

Abstract
Background
Anopheles sinensis is an important mosquito vector of Plasmodium vivax, which is the most frequent and widely distributed cause of recurring malaria throughout Asia, and particularly in China, Korea, and Japan.
Results
We performed 454 next-generation sequencing and obtained a draft sequence of A. sinensis assembled into scaffolds spanning 220.8 million base pairs. Analysis of this genome sequence, we observed expansion and contraction of several immune-related gene families in anopheline relative to culicine mosquito species. These differences suggest that species-specific immune responses to Plasmodium invasion underpin the biological differences in susceptibility to Plasmodium infection that characterize these two mosquito subfamilies.
Conclusions
The A. sinensis genome produced in this study, provides an important resource for analyzing the genetic basis of susceptibility and resistance of mosquitoes to Plasmodium parasites research which will ultimately facilitate the design of urgently needed interventions against this debilitating mosquito-borne disease.
Keywords: Genome, Anopheles sinensis, Malaria
Background
Malaria is caused by infection with Plasmodium parasites, which are transmitted via the bites of infected female Anopheles mosquitoes [1]. Malaria is prevalent and widely distributed in tropical and subtropical regions, including much of sub-Saharan Africa, Asia, and the Americas [2,3]. Indeed, according to the latest World Malaria Report, in 2010 malaria caused an estimated 216 million clinical episodes and 655,000 deaths worldwide [4]. Of the few available management strategies for this disease, vector control offers an important means of limiting the spread of malaria. The effective control of mosquito vectors, however, requires information on their genetic structure, because the biology and physiology of infections, the development of insecticide resistance, and the epidemiology of malaria in the human host can all be affected by genetic variation in the mosquito vector populations. To date, our understanding of the role of vector genetics in the dynamics of malaria transmission is poor. In particular, the function and evolutionary aspects of important genes, such as those associated with vector competence, remains unclear. The paucity of genetic information on Plasmodium-susceptible mosquitoes is a major obstacle to the development of appropriate diagnostic and therapeutic tools against malaria.
All malaria vectors belong to the subfamily Anophelinae. Mosquitoes of the subfamily Culicinae are not susceptible to infection by Plasmodium parasites and thus, do not transmit Plasmodium. The genomes of A. gambiae, Aedes aegypti and Culex quinquefasciatus were sequenced in 2002, 2007 and 2010, respectively. Comparative genomic studies of these three species have provided important genetic insights into this vector-disease system including the identification of conserved gene regions; the identification of highly diverged genes; recognition of gene families that have expanded or contracted; and the evolution of species-specific physiological or behavioral genetic variations. Nevertheless, information provided by these genome sequences has provided only a limited understanding of the genetic basis of species-specific susceptibility to Plasmodium.
In this study, we sequenced the genome of A. sinensis, a malaria vector within the subfamily Anophelinae. A. sinensis is an Asiatic mosquito species with a wide geographical distribution in East Asia region, ranging from the Philippines to Japan [5]. While A. gambiae is considered to be an efficient vector of P. falciparum[6], A. sinensis is suspected to be the most dominant and important vector of P. vivax[7]. In addition, A. sinensis was found to be solely responsible for the recent outbreaks of malaria in China [8]. Contrasting the genetic composition of these two anopheline mosquitos with that of culicine mosquitos offers a means of investigating the genetic basis of their phenotypic differences to Plasmodium susceptibility, which is a critical step in developing novel ways to reduce human malaria transmission.
Traditional methods of gene detection are costly and time consuming and typically require prior knowledge of target gene regions, as they rely on specific primers. Therefore, these techniques are unsuitable for analyzing large numbers of unknown sequences. The development of next-generation sequencing (NGS) technologies provides an ideal method for rapid and reliable genomic exploration of mosquitoes.
In this study, we employed Roche/454 GS FLX sequencing technology to produce the first genome sequences of A. sinensis. A single-end 454 Jr. run combined with a paired-end 454 Jr. run (3, 8 and 20 Kb libraries) provided a cost-effective solution that produced high quality draft assemblies, and allowed us to obtain detailed gene annotations and meaningful results. Our comparative genomic analyses of the genomes of anopheline and culicine mosquitoes revealed key genetic difference that may underlie important species-specific biological functions in these two groups. This study provides critical genomic information that will pave the way for further in-depth molecular investigations into the biological and vector competency of A. sinensis.
Results and discussion
Sequencing and assembly
We sequenced the whole-genome of A. sinensis using the Roche/454 GS FLX sequencing approach. A total of 5,171,177 single-end reads, 6,302,769 3 Kb mate-pair reads, 2,829,232 8 Kb mate-pair reads and 864,365 20 Kb mate-pair reads were generated (Table 1). After adaptor trimming and low quality reads filtering, a total of 2.7 G single-end sequences and 0.6 G mate-pair sequences were obtained. The genome size of A. sinensis was estimated 267.7 Mb based on K-mer statistics (Table 2), supporting previous estimates of genome size in this mosquito subfamily (230-284 M) [9].
Table 1.
Summary of the raw reads of the sequencing analysis of A. sinensis
| Library | Raw reads | Trimmed reads | Reads used in assembly | Average read length (bp) |
|---|---|---|---|---|
| Single-end |
5,171,177 |
153,380 |
5,017,797 |
383 |
| 3 K paired end |
6,302,769 |
270,609 |
6,032,160 |
205 |
| 8 K paired end |
2,829,232 |
80,660 |
2,748,572 |
207 |
| 20 K paired end | 864,365 | 14,201 | 850,164 | 347 |
Table 2.
Estimated genome size of A. Sinensis based on K-mer analysis
| K-mer value | K-mer number | Depth | Genome size (bp) | Used bases | Used reads |
|---|---|---|---|---|---|
| 13 | 1,874,145,919 | 7 | 267,735,131 | 1,934,235,439 | 5,007,460 |
The whole-genome assembly initially resulted in 9597 scaffolds. After screening for contamination, three scaffolds were identified as putative contaminating sequence of possible bacterial origin and removed (Additional file 1: Table S1). The final 9594 scaffolds spanned 220.8 M with an N50 scaffold size of 814.2 Kb, and contained approximately 82.5% of the A. sinensis genome, based on a genome size of 267.7 Mb. Contig sizes ranged from 65 bp to 357,810 bp, while scaffold sizes ranged from 75 bp to 5,918,260 bp (Table 3). Assembly quality was assessed by aligning the transcripts onto the scaffolds, and 97.5% mapping rate was observed (Additional file 1: Table S2). Assembly quality was also assessed by aligning 454 single reads to the scaffolds. Approximately 99.2% of single 454 data with depth over 3X can be mapped. Further analysis of single nucleotide variants (SNVs) and insertion and deletion (INDEL) variation revealed base error rate was 0.015% and short indel error rate was 0.011%, which supported the high quality of genome assembly (Additional file 1: Table S3). Additionally, analysis of the draft genome assembly for core eukaryotic genes (CEGs) revealed almost all of 458 CEGs (446 out of 458, 97.4%), complete 248 highly conserved CEGs (239 out of 248, 96.4%) and partial 248 highly conserved CEGs (244 out of 248, 98.4%) were found, again confirming the assembly quality of A. sinensis. This Whole Genome project has been deposited at DDBJ/EMBL/GenBank under the accession ATLV00000000. The version described in this paper is version ATLV01000000.
Table 3.
Statistics for the assembly of the A. sinensis genome
| |
Contig |
Scaffold |
||
|---|---|---|---|---|
| Size (bp) | Number | Size (bp) | Number | |
| N90 |
2,384 |
10,962 |
30,600 |
582 |
| N80 |
7,384 |
6,003 |
149,975 |
249 |
| N70 |
13,407 |
3,858 |
338,010 |
149 |
| N60 |
20,357 |
2,558 |
537,812 |
98 |
| N50 |
30,137 |
1,685 |
814,231 |
66 |
| Longest |
357,810 |
|
5,918,260 |
|
| Total Size |
214,524,114 |
|
220,784,734 |
|
| Total Number (>100 bp) |
|
27,488 |
|
9,596 |
| Total Number (>2 Kb) | 12,156 | 2,038 | ||
This genome had a GC percentage of 42.6%, which was higher than the mean GC content in the other three sequenced mosquito species (Table 4). Earlier study suggested that the amount of introns may have an association with differential GC content among these three sequenced mosquito species [10]. Here, we found relatively lower introns in A. sinensis (~32,000 introns) than other three mosquito species (A. gambiae ~ 38,000 introns, A. aegypti ~ 51,000 introns, and C. quinquefasciatus ~ 52,000 introns). This result strongly suggested a negative correlation between the GC content and intron numbers. Haddrill et al. [11] found a strongly negative correlation between intron length and rate of divergence of the genes. Also, a positive correlation between the recombination rate and GC content has been found in many species, such as yeast [12], birds [13], insect [14], plants [15] and mammals [16,17]. Recombination acquire a larger amount of genetic diversity [18]. Both lower intron length (Additional file 1: Table S4) and higher GC content in An. sinensis and An. gambiae may indicate high genetic diversity rate than other two subfamily Culicinae mosquitoes. Interestingly, genetic diversity in the susceptibility to malaria parasites in mosquitoes has been already amply confirmed [19,20]. However, we also recognized this estimate of GC content was susceptible of a non trivial error bar, because nearly 20% of the genome was missing from the draft assembly.
Table 4.
Characteristics of the genomes of A.sinensis , A.gambiae , Ae.aegypti , and C.quinquefasciatus
| A. sinensis | A. gambiae | Ae. aegypti | C. quinquefasciatus | |
|---|---|---|---|---|
| Genome size (Mbps) |
220.8 |
278 |
1376 |
540 |
| Genome coverage (×) |
18.8 |
10.2 |
7.6 |
6.1 |
| Number of contig |
27,497 |
18,962 |
36,206 |
48,671 |
| Number of scaffold |
9,594 |
8,987 |
4,758 |
3,171 |
| Contig N50 (Kbps) |
30.1 |
--- |
82.6 |
28.6 |
| Scaffold N50 (Kbps) |
814.2 |
--- |
1.5 |
486.8 |
| Average contig size (bps) |
7800 |
13878 |
36184 |
--- |
| Average scaffold size (bps) |
23012 |
30930 |
290873 |
--- |
| GC (%) |
42.6 |
40.9 |
38.2 |
37.4 |
| Number of gene (protein-coding) |
16766 |
12457 |
15419 |
18883 |
| Percentage of gene Length (%) |
26.4 |
23.1 |
17.4 |
18.5 |
| Percentage of exon region length (%) | 11.0 | 7.2 | 1.9 | 4.4 |
Repetitive elements analysis
We estimated 15,200,821 nt repetitive elements, which accounted for approximately 6% of the A. sinensis genome. The most abundant of repetitive elements were transposable elements (TEs) or potential TEs (Figure 1). These constituted about 97.9% of the repetitive elements(70.4% potential TEs and 27.5% TE) and 6% of the genome (Figure 1). Of the remaining repetitive elements, 0.5% were unclassified repeats, 0.3% were satellites, 1.2% were simple repeats and 0.1% were low complexity. In all TEs classification, 19% were retroelements (Class I elements), 9% were DNA transposon elements (Class II elements) and 72% were potential TEs. Class I elements consisted of five clades (L2/L3/CR1/Rex, R1/LOA/Jockey, R2/R4/NeSL, RTE/Bov-B and L1/CIN4), while Class II elements also consisted of five clades (hobo-Activator, Tc1-IS630-Pogo, PiggyBac, Tourist/Harbinger and Mirage/P-element/Transib). Three further clades (BEL/Pao, Ty1/Copia and Gypsy/DIRS1) were identified from long interspersed elements (LINEs) and long terminal repeat (LTR) retrotransposon elements.
Figure 1.

Repetitive elements in A. sinensis .
Compared with published mosquito genome sequences, the TEs content of Anophelinae (A. gambiae, 11% to 16%) were far less than Culicinae (Ae. aegypti, 42% to 47%; C. quinquefasciatus, 29%) [21-23]. TEs content could be a leading factor influencing genome size in many species [24,25]. For example, studies have shown that the genome of Ae. aegypti has doubled its size as a result of TEs [22]. Thus, the differences in the genome size of A. sinensis and other mosquito species could in part be due to the accumulation or loss of TEs in the different species.
Gene prediction
Based on homology and de novo predictions, we identified 16,766 protein-coding genes with an average transcript length of 2608 bp, a coding sequence size of 1083 bp and 2.9 exons per gene (Additional file 1: Table S4). Given the high conservation of single-copy orthologs, protein lengths should have a high coherence between A. sinensis and D. melanogaster. We found that A. sinensis proteins exhibited slightly lower than expected concordance values with D. melanogaster (0.92, see Figure 2), but that this concordance value was similar to that reported for the three other sequenced mosquitoes (A. gambiae, 0.92; Ae. Aegypti, 0.93; C. quinquefasciatus, 0.90) [21-23]. This finding indicates that the gene prediction analysis for A. sinensis was robust.
Figure 2.

D. melanogaster protein length plotted against the orthologous protein length for A. sinensis. The red dashed line shows the results of a linear regression. The concordance of the two variables is presented with 95% confidence intervals. Perfect concordance (1.0) would indicate that all points fall on the line where x = y, depicted by the edge of the grey shading.
Although the predicted genome size of A. sinensis was smaller than that of A. gambiae, we found a greater number of predicted genes in the genome of A. sinensis (Table 4). This discrepancy was consistent with the results of the genome-wide analysis that revealed a higher percentage of exon region length and less TEs content in the genome of A. sinensis. Differences in the numbers of predicted genes in these two species may be consequence of species-specific genetic differences that have arisen from biological adaptations to the different environmental challenges faced by these two mosquitoes. However, it is also possible that gene numbers were overestimated in the genome of A. sinensis because of false-positive gene predictions. These can arise because of inaccurate annotation of the automated consensus gene set or because of putative TEs and bacterial contaminates which escaped earlier detection. Overestimation of the number of predicted genes has been reported for other mosquito species [23]. The third possibility is genes were under-prediction in A. gambiae. It was the second insect genome to be sequenced, and like the initial D. melanogaster, the used consortia eliminated most ab initio gene models without comparative or experimental support, which may cause under-prediction. For example, a recent genomic comparative paper on orphan genes in insects does not even include the A. mellifera gene set, and notes that the A. gambiae gene count is abnormally low for orphan genes [26]. Manual examination of the output will be required to assure the accuracy of the predicted genes of A. sinensis found in this study.
In addition to protein-coding genes, we also identified 41 microRNA (miRNA), 348 tRNA and 2017 rRNA genes in the A. sinensis genome (Additional file 1: Table S5; see the Additional file 2: “MiRNA list” for a list of all predicted A. sinensis miRNA target genes and their annotations). At present, 67 miRNAs have been described for A. gambiae, which is almost 1.5 times that found for A. sinensis. Our finding of just 41 miRNA for A. sinensis might be an underestimation as the target prediction was based on an imperfect match between known miRNA and our genomic sequence. It is also possible that some target genes were missed during the alignment due to the differences between the two genomes of these two anopheline species. Another possible factor that may contribute to lower miRNA genes in A. sinensis is that A. sinensis may have unidentified miRNAs.
Functional annotation and gene family analysis
For all predicted protein-coding genes, 93.8% had matches in the non-redundant (NR) databases, 64.6% were similar to entries in the InterPro database, 67.7% were assigned GO terms, 14.2% were mapped to known pathways, 14.0% had signal peptides and 21.4% had transmembrane regions (Additional file 1: Table S6). There were several domains (fibrinogen, protein kinase and six-bladed beta-propeller) and repeats (LDLR class B repeat and Leucine-rich repeat) overrepresented in A. sinensis compared to A. gambiae (Additional file 1: Table S7). However, no significant differences of PTMs were observed between A. sinensis and A. gambiae. Several domains (histone, F-box and Zinc finger) were down-represented in anopheline species compared with the culicine species, though no significant differences of translational modification (PTM) and repeat were observed between these two subfamilies (Additional file 1: Table S8).
We assessed the functional predictions of proteins according to broad GO categories standardized to level 2 terms (Figure 3). Additional GO analysis of the proteomes revealed differences between A. sinensis and A. gambiae (Additional file 1: Table S9), and between the anopheline and the culicine species (Additional file 1: Table S10). In the biological process category, proteins involved with signaling processes (GO:0023052) in were expanded in anopheline species compared with the culicine species (Additional file 1: Table S10). While, in the molecular function category, proteins involved in molecular transducer activity (GO:0060089) was expanded in anopheline species compared with culicine species (Additional file 1: Table S10).
Figure 3.

Gene ontology classifications for A. sinensis at level two. Gene ontology classifications included three components: biological processes, cellular components and molecular functions.
Of the entire A. sinensis gene set, 2377 genes had an ortholog belonging to one of the 235 known biological pathways. There were no significant differences in the mapping of genes to pathways between A. sinensis and A. gambiae or between the anopheline and culicine subfamilies.
The frequencies of transmembrane regions in A. sinensis were relative lower than other three mosquito species (Additional file 1: Table S11). With just three exceptions (6, 7 and 10), protein numbers tended to decrease with the increasing transmembrane helices (Additional file 1: Table S12). InterPro analysis revealed that olfactory receptors (14.29%), G-protein coupled receptors (GPCRs, 34.91%) and major facilitator superfamily domain (16.25%) accounted for the largest proportion of the predicted proteins of the 6, 7 and 10 transmembrane helices, respectively.
The A. sinensis genome revealed 3,972 gene clusters containing 11,300 genes that were common to the genomes of the three previously sequenced mosquito species. There were 4,065 gene clusters containing 10,465 genes in A. gambiae, 4,064 gene clusters containing 12,608 genes in Ae. aegypti, and 4,073 gene clusters containing 14,827 genes in C. quinquefasciatus. 109 clusters found only in the four mosquito genomes, 34 clusters found specific to the Anophelinae, and 29 clusters containing 30 genes found specific to A. sinensis.
Gene orthology prediction
Consistent with evolutionary distance estimates, we observed a higher degree of genetic similarity between A. sinensis and other mosquito species proteomes than between A. sinensis and D. melanogaster proteomes (Figure 4). A. sinensis and A. gambiae shared the highest number of orthologous genes (48.3%) while A. sinensis and D. melanogaster shared the lowest number of orthologous genes (36.8%, Figure 4). A total of 4727 orthologous genes were shared only among the mosquitoes.
Figure 4.
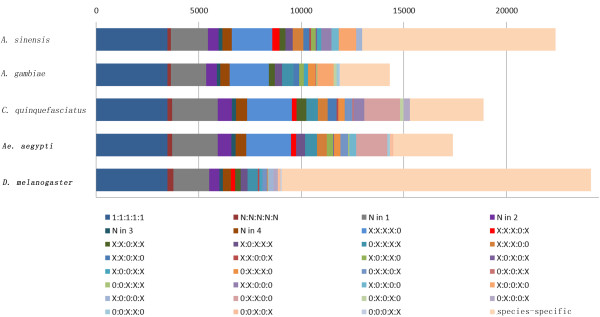
Ortholog delineation among the protein-coding gene repertoires of the four mosquito species and D. melanogaster. Membership of the categories of orthologous groups are depicted as follows: (i) 1:1:1:1:1 indicates single-copy orthologs in all species; (ii) N:N:N:N:N indicates multi-copy orthologs in all species; (iii) N in 1, N in 2, etc. indicates multi-copy orthologs in one or two species, etc; (iv) x:x:x:x:0, x:0:x:x:x, x:x:0:x:0 etc. indicates (by a 0) which of the five species, in the order listed above, did not contain single-copy or multi-copy orthologs. The remaining proportion of the sequence for each species exhibited no orthologs with genes in the other species (depicted as specific-specific in the figure).
Analysis of InterPro in these 4727 orthologous genes revealed the most gene-enriched domain and family were peptidase (Additional file 1: Table S13), while analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway revealed that genes were most enriched in metabolic pathways (Additional file 1: Table S14), both indicating functions central to mosquito biology, such as feeding behavior. Feeding releases peptidase in the midgut and assists in the degradation of blood meal proteins into peptides and amino acids [27].
Microsynteny with sequenced mosquito genomes
A genome-wide analysis revealed a significantly higher microsynteny between A. sinensis and A. gambiae (59.8%) than between A. sinensis and Ae. aegypti (42.1%) or between A.sinensis and C. quinquefasciatus (39.9%), or A. sinensis and D. melanogaster (20.4%, Table 5). The largest microsynteny, between A. sinensis and A. gambiae, also included the most shared gene families (8,457) and the largest coverage of the A. sinensis genome (132 M, Table 5). These findings are consistent with our present knowledge of the evolutionary relationship among these species. Given the close relationship between A. sinensis and A. gambiae, we took the chromosomes of A. gambiae as a reference for alignment, and aligned A. sinensis to the 2nd, 3rd and X chromosomes of A. gambiae. Coloring inside the schematic chromosome arms indicated microsynteny matches to a microsynteny block of A. sinensis (Figure 5). Chromosomal rearrangements in A. sinensis were observed, most obviously with respect to the 2 L chromosome arm of A. gambiae. In contrast, chromosomal rearrangements were relatively rare in other chromosomes arms of A. gambiae (Additional file 3: Figure. S1). In the genus Drosophila, the interspecies chromosomal rearrangements can be caused by the occurrence of paracentric inversions, Robertsonian translocations or transposon [28]. Such genetic changes may also have contributed to the chromosomal rearrangements observed in A. sinensis.
Table 5.
Characteristics of microsynteny blocks between A. sinensis , A. gambiae , Ae. aegypti , C. quinquefasciatus , and D. melanogaster
| Microsynteny blocks | Numbers of microsynteny blocks | Total length of microsynteny (Mbps) | Shared gene families |
|---|---|---|---|
|
A. sinensis/A. gambiae |
927 |
A. sinensis:132 |
8,457 |
|
A. gambiae:157 | |||
|
A. sinensis/Ae. aegypti |
1,668 |
A. sinensis:93 |
6,792 |
|
Ae. aegypti:398 | |||
|
A. sinensis/C. quinquefasciatus |
1,690 |
A. sinensis:88 |
7,087 |
|
C. quinquefasciatus:185 | |||
| A. sinensis/D. melanogaster | 1,031 |
A. sinensis:45 |
2,658 |
| D. melanogaster:38 |
Figure 5.

The coverage of the microsynteny block of A. sinensis on the chromosome of A. gambiae .
Divergence time
We calibrated the remaining 2,348 linear trees assuming a divergence time of ~260 million years ago (Mya) between Drosophila and Anopheles. This is the most rigorously calculated date available for the most recent split involving a mosquito lineage and its sister taxon [29,30]. Based on this basal divergence time, we obtained an estimate of the split between the Anophelinae and the Culicinae of approximately122 Mya (Figure 6). This is slightly later than a previous estimate of 145-200 Mya, which was inferred from mitochondrial sequences [8]. We estimated the split between A. sinensis and A. gambiae to have occurred ~52 Mya. This date of divergence was earlier than the split between A. funestus, another member in anopheline group, and A. gambiae (15–25 Mya) [31].
Figure 6.
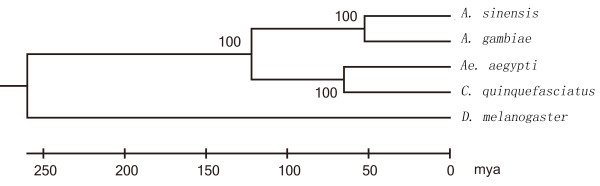
The inferred supertree for four mosquito species and D. melanogaster. The topology of the supertree was evaluated by bootstrap percentages. Distances are in millions of years.
Few immune-related gene sets may be associated with malaria vectorial capacity
Anophelinae are recognized as major vectors of human malaria, while culicine species are the principal etiological agents of mosquito-borne viruses. It is not surprising that genetic factors play decisive roles in determining vectorial capacity [32]. Previous studies regarding the immune system of Anophelinae have shown that changes in certain aspects can affect the development of Plasmodium either positively or negatively [33]. As shown in Additional file 1: Table S15, relative to Culicinae, C-type lectins (CTLs), serine protease inhibitors (serpins, SRPNs) and MD2-like gene (ML) families have contracted in the Anophelinae, whereas the thioester-containing protein (TEP) and peroxidase gene families have expanded, which may result from the differential duplication and/or loss of genes among these evolutionary lineages. Although comparative immune-related gene families in C. quinquefasciatus, Ae. aegypti, and A. gambiae have been studied, limited information is available due to limited numbers of anopheline species. With the discovery of the second anopheline mosquito, A. sinensis, we may reveal the Plasmodium-susceptible genotype, which will help to understand the details of the relationships between anopheline mosquito vectors and malarial pathogens.
Both the ML and serpin gene families have been shown obviously interfere with malarial infection. AgMDL1, an MD2-like receptor, showed specificity in regulating resistance to P. falciparum and O’nyong-nyong virus [34,35]. These overlaps between the effects of MLs on Plasmodium spp. and other viruses suggest that MLs are a universal defense mechanism for mosquitoes against invading pathogens. Expression of the SRPN6 gene can limit the number of rodent malarial oocysts in A. stephensi[36]. Thus, the significant contraction of MLs and Serpins can help the malarial parasite to survive in Anophelinae.
The TEP [37] and C-type lectins (CTLs) [38] gene families are both involved in pathogen recognition (PRRs) and immune response activation. One TEP family gene in particular (TEP1), can be upregulated after malarial infection and strongly inhibit the development of infection in both rodents and humans by binding to Plasmodium parasite surfaces [33]. In contrast, two circulating CTLs from A. gambiae (C-type lectin 4 [CTL4] and CTL mannose binding 2 [CTLMA2]) have been identified as agonists of the rodent malaria species, P. berghei, which can induce massive ookinete melanization when silenced [39]. Consequently, the downregulation of CTL members and the upregulation of TEP members in Anophelinae are likely to depend on their relative roles in promoting or inhibiting the development of malarial parasites. Putative HPX (HPX2, HPX7 and HPX8) can be induced in the mosquitoes midgut in response to Plasmodium infection, in order to potentiate nitric oxide toxicity and improve antiplasmodial effects [40,41]. Thus, HPX enzymes have been considered as key enzymes induced in the midgut cells of A. gambiae invaded by Plasmodium ookinetes.
The observed contraction of these two immune gene families (MLs and serpins) could be explained as important genetic components in the Plasmodium-susceptible phenotype. Such gene expression changes occur earlier than the invasion of Plasmodium, whereby Anophelinae are able to transmit Plasmodium. However, Plasmodium infection may induce the activation (and subsequent expansion or contraction) of immune-related genes involved in pathogen defense in mosquitoes. The two expanded gene families (TEP and peroxidases) and one contracted gene family (CTLs) observed in this study could have formed gradually as a long-term adaptive immune response against Plasmodium infection, and been expanded or contracted under positive selection in Anophelinae. Consequently, this immune-related gene set analysis at the theoretical level can provide clues for understanding the genetic basis of a Plasmodium-susceptible phenotype. These selective genes may serve as valuable potential targets for future malarial control strategies.
Methods
Strain selection and DNA extraction
The laboratory strain of A. sinensis used in this study has been inbred within the lab since 1984 and, never been exposed to pesticides. These mosquitoes were reared at 26 ± 1°C and 75 to 85% humidity, under a 10:14 h light:dark cycle. Genomic DNA was extracted from 300 adult females and 300 males (2 to 3 days post adult emergence) according to methods described in [42]. To prevent RNA and protein contamination, extracted DNA was treated with RNase A and proteinase K and, subsequently, precipitated with ethanol.
Whole-genome sequencing and assembly
We employed a whole-genome sequencing strategy with Roche/454 GS FLX. We constructed a total of five single-end and seven mate-pair sequencing libraries with insert sizes of about 3 Kb, 8 Kb and 20 Kb from 1 μg, 5 μg, 30 μg and 60 μg of starting DNA. In total, we generated 4.16 Gb of data of sequencing reads ranging from 40 to 1196 bp. To reduce the effect of sequencing error during assembly, we undertook a series of checking and filtering steps in assembling the reads generated. By using stringent criteria, 3.34 G of high quality data were incorporated into the final de novo genome assembly.
The Lander-Waterman algorithm [43] were used to estimate the genome size of A. sinensis. K-mer analysis for single-end reads [44] revealed a frequency distribution that conformed to the Poisson expectation when K-mer was equal to 13. The value of expected depth was calculated based on the lambda, a parameter of possion distribution. The genome size of A. sinensis was then calculated using the total K-mer number divided by the expected depth value.
Whole-genome assembly was carried out with a Celera Assembler V6.1 for the remaining 454 reads [45]. The revised pipeline (called Celera Assembler with the Best Overlap Graph, CABOG) was robust to uncertainty in homopolymer run length, high read coverage and heterogeneous read lengths. We utilized the following modules of the Celera Assembler software for successive phases of the assembly: pairwise overlap detection; initial ungapped multiple sequence alignments, called unitigs; unitig consensus calculation; combining unitigs with mate constraints to form contigs and scaffolds that were ungapped and gapped multiple sequence alignments; and, finally, scaffold consensus determination. Because the genome used for sequencing were constructed from whole adult mosquitoes, contamination from bacteria in gut or adhering on the surface were inevitable. To check for possible microbial contamination of the assembly, we screened scaffolds against the NCBI NT database using query alignment and identity cut-off of 90% and e-value cut-off of 1e-6. When the top hit was bacterial species, this scaffold was removed.
In order to assess the assembly quality, the transcriptome was sequenced and aligned to the scaffold sequences using Blat with default parameters [46]. Assembly quality was also assessed by mapping the 454 Single reads to the scaffolds using BWA. The mapped regions (consensus sequences) with depth over 3X were extracted for SNVs and INDEL variation analysis, which represent potential base error and short indel error rate in the genome, respectively [46]. Additionally, presence of CEGs was evaluated for the genome assembly (http://korflab.ucdavis.edu/Datasets/cegma/submit.html) [47,48].
Identification of repetitive elements
The identification of repetitive elements is essential for genome sequencing, as unidentified repetitive elements can affect the quality of gene predictions, annotation and annotation-dependent analyses [49]. Two methods were adopted for masking repeat regions in A. sinensis. First, RepeatMasker V3.3.0 (http://www.repeatmasker.org/) was applied against the Repbase library (species Anopheles) based on the scaffolds. Then, RepeatScout V1.0.5 [50] software was used (with frequency set to ≥50) to build a repeat regions database by providing scaffolds and potentially repeat sequences. These results were merged with the results of the transposable elements for mosquitoes, which were downloaded from TEfam database (http://tefam.biochem.vt.edu/tefam/). Finally, these merged results were reprocessed with RepeatMasker.
Gene prediction
To predict genes, we used two independent approaches: a homology-based method and a de novo method. The results of these two methods were integrated by the EVidenceModeler utility and then filtered multiple times and also checked manually. The reference protein sequences for protein alignment were obtained from VectorBase (for the aforementioned three sequenced mosquito species) and the NCBI database (for Culicidae species). CD-HIT software was used to cluster these protein sequences with 100% global similarity [51]. AAT [52] and Genewise [53] software were used to align the protein data to the masked scaffolds. By comparing the databases, we obtained the number of protein distributions.
Four ab initio gene prediction programs were run on the genome: SNAP [54], Augustus [55], GlimmerHMM [56], and Genezilla [57] with the model trained using the published mosquito gene information (A. gambiae, Ae. aegypti and C. quinquefasciatus).
Quality of protein-coding gene predictions
To estimate the accuracy of gene prediction, we undertook a consistency check for the protein length of single-copy orthologs between A. sinensis and D. melanogaster. Considering the high conservation of single-copy orthologs, the protein length should have a high coherence between two species [23]. The protein lengths of the two species were plotted as a scatter diagram and analyzed with a regression analysis. We compared the results of this regression analysis with results from the published literature.
Identification of noncoding RNA genes
tRNA genes were predicted by tRNAscan-SE-1.23 with eukaryote parameters [58]. The rRNA fragments were identified by aligning the rRNA template sequences from the SILVA database [59] and RNAmmer database, by using BlastN at E-value 1e-5 with cutoff of identity ≥95% and match length ≥50 bp. It is important to note that rRNA genes in the A. sinensis genome were combined by aligning the 5.8S, 18S, 25S and 28S regions of databases using BlastN. miRNA was predicted by BlastN against the hairpin sequences from miRBase database (RELEASE 17) with E-value 1e-3, allowing no less than 70 bp alignment length, and requiring no less than 85% overall identity and 80% coverage.
Functional annotation
Gene functions were assigned according to the best match of the alignments using Blast and BlastP (query coverage ≥50%; E-value: 1e-10) against the NCBI NR protein database. All predicted protein-coding genes were obtained with the InterProScan analysis tool [60]. According to features of the predicted protein sequences, the InterProScan analysis was based on the active site, the binding site, the conserved site, the domain, the family, the PTM, and the repeat. Gene Ontology (GO) IDs for each gene were obtained from the corresponding InterProScan entry. All genes were aligned against the KEGG proteins, and the pathway in which the gene might be involved was derived from the matching genes in the KEGG. SignalP 4.0 server was used to predict the presence and location of signal peptide cleavage sites in the amino acid sequences [61]. This method incorporates a prediction of cleavage sites and a signal peptide/non-signal peptide prediction based on a combination of several artificial neural networks. TMHMM software [62] was used with default values to predict the transmembane region based on a hidden Markov model.
Gene orthology prediction
The gene orthology predictions were generated by the Ensemble Gene Tree method [63], which is based on the PHYML algorithm for multiple protein sequence alignments, and uses MUSCLE for each gene family that contains sequences from all five species (A. sinensis, A. gambiae, Ae. aegypti, C. quinquefasciatus and D. melanogaster). Gene trees were reconciled with the species trees using the RAL algorithm to call duplication events on internal nodes and to root the trees. The relations of orthology were inferred from the results of each gene tree.
Defining gene families
The PANTHER hidden Markov models V7.2, annotated to different functional gene families, were used with default parameters (i.e. E-value: 1e-3) to classify all gene models of A. sinensis. Immune-related gene sets were downloaded from ImmunoDB resource (http://cegg.unige.ch/Insecta/immunodb) and subjected to inspection, curation, and phylogenetic analysis. Based on these gene sets, we re-annotated the proteins in the A. sinensis genome by Blast search, and counted the number of A. sinensis genes in each functional gene set. The threshold E-value in the Blast search was set to 1e-3, while the similarity was set to 0.35.
Construction of microsyntenic blocks
CHSMiner V1.1 [64] was used to construct the microsynteny map for A. sinensis and the other three previously sequenced mosquito species. Briefly, the program used the orthologs between two genomes as anchors, and merged two anchors into a block if they were located less than a specified gap size apart. We used default values for parameters and set the minimum length to 100 Kb. Each microsynteny detected was evaluated by corrected P-values; only those results with the P-values less than 1e-5 were preserved.
Phylogeny construction
M-Coffee V9.0 program [65] was used to perform the multiple alignment of proteins in each family. A phylogeny tree was constructed based on the 3,470 single-copy families in the five species (A. sinensis, A. gambiae, Ae. aegypti, C. quinquefasciatus and D. melanogaster). We used the Phylip package V3.69 [66] to build the maximum likelihood (ML) tree for each protein family under the JTT substitution model. Then the SuperTree software was used to get an integrated supertree. To evaluate the topology of the supertree, we performed a bootstrap resample analysis using 100 resamples from the original tree.
Conclusions
Malaria is caused by infection with Plasmodium parasites that are transmitted via the bites of infected female Anopheles mosquitoes. Vector control offers an important means of limiting the spread of malaria; however, the lack of genetic information on Plasmodium-susceptible anopheline mosquitoes is a major obstacle to the development of effective vector management. We generated the first draft genome sequence of Anopheles sinensis, an Asiatic mosquito species suspected to be the most important vector of P. vivax. We compared the genetic composition of this species to that of other sequenced mosquito species in the subfamily Anophelinae and the subfamily Culicinae (the latter are not susceptible to Plasmodium infection). The results of these comparisons provide important genetic insights into this vector-disease system. In particular,we observed the expansion and contraction of several important immune-related gene families known to influence aspects of Plasmodium development, in the anopheline species relative to the culicine species. These differences suggest that species-specific immune responses to Plasmodium infection underpin the biological differences in Plasmodium susceptibility that characterize these two mosquito subfamilies. This study provides critical genomic information that will pave the way for analyses investigating the genetic basis of mosquito susceptibility and resistance to Plasmodium parasites.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BS and CLZ are the principal investigators and project managers in this work. HYZ and CRX conducted the sample collection. DZ, LNS and BS extracted DNA. DZ, YX and QH edited tables and figures. DZ, GHD, BS, CLZ, DHZ, SDL, SCH, XYY, PZ, CC, XLC, WJW, YL, YTY, YS, LM did the genomics analysis. DZ, DHZ, JY, and BS wrote and edited the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Putative contaminating scaffold sequence of possible bacterial origin. Table S2: Assessment of gene coverage by assembled transcripts of A. sinensis. Table S3: Variation statistics regarding mapping of raw reads to the scaffolds. Table S4: Results of gene prediction and predicted protein-coding genes for A. sinensis. Table S5: Identification of non-coding RNA genes in the A. sinensis genome. Table S6: Functional annotation of predicted genes for A. sinensis. Table S7: Occurrence of the over-represented InterPro domains and repeats in the genome of A. sinensis compared with the genome of A.gambiae. Table S8: Occurrence of the down-represented InterPro domains in the genomes of the Anopheline species compared with the Culicine species. Table S9: Occurrence of the over-represented level 2 GO terms in the genome of A. sinensis compared with the genome of A.gambiae. Table S10: Occurrence of the over-represented level 2 GO terms in the genomes of the Anopheline species compared with the Culicine species. Table S11: Number of transmembrane regions in the four mosquito species. Table S12: Distribution results of the transmembrane regions in the four mosquito species. Table S13: Occurrence of the top 10 domains and families enriched in orthologous genes that were shared only among the four mosquito species. Table S14: Occurrence of the top 12 pathways enriched in orthologous genes that were shared only among the four mosquito species. Table S15: Number of selected immune-related gene sets in the four mosquito species.
MiRNA list of all predicted A. sinensis miRNA target genes and their annotations.
Microsynteny between the genomes of A. sinensis and A. gambiae. Genomic scaffolds of A. sinensis inferred to be syntenic were linked (blue lines) to A. gambiae chromosomes.
Contributor Information
Dan Zhou, Email: zdd1209love@163.com.
Donghui Zhang, Email: donghuizhang2004@gmail.com.
Guohui Ding, Email: gwding@sibs.ac.cn.
Linna Shi, Email: selina-007-520@163.com.
Qing Hou, Email: houqingradiology@163.com.
Yuting Ye, Email: she.jueting@163.com.
Yang Xu, Email: xylocaine@126.com.
Huayun Zhou, Email: hyzhou88@163.com.
Chunrong Xiong, Email: crxiong@hotmail.com.
Shengdi Li, Email: lishengdi@sibs.ac.cn.
Jing Yu, Email: xiaoyu_7021@126.com.
Shanchao Hong, Email: hongshanchao1@163.com.
Xinyou Yu, Email: yxy_nxmc@163.com.
Ping Zou, Email: yezihenhaohenhaode@126.com.
Chen Chen, Email: dawn_draw@163.com.
Xuelian Chang, Email: xuelianchang@126.com.
Weijie Wang, Email: wwjsix@163.com.
Yuan Lv, Email: lvyuan19870212@163.com.
Yan Sun, Email: sunyan@njmu.edu.cn.
Lei Ma, Email: malei@njmu.edu.cn.
Bo Shen, Email: shenbo@njmu.edu.cn.
Changliang Zhu, Email: clzhu@njmu.edu.cn.
Acknowledgments
This work was supported by the National Institutes of Health of US (NIH)(Grant No. 2R01AI075746-05), the National Natural Science Foundation of China (Grant No. 30901244, 30972564, 81171900 and 81101279), the National S & T Major Program (Grant No. 2012ZX10004-219, 2008ZX10004-010, 2012ZX10004-220 and 2008ZX10004-011), Specialized Research Fund for the Doctoral Program of Higher Education of China (Grant No.20113234120007), Natural Science Foundation of Jiangsu Province (Grant No.81101279), and Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
- Kiang KM, Bryant PA, Shingadia D, Ladhani S, Steer AC, Burgner D. The treatment of imported malaria in children: an update. Arch Dis Child Educ Pract Ed. 2013;15:7–15. doi: 10.1136/archdischild-2011-301530. [DOI] [PubMed] [Google Scholar]
- Patarroyo MA, Calderon D, Moreno-Perez DA. Vaccines against Plasmodium vivax: a research challenge. Expert Rev Vaccines. 2012;15(10):1249–1260. doi: 10.1586/erv.12.91. [DOI] [PubMed] [Google Scholar]
- Eastman RT, Pattaradilokrat S, Raj DK, Dixit S, Deng B, Miura K, Yuan J, Tanaka TQ, Johnson RL, Jiang H. et al. A class of tricyclic compounds blocking malaria oocyst development and transmission. Antimicrob Agents Chemother. 2013;15(1):425–435. doi: 10.1128/AAC.00920-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organization WH. World malaria report 2011. Geneva: World Health Organization; 2011. [Google Scholar]
- Jung J, Lee E, Kim W. Isolation and characterization of polymorphic microsatellite markers of Anopheles sinensis, a malaria vector mosquito in the East Asia region. Mol Ecol Notes. 2006;15(4):1272–1274. doi: 10.1111/j.1471-8286.2006.01512.x. [DOI] [Google Scholar]
- Welch SG, McGregor IA, Williams K. The Duffy blood group and malaria prevalence in Gambian West Africans. Trans R Soc Trop Med Hyg. 1977;15(4):295–296. doi: 10.1016/0035-9203(77)90102-X. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Klein TA, Kim HC, Choi YM, Yoon SH, Chang KS, Chong ST, Lee IY, Jones JW, Jacobs JS. et al. Anopheles kleini, Anopheles pullus, and Anopheles sinensis: potential vectors of Plasmodium vivax in the Republic of Korea. J Med Entomol. 2007;15(6):1086–1090. doi: 10.1603/0022-2585(2007)44[1086:AKAPAA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Zhou SS, Huang F, Wang JJ, Zhang SS, Su YP, Tang LH. Geographical, meteorological and vectorial factors related to malaria re-emergence in Huang-Huai River of central China. Malar J. 2010;15:337. doi: 10.1186/1475-2875-9-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai KS, Black WC. Mosquito genomes: structure, organization, and evolution. Adv Genet. 1999;15:1–33. doi: 10.1016/s0065-2660(08)60149-2. [DOI] [PubMed] [Google Scholar]
- Behura SK, Singh BK, Severson DW. Antagonistic relationships between intron content and codon usage bias of genes in three mosquito species: functional and evolutionary implications. Evol Appl. 2013;15(7):1079–1089. doi: 10.1111/eva.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill PR, Charlesworth B, Halligan DL, Andolfatto P. Patterns of intron sequence evolution in Drosophila are dependent upon length and GC content. Genome Biol. 2005;15(8):R67. doi: 10.1186/gb-2005-6-8-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerton JL, DeRisi J, Shroff R, Lichten M, Brown PO, Petes TD. Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2000;15(21):11383–11390. doi: 10.1073/pnas.97.21.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, Brunton CF, Smith NG. Small introns tend to occur in GC-rich regions in some but not all vertebrates. Trends Genet. 1999;15(11):437–439. doi: 10.1016/S0168-9525(99)01832-6. [DOI] [PubMed] [Google Scholar]
- Takano-Shimizu T. Local changes in GC/AT substitution biases and in crossover frequencies on Drosophila chromosomes. Mol Biol Evol. 2001;15(4):606–619. doi: 10.1093/oxfordjournals.molbev.a003841. [DOI] [PubMed] [Google Scholar]
- Birdsell JA. Integrating genomics, bioinformatics, and classical genetics to study the effects of recombination on genome evolution. Mol Biol Evol. 2002;15(7):1181–1197. doi: 10.1093/oxfordjournals.molbev.a004176. [DOI] [PubMed] [Google Scholar]
- Berglund J, Pollard KS, Webster MT. Hotspots of biased nucleotide substitutions in human genes. PLoS Biol. 2009;15(1):e26. doi: 10.1371/journal.pbio.1000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Arndt PF. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 2008;15(5):e1000071. doi: 10.1371/journal.pgen.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CC, Deloukas P, Hunt S, Mullikin J, Myers S, Silverman B, Donnelly P, Bentley D, McVean G. The influence of recombination on human genetic diversity. PLoS Genet. 2006;15(9):e148. doi: 10.1371/journal.pgen.0020148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle MM, Markianos K, Niare O, Xu J, Li J, Toure AM, Podiougou B, Oduol F, Diawara S, Diallo M. et al. Natural malaria infection in Anopheles gambiae is regulated by a single genomic control region. Science. 2006;15(5773):577–579. doi: 10.1126/science.1124153. [DOI] [PubMed] [Google Scholar]
- Lehmann T, Hume JC, Licht M, Burns CS, Wollenberg K, Simard F, Ribeiro JM. Molecular evolution of immune genes in the malaria mosquito Anopheles gambiae. PLoS One. 2009;15(2):e4549. doi: 10.1371/journal.pone.0004549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R. et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002;15(5591):129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- Nene V, Wortman JR, Lawson D, Haas B, Kodira C, Tu ZJ, Loftus B, Xi Z, Megy K, Grabherr M. et al. Genome sequence of Aedes aegypti, a major arbovirus vector. Science. 2007;15(5832):1718–1723. doi: 10.1126/science.1138878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arensburger P, Megy K, Waterhouse RM, Abrudan J, Amedeo P, Antelo B, Bartholomay L, Bidwell S, Caler E, Camara F. et al. Sequencing of Culex quinquefasciatus establishes a platform for mosquito comparative genomics. Science. 2010;15(6000):86–88. doi: 10.1126/science.1191864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM. What controls the length of noncoding DNA? Curr Opin Genet Dev. 2001;15(6):652–659. doi: 10.1016/S0959-437X(00)00249-5. [DOI] [PubMed] [Google Scholar]
- Fernandez-Medina RD, Struchiner CJ, Ribeiro JM. Novel transposable elements from Anopheles gambiae. BMC Genomics. 2011;15:260. doi: 10.1186/1471-2164-12-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissler L, Gadau J, Simola DF, Helmkampf M, Bornberg-Bauer E. Mechanisms and dynamics of orphan gene emergence in insect genomes. Genome Biol Evol. 2013;15(2):439–455. doi: 10.1093/gbe/evt009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascon AA Jr, Gearin J, Isoe J, Miesfeld RL. In vitro activation and enzyme kinetic analysis of recombinant midgut serine proteases from the Dengue vector mosquito Aedes aegypti. BMC Biochem. 2011;15:43. doi: 10.1186/1471-2091-12-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Von Mering C, Letunic I, Torrents D, Suyama M, Copley RR, Christophides GK, Thomasova D, Holt RA, Subramanian GM. et al. Comparative genome and proteome analysis of Anopheles gambiae and Drosophila melanogaster. Science. 2002;15(5591):149–159. doi: 10.1126/science.1077061. [DOI] [PubMed] [Google Scholar]
- Gaunt MW, Miles MA. An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol Biol Evol. 2002;15(5):748–761. doi: 10.1093/oxfordjournals.molbev.a004133. [DOI] [PubMed] [Google Scholar]
- Krzywinski J, Grushko OG, Besansky NJ. Analysis of the complete mitochondrial DNA from Anopheles funestus: an improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol Phylogenet Evol. 2006;15(2):417–423. doi: 10.1016/j.ympev.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Garros C, Harbach RE, Manguin S. Systematics and biogeographical implications of the phylogenetic relationships between members of the funestus and minimus groups of Anopheles (Diptera: Culicidae) J Med Entomol. 2005;15(1):7–18. doi: 10.1603/0022-2585(2005)042[0007:SABIOT]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Beerntsen BT, James AA, Christensen BM. Genetics of mosquito vector competence. Microbiol Mol Biol Rev. 2000;15(1):115–137. doi: 10.1128/MMBR.64.1.115-137.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandin S, Shiao SH, Moita LF, Janse CJ, Waters AP, Kafatos FC, Levashina EA. Complement-like protein TEP1 is a determinant of vectorial capacity in the malaria vector Anopheles gambiae. Cell. 2004;15(5):661–670. doi: 10.1016/S0092-8674(04)00173-4. [DOI] [PubMed] [Google Scholar]
- Dong Y, Aguilar R, Xi Z, Warr E, Mongin E, Dimopoulos G. Anopheles gambiae immune responses to human and rodent Plasmodium parasite species. PLoS Pathog. 2006;15(6):e52. doi: 10.1371/journal.ppat.0020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldock J, Olson KE, Christophides GK. Anopheles gambiae antiviral immune response to systemic O’nyong-nyong infection. PLoS Negl Trop Dis. 2012;15(3):e1565. doi: 10.1371/journal.pntd.0001565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham EG, Pinto SB, Ghosh A, Vanlandingham DL, Budd A, Higgs S, Kafatos FC, Jacobs-Lorena M, Michel K. An immune-responsive serpin, SRPN6, mediates mosquito defense against malaria parasites. Proc Natl Acad Sci USA. 2005;15(45):16327–16332. doi: 10.1073/pnas.0508335102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obbard DJ, Callister DM, Jiggins FM, Soares DC, Yan G, Little TJ. The evolution of TEP1, an exceptionally polymorphic immunity gene in Anopheles gambiae. BMC Evol Biol. 2008;15:274. doi: 10.1186/1471-2148-8-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambi A, Gijzen K, De Vries JM, Torensma R, Joosten B, Adema GJ, Netea MG, Kullberg BJ, Romani L, Figdor CG. The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur J Immunol. 2003;15(2):532–538. doi: 10.1002/immu.200310029. [DOI] [PubMed] [Google Scholar]
- Osta MA, Christophides GK, Kafatos FC. Effects of mosquito genes on plasmodium development. Science. 2004;15(5666):2030–2032. doi: 10.1126/science.1091789. [DOI] [PubMed] [Google Scholar]
- Oliveira Gde A, Lieberman J, Barillas-Mury C. Epithelial nitration by a peroxidase/NOX5 system mediates mosquito antiplasmodial immunity. Science. 2012;15(6070):856–859. doi: 10.1126/science.1209678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi H, Grambihler A, Canbay A, Bronk SF, Gores GJ. Bile acids up-regulate death receptor 5/TRAIL-receptor 2 expression via a c-Jun N-terminal kinase-dependent pathway involving Sp1. J Biol Chem. 2004;15(1):51–60. doi: 10.1074/jbc.M309476200. [DOI] [PubMed] [Google Scholar]
- Collins FH, Mendez MA, Rasmussen MO, Mehaffey PC, Besansky NJ, Finnerty V. A ribosomal RNA gene probe differentiates member species of the Anopheles gambiae complex. Am J Trop Med Hyg. 1987;15(1):37–41. doi: 10.4269/ajtmh.1987.37.37. [DOI] [PubMed] [Google Scholar]
- Lander ES, Waterman MS. Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics. 1988;15(3):231–239. doi: 10.1016/0888-7543(88)90007-9. [DOI] [PubMed] [Google Scholar]
- Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;15(6):764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics. 2008;15(24):2818–2824. doi: 10.1093/bioinformatics/btn548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Hu L, Hou H, Lee H, Xu J, Kwon S, Oh S, Kim HM, Jho S, Kim S. et al. The tiger genome and comparative analysis with lion and snow leopard genomes. Nat Commun. 2013;15:2433. doi: 10.1038/ncomms3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;15(9):1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, Boisvert S, Chapman JA, Chapuis G, Chikhi R. et al. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. GigaScience. 2013;15(1):10. doi: 10.1186/2047-217X-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi H, Thiagarajan M, Haas B, Wortman J, Hall N, Caler E. Genome wide survey, discovery and evolution of repetitive elements in three Entamoeba species. BMC Genomics. 2008;15:595. doi: 10.1186/1471-2164-9-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;15(Suppl 1):i351–i358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;15(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Huang X, Adams MD, Zhou H, Kerlavage AR. A tool for analyzing and annotating genomic sequences. Genomics. 1997;15(1):37–45. doi: 10.1006/geno.1997.4984. [DOI] [PubMed] [Google Scholar]
- Birney E, Clamp M, Durbin R. Genewise and genomewise. Genome Res. 2004;15(5):988–995. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korf I. Gene finding in novel genomes. BMC Bioinforma. 2004;15:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Schoffmann O, Morgenstern B, Waack S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinforma. 2006;15:62. doi: 10.1186/1471-2105-7-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;15(16):2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Delcher AL, Salzberg SL. Efficient decoding algorithms for generalized hidden Markov model gene finders. BMC Bioinforma. 2005;15:16. doi: 10.1186/1471-2105-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;15(5):955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glockner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;15(21):7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;15(9):847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, Von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;15(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;15(7):646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009;15(2):327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ding G, Yu Z, Liu L, Li Y. CHSMiner: a GUI tool to identify chromosomal homologous segments. Algorithms Mol Biol. 2009;15:2. doi: 10.1186/1748-7188-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;15(1):205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996;15:418–427. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Putative contaminating scaffold sequence of possible bacterial origin. Table S2: Assessment of gene coverage by assembled transcripts of A. sinensis. Table S3: Variation statistics regarding mapping of raw reads to the scaffolds. Table S4: Results of gene prediction and predicted protein-coding genes for A. sinensis. Table S5: Identification of non-coding RNA genes in the A. sinensis genome. Table S6: Functional annotation of predicted genes for A. sinensis. Table S7: Occurrence of the over-represented InterPro domains and repeats in the genome of A. sinensis compared with the genome of A.gambiae. Table S8: Occurrence of the down-represented InterPro domains in the genomes of the Anopheline species compared with the Culicine species. Table S9: Occurrence of the over-represented level 2 GO terms in the genome of A. sinensis compared with the genome of A.gambiae. Table S10: Occurrence of the over-represented level 2 GO terms in the genomes of the Anopheline species compared with the Culicine species. Table S11: Number of transmembrane regions in the four mosquito species. Table S12: Distribution results of the transmembrane regions in the four mosquito species. Table S13: Occurrence of the top 10 domains and families enriched in orthologous genes that were shared only among the four mosquito species. Table S14: Occurrence of the top 12 pathways enriched in orthologous genes that were shared only among the four mosquito species. Table S15: Number of selected immune-related gene sets in the four mosquito species.
MiRNA list of all predicted A. sinensis miRNA target genes and their annotations.
Microsynteny between the genomes of A. sinensis and A. gambiae. Genomic scaffolds of A. sinensis inferred to be syntenic were linked (blue lines) to A. gambiae chromosomes.