide treatment increased promoter methylation and silenced homeobox-1(CDX1), a tumor suppressor gene which enhanced the progression of colorectal cancer. Moreover hydrogen peroxide caused the upregulation of DNA methyltransferase 1 (DNMT1) and enhanced the expression and activity of histone deacetylase 1 (HDAC1) [19].
These data demonstrate that ROS stimulate cancer development through DNA hypermethylation. However DNA methylation can also decrease ROS levels in cells through downregulation of NADPH oxidases DUOX1 and DUOX2. In the same way the aberrant hypermethylation of DUOX1 and DUOX2 promoters might lead to the reduction of ROS levels in cancer [20]. ROS formation induced by high glucose also resulted in the significant hypomethylation of CpG dinucleotides in p66Shc promoter [21]. Significant increase in ROS generation decreased histones H3K4 and H3K9 methylation and histone H3 acetylation after insulin treatment [22].
ROS-induced DNA Methylation in Aging
DNA methylation is changed in the age and it is possible that DNA hypomethylation is a typical aspect of aging processes. There are numerous examples of DNA hypomethylation in age. For example it has been shown that aging changes distribution of the product of DNA methylation 5-methylcytosine across the genome that leads to a decrease in global DNA methylation [23,24]. The age-related reduction of global DNA methylation was observed in peripheral leukocytes of female centenarians [25]. It is interesting that aging stimulated global hypomethylation with hypermethylation of specific gene promoters in cancer patients [26].
Unfortunately ROS signaling in DNA methylation during aging processes is not well studied and understood. However it is interesting to look at the effect of caloric restriction (CR) on DNA methylation in the age [27]. It is believed that CR reduces ROS levels and therefore is able to influence life aging and vascular aging by diminishing ROS formation [28]. For example an increase in the methylation of proto-oncogenes in CR-fed rats might be caused by decreasing ROS levels [29]. It should be mentioned that Ford [30] found that the diet containing antioxidant components affected DNA methylation in honeybees and honeybee cells lines with the age. Similarly, methionine dietary restriction increases rat longevity through the reduction of ROS production and genomic DNA methylation in the rat heart [31].
Ros signaling in histone modification
It has been shown that two processes: the acetylation of lysine residues on the N-terminal tails of core histones by histone acetyltransferases (HAT) and the deacetylation of histones by histone deacetylases (HDAC) depend on ROS signaling. Both processes are linked and are very important in chromatin remodeling. However ROS can participate at various stages of histone acetylation/deacetylation by HATs and HDACs or in the competition between HATs and HDACs that creates additional difficulties in the investigation of ROS signaling.
ROS are able to influence histone acetylation directly and indirectly (in the reactions catalyzed by HATs and HDACs). In 2002 Rahman et al. [32] showed that hydrogen peroxide enhanced histone acetylation in alveolar epithelial cells. Later on these authors also demonstrated that co-treatment by hydrogen peroxide and environmental particles with a diameter of <10 μm (PM 10) in human lung alveolar-like epithelial cells enhanced the acetylation of histone 4 (H4) and HAT activity. Enhanced H4 acetylation depended on the level of oxidative stress and was inhibited by thiol antioxidants [33]. Similarly, hydrogen peroxide increased histone H4 acetylation and HAT activation in human cells [34].
Thalidomide-induced ROS formation induced histone H4 acetylation after an increase in the expression of γ-globin gene and the activation of p38 MAPK kinase [35]. ROS produced in endothelial cells by high levels of glucose promoted acetylation of histone H3 [21]. Glucose deprivation-induced oxidative stress led to the accumulation of ROS and the depletion of reduced glutathione (GSH) which inhibited HDAC activity, increased acetylation in miR-466h-5p promoter region, and the activation of miRNA [36]. ROS generated by both mitochondria and NADPH oxidase participated in the ethanol induced acetylation of histone H3 at Lys9 in the primary cultures of rat hepatocytes [37]. Histone acetyltransferase GCN5 controlled superoxide-generating system in leukocytes through the regulation of gp91-phox gene expression. GCN5-depended superoxide production probably increased the acetylation of H2BK16 and H3K9 histones around the promoter of gp91-phox gene [38].
Similarly to the dual effects of ROS on DNA methylation, ROS can also inhibit histone acetylation. For example it was found that cupric ions inhibited histone acetylation by triggering oxidative stress. Histone acetylation was also efficiently suppressed by exogenous hydrogen peroxide and enhanced by SOD and catalase [39]. Likewise, exposure of human hepatoma cells to nickel ions induced ROS generation which inhibited histone acetylation [40]. ROS generation reduced histone H3 acetylation under hyperglycemic condition in preadipocytes [22]. The treatment of human hepatoma cells with curcumin (Cur) (dietary pigment derived from Curcuma longa) led to the significant ROS-dependent HAT-catalyzed decrease at histone acetylation [41]. The HAT inhibitor chaetocin suppressed the trimethylation of histone H3 and induced apoptosis in leukemia cells by a ROS-dependent mechanism [42]. TGF-β1-mediated ROS formation decreased histone acetylation by increasing HDAC-2 activity [43].
There could be various reasons of ROS opposite effects on histone acetylation. For example it has been suggested that enhanced oxidative stress might lead to the imbalance of HAT and HDAC stoichiometry and contribute to the heightened inflammatory response increasing the acetylation of inflammatory gene promoters in the cystic fibrosis (CF) airways [44].
ROS Signaling in Regulation of Transcription Factor Foxo and Sirtuin Histone Deacetylases
Sirtuin histone deacetylases regulate FOXO transcription factors responsible for cell growth, proliferation, and longevity. It has been found that these regulatory processes as a rule depend on ROS signaling. In addition ROS effects on Foxo and HDACs Sirt1 and Sirt2 are frequently linked. In 2002 Korp et al. demonstrated that protein kinase B (Akt) regulated Foxo3a by reducing ROS generation in quiescent cells through an increase in messenger RNA and the MnSOD protein [45]. Similarly Akt activation and hydrogen peroxide are able to upregulate Foxo3a on both transcriptional and protein levels [46]. It was also found that hydrogen peroxide can enhance Foxo reversible acetylation which was inhibited by Sirt2 [47].
Treatment with hydrogen peroxide caused the upregulation and nuclear translocation of FoxO1 in mouse follicular granulosa cells (MGCs) [48]. Activation of Foxo3 induced two sequential ROS waves as a result of Bim-dependent impairment of mitochondrial respiration in neuronal cells with following apoptosis [49]. Induction of oxidative stress in mouse cardiomyocytes promoted Foxo1 and Foxo3 nuclear localization and gene activation. Enhanced Foxo1 or Foxo3 expression reduced ROS formation and diminished cell death. These findings supported a critical role for Foxo1 and Foxo3 in cardiomyocyte survival during oxidative stress and possible effectiveness of antioxidants [50].
As it was mentioned above, Foxo and Sirtuin can interact during various epigenetic modifications. It has been found that the activity of Sirt1 declines with age and that pharmacological activators of Sirt1 confer significant anti-aging cardiovascular effects. Deregulation of Sirt1 resulted in the NADPH oxidase-dependent ROS production and impaired endothelial function [51]. It was also found that ROS induced the formation of cysteine-thiol disulfide–dependent complexes of Foxo and the p300/CBP acetyltransferase. Foxo modulation of p300/CBP-mediated acetylation was fully dependent of the formation of this redox-dependent complex [52]. Deacetylation of Foxo by Sirt1 influenced the Foxo target gene expression, subcellular localization, and protein stability [53]. Sirt1 also protected against emphysema (chronic obstructive pulmonary disease, GOPD) through Foxo3-mediated reduction of cellular senescence [54].
It has been suggested that Sirtuim HDACs might exhibit both oxidant and antioxidant effects at epigenetic modifications including acetylation/deacetylation. For example tumor suppressor histone deacetylase Sirt3 diminished superoxide production through the deacetylation and activation of MnSOD [55]. At the same time hydrogen peroxide treatment increased Sirt2 expression in the cells of caloric-restricted mice. However attaching Sirt2 to Foxo3a reduced its acetylation and decreased ROS production. As a result, mammalian Sirt2 responded to caloric restriction and oxidative stress by the deacetylation of Foxo transcription factors [56].
It is known that the enzymatic activity of MnSOD is regulated by the reversible acetylation of evolutionarily conserved lysines. Mitochondrial anti-aging protein Sirt3 depends on the changes in mitochondrial nutrients or redox status. Sirt3 is able to modify MnSOD activity and change the levels of superoxide production [57]. For example it has been shown that Sirt3 reduces MnSOD-depended superoxide levels by deacetylation of two critical lysine residues on MnSOD. Owing to this Sirt3 can increase antioxidative activity and oxidative stress resistance in cells [58].
The key tumor suppressor gene p53 plays critical role in tumor prevention. It has been shown that p53 can exhibit both prooxidant and antioxidant properties. For example high ROS levels can activate p53 resulting in p53-mediated apoptosis and senescence. On the other hand under conditions of low oxidative stress p53 can reduce ROS levels in cells, prevent oxidative stress-induced DNA damage and promote cell survival [59]. There are numerous data on Sirt1-depended deacetylation of p53 although ROS signaling in this process is not extensively studied. However Xu et al. showed that high-fat diet and exposure to hydrogen peroxide increased p53 lysine-382 acetylation which was inhibited by Sirt1in mice [60].
ROS Signaling in Acetylation and Methylation of Histones and Other Proteins in Aging
ROS-induced DNA methylation in aging has already been considered above. Similarly histone methylation and acetylation can be changed in aging. For example H4K16 hypoacetylation (AcH4K16, the acetylated lysine 16 of histone H4) which is associated with normal aging can contribute to genomic instability by reducing DSB repair [61]. The loss of H3K4 methylation was found in retinoblastoma tumor suppressor gene. H3K4 demethylases Jarid1a and Jarid1b catalyzed H3K4 demethylation contributing to silencing of retinoblastoma target genes in senescent cells. Therefore Jarid1a and Jarid1b are tumor-suppressors controlling cellular senescence [62]. ROS significantly increased the acetylation levels of H4K12 in porcine oocytes during porcine oocyte aging in vitro [63]. Antioxidants such as epigallocatechin-3-O-gallate (EGCG) are apparently able to suppress hydrogen peroxide-induced cellular senescence by reducing p53 acetylation in human dermal fibroblasts [64]. On the other hand resveratrol (3,5,4’-trihydroxy-trans-stilbene, a natural phenol and cancer suppressor) inhibited cancer cell growth and stimulated premature senescence by ROS-dependent DNA interaction [65]. Cellular senescence might be regulated by histone modification of p66She gene. It is possible that epigenetic enhancement of p66Shc depends on the increased histone acetylation and methylation and contributes to cellular replicative senescence or premature senescence [66].
Nucleophilic mechanism of superoxide-dependent epigenetic processes
The quoted above examples of superoxide-dependent DNA methylation and histone modification demonstrate an importance of superoxide signaling in these processes. One principal characteristic of these findings is the possibility of superoxide signaling to influence both the acceleration and suppression of epigenetic modifications. Such a behavior is not typical for free radicals which usually suppress chemical and biochemical reactions by damaging of biomolecules. Therefore we propose that the role of superoxide in epigenetic processes is not a typical for free radicals.
As it was noted above, superoxide is actually a nucleophilic agent and therefore is able to accelerate or decelerate the reactions of hydrolysis and etherification. However there is another important characteristic of superoxide signaling in biochemical processes. Both superoxide and hydrogen peroxide responsible for ROS signaling are free diffusible species which are not principally bound to enzymes or substrates. Therefore biological systems are really saturated with these species which concentrations are changed or unchanged during epigenetic processes (or any other biochemical processes). Correspondingly ROS signaling cannot influence only one stage of a multistage process but ought to affect all of them simultaneously. Of course, ROS are able to participate only on those stages where they can react with some molecules or participate in redox reactions. Nonetheless it is wrong to consider ROS signaling able to influence only one stage of multistage process without affecting the other stages.
DNA Methylation
DNA methylation and histone modification are processes catalyzed by positive charged species. It has been shown that CpG sites, the DNA regulatory sites of gene promoter regions (the sites containing cytosine nucleotides next to guanine nucleotides), are major targets of DNA methylation. The principal intermediate (the catalyst) of the methyltransferase (DNMT)-catalyzed methylation of CpG-cytosines is S-adenosyl-L-methionine (SAM) having a positive charge at the sulfur atom. In 1987 Wu and Santi [67] proposed that the mechanism of DNA methylation catalyzed by (cytosine-5)-DNMT involved the attack on cytosine C5 by the enzyme nucleophile with the subsequent formation of a transient covalent complex. Subsequent works confirmed the formation of the DNA-SAM complex and supported the nucleophilic mechanism of this reaction (Fig.2). It was also proposed that nucleophilic insertion of methyl group at the C5 carbon atom of cytosine molecule can take place only after attachment of negative-charged cysteine residue to C6 carbon in order to supply nucleophilic properties to the C5 position [68,69].
Figure 2.
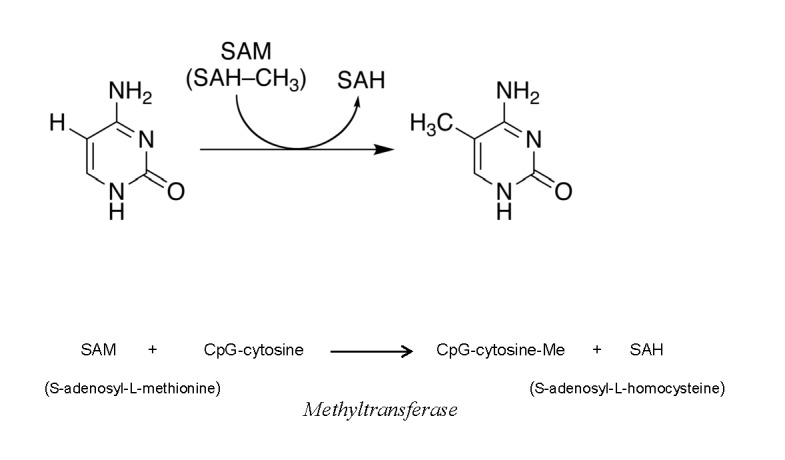
Mechanism of DNA methylation catalyzed by (cytosine-5)-DNMT in which cytosine C5 is attacked by the enzyme nucleophile with the subsequent formation of a transient covalent complex.
The attachment of cysteine residue to the C6 carbon atom makes possible a nucleophilic attack by positive charged S-adenosyl-L-methionine because it converts cytosine molecule into a negatively charged one. Therefore the formation of the DNA-SAM complex containing cysteine residue is the first step of DMNT-catalyzed DNA methylation. However there is another pathway for the SAM nucleophilic attack. As we have mentioned above, a free diffusible superoxide always presents in cellular systems. Therefore we can suggest that the first step of DNA methylation will be the deprotonation by superoxide of cytosine at C5 position. In this case the direct nucleophilic attack of SAM on cytosine molecule with the formation of DNA-SAM complex and its subsequent dissociation and formation of methylcytosine becomes possible (Fig. 3). This new mechanism is not excluded a previous one. It is possible that the competition between two mechanisms might depend on superoxide concentration under physiological or pathological conditions.
Figure 3.
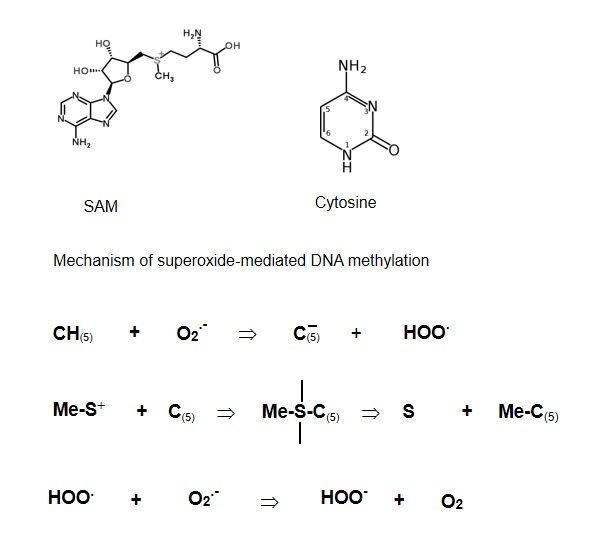
Superoxide-dependent mechanism of DNA methylation.
Histone Modification
During histone modification lysine residues in the N-terminal tails are acetylated and deacetylated via reactions catalyzed by histone acetyltransferases (HAT) and histone deacetylases (HDAC). In these reactions the positively charged protonated lysine residues interact with acetyl-coenzyme A (AcCoA), a supplier of acetyl group (Fig. 4). It has been proposed that similarly to DNA methylation this process proceeds by a nucleophilic mechanism and consists of the stages of protonation, formation of the (lysineNH3+)-AcCoA-HAT complex, and its dissociation to lysine-NHAc [70–72]. As in the case of DNA methylation, it was assumed that the deprotonation of histone molecule is catalyzed by a negatively charged base, the glutamic acid-173 residue of HAT. We now suggest that superoxide can again carry out the function of a negatively charged base (Fig.4).
Figure 4.
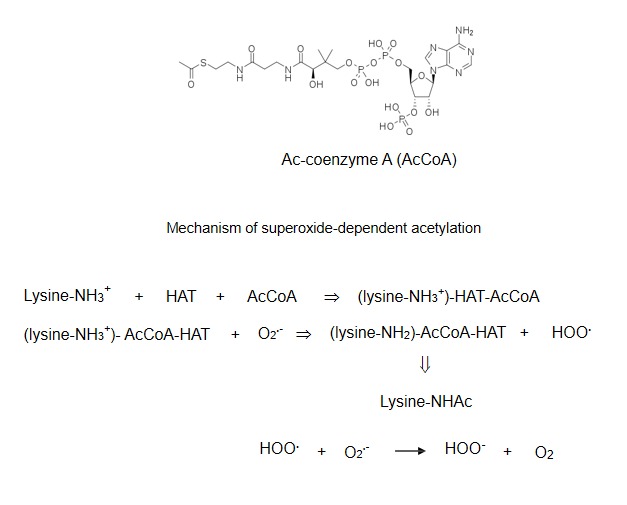
Mechanism of superoxide-dependent histone acetylation
New nucleophilic mechanisms of DNA methylation and histone modification are of course hypothetic ones and need additional experimental evidences. However for the first time these hypotheses are able to explain the role of ROS in epigenetic processes.
Ros signaling in DNA methylation and histone modification in cancer and aging
As it was noted above, major nucleophilic agents, superoxide and hydrogen peroxide are free diffusible molecules which are always formed in cells. Therefore ROS are capable of affecting many stages of epigenetic modifications. It is quite possible that the effects of ROS in the cancer and senescence cells depend on their concentration. Furthermore the change in ROS concentrations under pathological conditions could increase or decrease these epigenetic processes.
It is known that cancer cells exist under the conditions of enhanced oxidative stress [1,10,73]. For this reason ROS effects on epigenetic modifications in cancer cells might be elevated. However it is of an utmost importance that ROS signaling results in silencing tumor suppressor genes and stimulates tumor progression. For example it was shown above that ROS-dependent DNA methylation inactivated tumor suppressor gene p16 [16], downregulated catalase gene in hepatocellular carcinoma through the methylation of catalase promoter [13], stimulated the cancer-specific aberrant DNA methylation and transcriptional silencing [16], and so on. Therefore silencing of tumor suppressor and antioxidant genes through DNA methylation might be the additional important cause of cancer cells survival under the conditions of enhanced oxidative stress (Fig.5).
Figure 5.
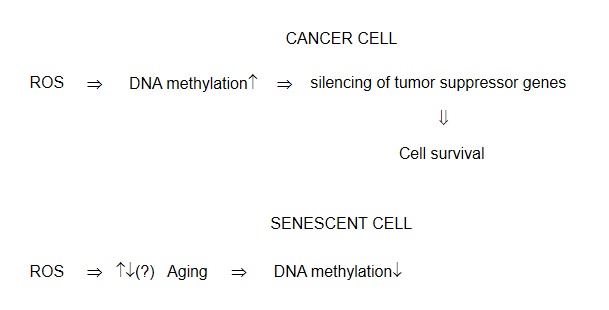
ROS-dependent DNA methylation in cancer and senescent cells
There are numerous data demonstrating an increase in ROS formation in the age (quoted in Ref.74). It is usually believed (the Harmon theory) that enhanced oxidative stress is a major cause of aging development. Is it accompanied by the change in DNA methylation? The above data on the role of reactive oxygen species in DNA methylation in aging are very tentative. It has been shown in some studies [23–25] that ROS stimulated DNA hypomethylation in the age but these data should be confirmed by future works.
ROS generation strongly affects histone modification although its effects can be of opposite directions in different systems. Numerous reasons might be responsible for these discrepancies. Therefore the role of ROS in histone acetylation/deacetylation in cancer and aging remains unclear. Again, additional future experimental data are needed for evaluation of ROS effects in histone modification.
Conclusions
ROS (superoxide and hydrogen peroxide) are the active intermediates of DNA methylation and histone modification. These reactive oxygen species can take part in epigenetic processes by the reactions of nucleophilic substitution. ROS signaling is of utmost importance during cancer development and probably should be taken into account under consideration of aging processes. The participation of ROS in nucleophilic reactions represents the first explanation of their role in epigenetic processes.
References
- [1].Afanas’ev I. Nucleophilic mechanism of ROS/RNS signaling in cancer epigenetic modifications. Am J Biomed Sci. 2012;4:285–306. [Google Scholar]
- [2].Afanas’ev IB. Superoxide Ion: Chemistry and Biological Implications. Vol. 1. CRC Press; Boca Raton, Florida: 1989. pp. 1–279. [Google Scholar]
- [3].Afanas’ev IB. On mechanism of superoxide signaling under physiological and pathophysiological conditions. Med Hypotheses. 2005;64:127–9. doi: 10.1016/j.mehy.2004.05.009. [DOI] [PubMed] [Google Scholar]
- [4].Afanas’ev IB. Competition between superoxide and hydrogen peroxide signaling in heterolytic enzymatic processes. Med Hypotheses. 2006;66:1125–8. doi: 10.1016/j.mehy.2005.11.046. [DOI] [PubMed] [Google Scholar]
- [5].Afanas’ev IB. Signaling functions of free radicals superoxide & nitric oxide under physiological & pathological conditions. Mol Biotechnol. 2007;37:2–4. doi: 10.1007/s12033-007-0056-7. [DOI] [PubMed] [Google Scholar]
- [6].Hayes P, Knaus UG. Balancing reactive oxygen species in the epigenome: NADPH oxidases as target and perpetrator. 2012. Antioxid Redox Signal, Nov 5. [DOI] [PubMed]
- [7].Xiong F, Xiao D, Zhang L. Norepinephrine causes epigenetic repression of PKC {varepsilon} gene in rodent hearts by activating Nox1-dependent reactive oxygen species production. FASEB J. 2012;26:2753–63. doi: 10.1096/fj.11-199422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patterson AJ, Xiao D, Xiong F, Dixon B, Zhang Hypoxia-derived oxidative stress mediates epigenetic repression of PKCɛ gene in foetal rat hearts. Cardiovasc Res. 2012;93:302–10. doi: 10.1093/cvr/cvr322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Nanduri J, Makarenko V, Reddy VD, Yuan G, Pawar A, Wang N, Khan SA, Zhang X, Kinsman B, Peng YJ, Kumar GK, Fox AP, Godley LA, Semenza GL, Prabhakar N. Epigenetic regulation of hypoxic sensing disrupts cardiorespiratory homeostasis. Proc Natl Acad Sci USA. 2012;109:2515–20. doi: 10.1073/pnas.1120600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Afanas’ev I. Reactive oxygen species in cancer: Comparison with aging. Aging & Disease. 2011;2:219–30. [PMC free article] [PubMed] [Google Scholar]
- [11].Lim SO, Gu LM, Kim MS, Kim HS, Park JN, Park CK, Cho JW, Park YM, Jung G. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135:2128–40. doi: 10.1053/j.gastro.2008.07.027. [DOI] [PubMed] [Google Scholar]
- [12].Hong J, Resnick M, Behar J, Wang LJ, Wands J, DeLellis RA, Souza RF, Spechler SJ, Cao W. Acid-induced p16 hypermethylation contributes to development of esophageal adenocarcinoma via activation of NADPH oxidase NOX5-S. Am J Physiol Gastrointest Liver Physiol. 2010;299:G697–706. doi: 10.1152/ajpgi.00186.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Min JY, Lim SO, Jung G. Downregulation of catalase by reactive oxygen species via hypermethylation of CpG island II on the catalase promoter. FEBS Lett. 2010;584:2427–32. doi: 10.1016/j.febslet.2010.04.048. [DOI] [PubMed] [Google Scholar]
- [14].Kang KA, Zhang R, Kim GY, Bae SC, Hyun JW. Epigenetic changes induced by oxidative stress in colorectal cancer cells: methylation of tumor suppressor RUNX3. Tumour Biol. 2012;33:403–12. doi: 10.1007/s13277-012-0322-6. [DOI] [PubMed] [Google Scholar]
- [15].He J, Xu Q, Jing Y, Agani F, Qian X, Carpenter R, Li Q, Wang XR, Peiper SS, Lu Z, Liu Z, Jiang BH. Reactive oxygen species regulate ERBB2 and ERBB3 expression via miR-199a/125b and DNA methylation. EMBO Rep. 2012;13:1116–22. doi: 10.1038/embor.2012.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste, Easwaran H, Casero RA, Sears CL, Baylin SB. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer Cell. 2011;20:606–19. doi: 10.1016/j.ccr.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Soberanes S, Gonsalez, Ulrich D, Chiarella SE, Radigan KA, Osornio-Vargas A, Joseph J, Kalyanaraman B, Ridge KM, Chandel NS, Mutlu GM, De Vizcaya-Ruiz A, Budinger GB. Particular matter air pollution hypermethylation of the 16 promoter via a mitochondrial ROS-JNK-DNMT 1 pathway. Sci Rep. 2012;2:275. doi: 10.1038/srep00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Peng DF, Hu TL, Schneider BG, Chen Z, Xu ZK, El-Rifai W. Silencing of glutathione peroxidase 3 through DNA hypermethylation is associated with lymph node metastasis in gastric carcinomas. PloS One. 2012;7:e46214. doi: 10.1371/journal.pone.0046214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang R, Kang KA, Kim KC, Na SY, Chang WY, Kim GY, Kim HS, Hyun JW. Oxidative stress causes epigenetic alteration SDX1 expression in colorectal cancer cells. Gene. 2013;524:214–9. doi: 10.1016/j.gene.2013.04.024. [DOI] [PubMed] [Google Scholar]
- [20].Luxen S, Belinsky SA, Knaus UG. Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res. 2008;68:1037–45. doi: 10.1158/0008-5472.CAN-07-5782. [DOI] [PubMed] [Google Scholar]
- [21].Paneni F, Mocharla P, Akhmedov A, Constantino S, Osto E, Volpe M, Luscher TF, Cosentino F. Gene silencing of the mitochondrial adaptor p66 (Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res. 2012;111:278–89. doi: 10.1161/CIRCRESAHA.112.266593. [DOI] [PubMed] [Google Scholar]
- [22].Gupta J, Kikoo Involvement of insulin-induced reversible chromatin remodeling in altering the expression of oxidative stress-responsible genes under hyperglycemia in 3T3-L1 preadipocytes. Gene. 2012;504:181–91. doi: 10.1016/j.gene.2012.05.027. [DOI] [PubMed] [Google Scholar]
- [23].Singhal RP, Mays-Hoopes LL, Eichhorn GL. DNA methylation in aging of mice. Mech Ageing Dev. 1987;41:199–210. doi: 10.1016/0047-6374(87)90040-6. [DOI] [PubMed] [Google Scholar]
- [24].Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–40. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- [25].Gentilini D, Mari D, Castaldi D, Remondini D, Ogliari G, Ostan R, Bucci L, Sirchia SM, Tabano S, Cavagnini F, Monti D, Franceschi C, Di Blasio AM, Vitale G. Role of epigenetics in human aging and longevity: genome-wide DNA methylation profile in centenarians and centenatian’s offspring. Age (Dordr) 2012 2012 Aug 25; doi: 10.1007/s11357-012-9463-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kurkjian C, Kummar S, Murgo AJ. DNA methylation: its role in cancer development and therapy. Curr Probl Cancer. 2008;32:187–235. doi: 10.1016/j.currproblcancer.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Li Y, Daniel M, Tollefsbol TO. Epigenetic regulation of caloric restriction in aging. BMC Med. 2011;9:98. doi: 10.1186/1741-7015-9-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ungvari Z, Parrado-Fernandez C, Csiszar A, de Cabo R. Mechanisms underlying caloric restriction and lifespan regulation: implications for vascular aging. Circ Res. 2008;102:519–28. doi: 10.1161/CIRCRESAHA.107.168369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li Y, Daniel M, Tollefsbol TO. Epigenetic regulation of caloric restriction in aging. BMC Med. 2011;9:98. doi: 10.1186/1741-7015-9-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ford D. Honeybeers and cells lines as models of DNA methylation and aging in response to diet. Exp Gerontol. 2012;pii doi: 10.1016/j.exger.2012.07.010. S0531-5565(12) 00200-8. [DOI] [PubMed] [Google Scholar]
- [31].Sanchez-Roman I, Gomez A, Gomez J, Suarez H, Sanchez C, Naudi A, Ayala V, Portero-Otin M, Lopez-Torres M, Pamplona R, Barja G. Forty percent methionine restriction lowers DNA methylation, complex IROS generation, and oxidative damage to mtDNA and mitochondrial proteins in rat heart. J Bioenerg Biomembr. 2011;43:699–708. doi: 10.1007/s10863-011-9389-9. [DOI] [PubMed] [Google Scholar]
- [32].Rahman I, Gilmour PS, Jimenez LA, MacNee W. Oxidative stress and TNF-alpha induce histone acetylation and NF-kappaB/AP-1 activation in alveolar epithelial cells: potential mechanism in gene transcription in lung inflammation. Mol Cell Biochem. 2002;234–235(1–2):239–48. [PubMed] [Google Scholar]
- [33].Gilmour PS, Rahman I, Donaldson K, MacNee W. Histone acetylation regulates epithelial IL-8 release mediated by oxidative stress from environmental particles. Am J Physiol Lung Cell Mol Physiol. 2003;284:L533–40. doi: 10.1152/ajplung.00277.2002. [DOI] [PubMed] [Google Scholar]
- [34].Tomita K, Barnes PJ, Adcock IM. The effect of oxidative stress on histone acetylation and IL-8 release. Biochem Biophys Res Commun. 2003;301:572–7. doi: 10.1016/s0006-291x(02)03029-2. [DOI] [PubMed] [Google Scholar]
- [35].Aerbajinai W, Zhu J, Gao Z, Chin K, Rodgers GP. Thalidomide induces gamma-globin gene expression through increased reactive oxygen species-mediated p38 MAPK signaling and histone H4 acetylation in adult erythropoiesis. Blood. 2007;110:2864–71. doi: 10.1182/blood-2007-01-065201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Druz A, Betenbaugh M, Shiloach J. Glucose depletion activates mmu-miR-4661-5p expression though oxidative stress and inhibition histone deacetylation. Nucleic Acids Res. 2012;40:7291–7302. doi: 10.1093/nar/gks452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Choudhury M, Park PH, Jackson D, Shukla SD. Evidence for the role of oxidative stress in the acetylation of histone H3 by ethanol in rat hepatocytes. Alcohol. 2010;44:531–40. doi: 10.1016/j.alcohol.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kikuchi H, Kuribayashi F, Kiwaki, Takami Y, Nakayama T. GCN5 regulates the superoxide-generating system in leukocytes via controlling gp91-phox gene expression. J Immunol. 2011;186:3015–3022. doi: 10.4049/jimmunol.1000364. [DOI] [PubMed] [Google Scholar]
- [39].Lin C, Kang J, Zheng R. Oxidative stress is involved in inhibition of copper on histone acetylation in cells. Chem Biol Interact. 2005;151:167–76. doi: 10.1016/j.cbi.2005.01.003. [DOI] [PubMed] [Google Scholar]
- [40].Kang J, Zhang Y, Chen J, Chen H, Lin C, Wang Q, Ou Y. Nickel-induced hypoacetylation: the role of reactive oxygen species. Toxicol Sci. 2003;74:279–86. doi: 10.1093/toxsci/kfg137. [DOI] [PubMed] [Google Scholar]
- [41].Kang J, Chen J, Shi Y, Jia J, Zhang Y. Curcumin-induced histone hypoacetylation: the role of reactive oxygen species. Biochem Pharmacol. 2005;69:1205–13. doi: 10.1016/j.bcp.2005.01.014. [DOI] [PubMed] [Google Scholar]
- [42].Chaib H, Nebbioso A, Prebet T, Castellano R, Garbit S, Restouin A, Vey N, Altucci L, Collette Y. Anti-leukemia activity of chaetocin via death receptor-dependent apoptosis and dual modulation of the histone methyltransferase SUV39H1. Leukemia. 2012;26:662–74. doi: 10.1038/leu.2011.271. [DOI] [PubMed] [Google Scholar]
- [43].Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, Lee HB. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am J Physiol Renal Physiol. 2009;297:F729–39. doi: 10.1152/ajprenal.00086.2009. [DOI] [PubMed] [Google Scholar]
- [44].Bartling TR, Drumm ML. Oxidative stress causes IL8 promoter hyperacetylation in cystic fibrosis airway cell models. Am J Respir Cell Mol Biol. 2009;40:58–65. doi: 10.1165/rcmb.2007-0464OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–21. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- [46].Van Gorp AGM, Pomeranz KM, Birkenkamp KU, Hui RC-Y, Lam EW-F, Coffer PJ. Chronic protein kinase B (PKB/c-akt) activation leads to apoptosis induced by oxidative stress–mediated Foxo3a transcriptional up-regulation. Cancer Res. 2006;66:10760–9. doi: 10.1158/0008-5472.CAN-06-1111. [DOI] [PubMed] [Google Scholar]
- [47].Van der Horst A, Tertoolen LGJ, de Vries-Smits LMM, Frye RA, Medema RH, Burgering BMT. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2SIRT1. J Biol Chem. 2004;279:28873–9. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- [48].Shen M, Lin F, Zhang J, Tang Y, Chen W-K, Liu H. Involvement of the up-regulated FoxO1 expression in follicular granulosa cell apoptosis induced by oxidative stress. J Biol Chem. 2012;287:25727–40. doi: 10.1074/jbc.M112.349902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hagenbuchner J, Kuznetsov A, Hermann M, Hausott B, Obexer P, Ausserlechner MJ. FOXO3-induced reactive oxygen species are regulated by BCL2L11 (Bim) and SESN3. J Cell Sci. 2012;125:1191–1203. doi: 10.1242/jcs.092098. [DOI] [PubMed] [Google Scholar]
- [50].Sengupta A, Molkentin JD, Paik J-H, DePinho RA, Yutzey KE. FoxO transcription factors promote cardiomyocyte survival upon induction of oxidative stress. J Biol Chem. 2011;286:7468–78. doi: 10.1074/jbc.M110.179242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zarzuelo MJ, Lopez-Sepulveda R, Sanchez M, Romero M, Gomez-Guzman M, Ungvary Z, Perez-Vizcaino F, Jimenez R, Duarte J. SIRT1inhibits NADPH oxidase activation and protects endothelial functions in the rat aorta: Implications for vascular aging. Biochemical Pharmacol. 2013;85:1288–96. doi: 10.1016/j.bcp.2013.02.015. [DOI] [PubMed] [Google Scholar]
- [52].Dansen TB, Smits LM, van Triest MH, de Keizer PL, van Leenen D, Koerkamp MG, Szypowska A, Meppelink A, Brenkman AB, Yodoi J, Holstege FC, Burgering BM. Redox-sensitive cysteines bridge p300/CBP-mediated acetylation and FoxO4 activity. Nat Chem Biol. 2009;5:664–72. doi: 10.1038/nchembio.194. [DOI] [PubMed] [Google Scholar]
- [53].Oellerich MF, Potente M. FOXOs and sirtuins in vascular growth, maintenance, and aging. Circ Res. 2012;110:1238–51. doi: 10.1161/CIRCRESAHA.111.246488. [DOI] [PubMed] [Google Scholar]
- [54].Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA, McBurney MW, Guarente L, Gu W, Rönty M, et al. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J Clin Invest. 2012;122:2032–45. doi: 10.1172/JCI60132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Tumor suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12:534–41. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–14. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- [57].Miller O, Park SH, KIM HS, Jiang H, Coleman MC, Spitz DR, Gius D. Acetylation of MnSOD directs enzymatic activity responding to cellular nutrition status or oxidative stress. Aging (Albany NY) 2011;3:102–7. doi: 10.18632/aging.100291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab 2010. 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- [59].Feng Z, Lin M, Wu R. The regulation of aging and longevity: a new and complex role of p53. Genes & Cancer. 2011;2:443–52. doi: 10.1177/1947601911410223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Xu S, Jiang B, Shi C, Bachschmid MM, Zang M, Verbeuren tJ, Cohen RA. High-fat diet increases and the polyphenol, S17834, decreases acetylation of the sirtuin-1-dependent lysine-382 on p53 and apoptotic signaling in atherosclerotic lesion-prone aortic endothelium of normal mice. J Cardiovasc Pharmacol. 2011;58:263–271. doi: 10.1097/FJC.0b013e3182239eb7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Krishnan V, Liu B, Zhou Z. “Relax and Repair” to restrain aging”. Aging (Albany NY) 2011;3:943–54. doi: 10.18632/aging.100399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, Garcia BA, Bernstein E, Lowe SW. H3K4 demethylation by Jard1a and Jard1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci USA. 2012;109:897–6. doi: 10.1073/pnas.1119836109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Cui MS, Wang XL, Tang DW, Zhang J, Liu Y, Zeng SM. Acetylation of H4K12 in porcine oocytes during in vitro aging: potential role of ooplasmic reactive oxygen species. Theriogenology. 2011;75:638–46. doi: 10.1016/j.theriogenology.2010.09.031. [DOI] [PubMed] [Google Scholar]
- [64].Han DW, Lee MH, Kim B, Lee JJ, Hyon SH, Park JC. Preventive effects of epigallocatechin-3-o-gallate against replicative senescence associated with p53 acetylation in human dermal fibroblasts. Oxid Med Cell Longev. 2012:13. doi: 10.1155/2012/850684. Article ID 850684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Luo H, Yang A, Schulte BA, Wargovich MJ, Wang GY. Resveratrol induces premature senescence in lung cancer cells via ROS-mediated DNA damage. PLoS One. 2013;8:e60065. doi: 10.1371/journal.pone.0060065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhang W, Ji W, Yang L, Xu Y, Yang J, Zhuang Z. Epigenetic enhancement of p66She during cellular replicative or premature senescence. Toxicology. 2010;278:189–94. doi: 10.1016/j.tox.2010.07.011. [DOI] [PubMed] [Google Scholar]
- [67].Wu LC, Santi DV. Kinetic and catalytic mechanism of HhaI methyltransferase. J Biol Chem. 1987;262:4778–86. [PubMed] [Google Scholar]
- [68].Vilkaitis G, Merkiene E, Serva S, Weinhold E, Klimauskas S. The mechanism of DNA cytosine-5 methylation. Kinetic and mutational dissection of hhai methyltransferase. J Biol Chem. 2001;276:20924–34. doi: 10.1074/jbc.M101429200. [DOI] [PubMed] [Google Scholar]
- [69].Gerasimaite R, Merkiene E, Klimasauskas S. Direct observation of cytosine flipping and covalent catalysis in a DNA methyltransferase. Nucleic Acids Res. 2011;39:377–80. doi: 10.1093/nar/gkq1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Tanner KG, Trievel RC, Kuo M-H, Howard RM, Berger SL, Allis CD, Marmorstein, Denu JM. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J Biol Chem. 1999;274:18157–60. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- [71].Schuetz A, Bernstein G, Dong A, Antoshenko T, Wu H, Loppnau P, Bochkarev A, Plotnikov AN. Crystal structure of a binary complex between human GCN5 histone acetyltransferase domain and acetyl coenzyme A. Proteins. 2007;68:403–7. doi: 10.1002/prot.21407. [DOI] [PubMed] [Google Scholar]
- [72].Jiang J, Lu J, Lu D, Liang Z, Li L, Ouyang S, Kong X, Jiang H, Shen B, Luo C. Investigation of the acetylation mechanism by GCN5 histone acetyltransferase. PLoS One. 2012;7:e36660. doi: 10.1371/journal.pone.0036660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Afanas’ev I. Enhanced free radical status of cancer cells: Success and failure of prooxidant/antioxidant treatment. Current Signal Transduction Therapy. 2012;7:228–36. [Google Scholar]
- [74].Afanas’ev I. Superoxide and nitric oxide in senescence and aging. Front Biosci. 2009;14:3899–912. doi: 10.2741/3499. [DOI] [PubMed] [Google Scholar]