

Abstract
Discussion of the challenges discerning the clinical significance of changes in CD73 expression, in human lymphocyte subsets.
Keywords: adenosine, adenosine 5′-monophosphate, AIDS, adenosine receptors, immunodeficiency, CD39, regulatory T cells
5′-NT grabbed the attention of immunologists long before it received a CD number in 1989 (CD73). The time was the late 1970s, shortly after mutations in the genes encoding ADA and PNP were discovered to cause severe primary immunodeficiency diseases. Although ADA and PNP are ubiquitously expressed and frequently thought of as housekeeping genes, the consequences of mutations that destroy enzyme activity are largely restricted to the immune system. Before the identification of patients with ADA and PNP deficiency, no one appreciated that normal purine metabolism was needed for immune-system development and function. Rheumatologists who investigated abnormalities in purine metabolism as a cause of gout expanded their focus to include the immune system. In 1977, Johnson et al. (reviewed in ref. [1]) reported a deficiency in 5′-NT enzyme activity in the PBMCs of patients with CVI, a type of hypogammaglobulinemia caused by a defect in the ability of B cells to make antibodies. In the following year, Edwards et al. (reviewed in ref. [1]) made a similar observation in patients with congenital XLA, in which peripheral blood B cells are absent. At the time, I was a postdoctoral fellow in the laboratory of Dr. J. Edwin Seegmiller at the University of California, San Diego, working with a new assistant professor of medicine, Dr. Gerry Boss. As 5′-NT is part of the same metabolic pathway as ADA and PNP (Fig. 1A), we were curious to know whether CVI and/or XLA might be caused by mutations in the gene encoding 5′-NT. As it eventually became clear, 5′-NT expression in lymphocytes is developmentally regulated, and patients who have blocks in early lymphocyte development have reduced 5′-NT expression as a consequence of their disease—not the cause [1]. Nevertheless, we spent several years measuring 5′-NT enzyme activity (this was long before mAb to 5′-NT were available) in the T and B cells of healthy subjects and patients with every type of immune deficiency we could find. When previously healthy gay men with Pneumocystis carinii pneumonia surfaced in Los Angeles in the early 1980s, we started measuring 5′-NT enzyme activity in men with AIDS. Much to our amazement, 5′-NT enzyme activity was low in the PBMCs of many of these subjects, much like what we had observed previously in patients with a variety of primary immunodeficiency diseases. Other groups made similar observations [2]. The finding of 5′-NT deficiency in patients with an acquired immunodeficiency suggested that assessment of 5′-NT expression might have broader clinical significance.
Figure 1. Postulated role of CD73 in Treg-mediated immunosuppression.
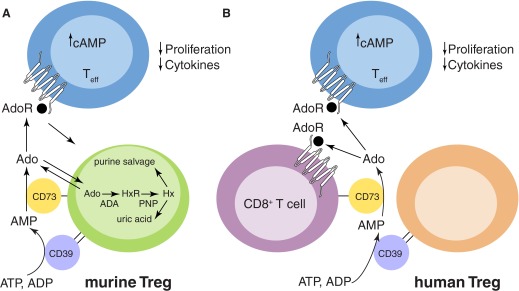
(A) Murine Tregs (green) express CD39 and CD73. Extracellular Ado, produced by murine Tregs, can bind to A2A AdoRs on Teffs, causing a rise in intracellular cAMP and suppression of proliferation and inflammatory cytokine production. The purine salvage pathway (which exists in the cytoplasm of all cells) is also illustrated in murine Tregs. HxR, Inosine; Hx, hypoxanthine. (B) Human Tregs (brown) express CD39. Extracellular AMP produced by human Tregs might be converted to extracellular Ado by CD73+CD8+ T cells. This extracellular Ado could then bind to A2A AdoR on CD8+ T cells or Teffs to inhibit their function.
In this issue of the Journal of Leukocyte Biology, Tóth and colleagues [3] have reinvestigated changes in lymphocyte 5′-NT (now called CD73) expression in HIV patients at various clinical stages using modern immunological approaches and with a focus on Tregs. They found significantly reduced percentages of CD73+ T cells in the CD8+ compartment, with the magnitude of the change increasing with the stage of the disease and the extent of viremia. Thus, elite controllers had the highest percentage of CD73+CD8+ T cells, and viremic patients had the lowest. Similar observations were made regarding the percentage of Tregs that express CD73. Interestingly, the percentage of CD73+CD8+ cells correlated positively with the CD4+ T cell count and inversely with expression of markers of T cell activation and exhaustion. The decrease in CD73+CD8+ T cells could be partially reversed by ART. CD73−CD8+ T cells from healthy subjects and HIV patients were functionally impaired, as shown by a decreased proliferative capacity and reduced secretion of IL-2 and TNF-α compared with CD73+CD8+ T cells.
Although the enzymatic reaction catalyzed by CD73 can be considered part of the purine salvage pathway (Fig. 1A), it is now appreciated that the primary function of CD73 is likely to act in concert with CD39 (another ecto-enzyme that converts ATP and ADP to AMP, the substrate of CD73) to provide extracellular Ado for the engagement of cell-surface AdoRs, which are are seven transmembrane-spanning GPCRs that regulate myriad physiological functions, including vascular tone, neurotransmission, heart rate, kidney function, and inflammation [4]. In mice, Tregs express CD39 and CD73, and it is believed that part of their immunosuppressive function comes from the generation of extracellular Ado [5]. For example, studies with our CD73 gene-targeted mice revealed that CD73 on Tregs helps to control mucosal inflammation and also contributes to the immunosuppressive microenvironment surrounding some tumors. Ado acting on A2A AdoRs increases intracellular cAMP and inhibits T cell proliferation and inflammatory cytokine production (Fig. 1A).
As in mice, CD39 is expressed on the majority of human Tregs. However, only a small minority of these also expresses CD73, as noted by Tóth et al. [3] and others. This raises the interesting question of whether the function of human Tregs can be linked to Ado production and AdoR signaling. Tóth et al. [3] make the provocative hypothesis that perhaps AMP produced by CD39 on Tregs can be further converted to extracellular Ado by CD73 expressed on neighboring CD8+ T cells (Fig. 1B). This Ado could then engage AdoRs on the same cell or on nearby Teffs. If this hypothesis is correct, then this pathway for dampening immune/inflammatory responses would be defective in HIV patients who show markedly reduced CD73 expression on their CD8+ cells. This situation would be exacerbated further by the degradation of extracellular Ado by ADA, which is bound to CD26 on the surface of activated T cells. Thus, it is possible that reduced expression of CD73 on CD8+ T cells contributes to the immune activation seen in HIV-infected patients.
Many interesting questions remain regarding the observation of reduced CD73 expression in T cells of HIV-infected patients:
Is CD73 down-regulated in HIV infection, and if so, by what mechanism? Tóth et al. [3] showed that loss of CD73 expression is not a universal consequence of T cell activation. Furthermore, an examination of the MFI of CD73 staining of CD8+ T cells in HIV-infected patients showed that the decrease in percentage of CD73+ cells is more profound than the decrease in MFI.
This raises important questions, such as: are there stable subsets of CD73+CD8+ and CD73−CD8+ cells, and do they have distinct functions?
Is the reduced CD73 expression in CD8+ T cells of HIV-infected individuals a consequence of the expansion of the CD73−CD8+ T cell subset found in healthy humans? (Approximately 50% of CD8+ T cells expresses high levels of CD73 in healthy humans [1]). CD73 “deficiency” also occurs in patients with acute, infectious mononucleosis [6]. It is possible that the CD8+ T cells that expand to fight viral infections derive from the CD73− subset. This could also be the case in HIV-infected patients.
Does the reduced expression of CD73 in HIV-infected patients contribute to their immune dysregulation, or is it simply a consequence of the infection? CD73 gene-targeted mice make good immune responses and actually show increased lymphocyte homing to draining LNs after an inflammatory stimulus [7]. Of course, mice are not men, and observations regarding the function of CD73 made in mice may not translate perfectly to humans.
Can the immunosuppressive function of human Tregs be explained, at least in part, by the production of extracellular AMP by CD39, which is then further converted to extracellular Ado by CD73+CD8+ T cells (or by other CD73+ cells)? This question could be addressed, in part, by in vitro assays in the presence and absence of the specific 5′-NT inhibitor α,β-methylene adenosine 5′-diphosphate.
In summary, there is much that we still do not know about the physiologic function of CD73 in humans. Studies with CD73 gene-targeted mice suggest a critical role in ischemic preconditioning, leukocyte migration, endothelial barrier function, bone formation, inflammation, and other critical physiological processes (reviewed in refs. [8, 9]). However, CD73-deficient humans suffer from extensive arterial calcifications, especially in the lower extremities [10], a phenotype that has not been observed in CD73-deficient mice, and appear to have normal immune function. We know that CD73 is an hypoxia-inducible gene, but other aspects of its regulation are less clear. Tóth et al. [3] document changes, which correlate with disease state and can be influenced by ART, in CD73 expression in lymphocytes of HIV patients, demonstrating that evaluation of CD73 expression may have clinical use. However, it is still an open question of whether the increased lymphocyte CD73 expression that occurs after ART contributes to the improved immune function in these patients or is simply a marker for the restoration of a more normal balance of CD73+ and CD73− lymphocyte subsets.
ACKNOWLEDGMENTS
Research on this topic in the author's laboratory is supported by a grant from the U.S. National Institutes of Health (R01 AI18220). L.F.T. holds the Putnam City Schools Distinguished Chair in Cancer Research.
SEE CORRESPONDING ARTICLE ON PAGE 551
- 5′-NT
- ecto-5′-nucleotidase
- ADA
- adenosine deaminase
- Ado
- adenosine
- ART
- antiretroviral therapy
- CVI
- common variable immunodeficiency
- MFI
- mean fluorescence intensity
- PNP
- purine nucleoside phosphorylase
- Teff
- effector T cell
- Treg
- regulatory T cell
- XLA
- X-linked agammaglobulinemia
REFERENCES
- 1. Resta R., Yamashita. Y., Thompson L. F. (1998) Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol. Rev. 161, 95–109 [DOI] [PubMed] [Google Scholar]
- 2. Salazar-Gonzalez J. F., Moody D. J., Giorgi J. V., Martinez-Maza O., Mitsuyasu R. T., Fahey J. L. (1985) Reduced ecto-5′-nucleotidase activity and enhanced OKT10 and HLA-DR expression on CD8 (T suppressor/cytotoxic) lymphocytes in the acquired immune deficiency syndrome: evidence of CD8 cell immaturity. J. Immunol. 135, 1778–1785 [PubMed] [Google Scholar]
- 3. Tóth I., Le A. Q., Hartjen P., Thomssen A., Matzat V., Lehmann C., Scheurich C., Beisel C., Busch P., Degen O., Lohse A. W., Eiermann T., Fätkenheuer G., Meyer-Olson D., Bockhorn M., Hauber J., van Lunzen J., Schulze zur Wiesch J. (2013) Decreased frequency of CD73+CD8+ T cells of HIV infected patients correlates with immune activation and T cell exhaustion. J. Leukoc. Biol. 94 [DOI] [PubMed] [Google Scholar]
- 4. Burnstock G. (2011) Introductory overview of purinergic signaling. Front. Biosci. (Elite Ed.). 3, 896–900 [DOI] [PubMed] [Google Scholar]
- 5. Ernst P. B., Garrison J. C., Thompson L. F. (2010) Much ado about adenosine: adenosine synthesis and function in regulatory T cell biology. J. Immunol. 185, 1993–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quagliata F., Faig D., Conklyn M., Silber R. (1974) Studies on the lymphocyte 5′-nucleotidase in chronic lymphocytic leukemia, infectious mononucleosis, normal subpopulations, and phytohemagglutinin-stimulated cells. Cancer Res. 34, 3197–3202 [PubMed] [Google Scholar]
- 7. Takedachi M., Ebisuno Y., McGee S. T., Maeda E., McEver R. P., Tanaka T., Miyasaka M., Thompson L. F. (2008) CD73-generated adenosine restricts lymphocyte migration into draining lymph nodes. J. Immunol. 180, 6288–6296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takedachi M., Colgan S. P., Thompson L. F. (2007) The role of CD73 in the generation of extracellular adenosine for adenosine receptor signaling. In Adenosine Receptors: Therapeutic Aspects for Inflammatory and Immune Diseases (Hasko G. B. N., Cronstein, Szabo C., eds.), CRC/Taylor & Francis, Boca Raton, FL, USA, 39–48 [Google Scholar]
- 9. Takedachi M., Oohara H., Smith B. J., Iyama M., Kobashi M., Maeda K., Long C. L., Humphrey M. B., Stoecker B. J., Toyosawa S., Thompson L. F., Murakami S. (2012) CD73-generated adenosine promotes osteoblast differentiation. J. Cell. Physiol. 227, 2622–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. St. Hilaire C., Ziegler S. G., Markello T. C., Brusco A., Groden C., Gill F., Carlson-Donohoe H., Lederman R. J., Chen M. Y., Yang D., Siegenthaler M. P., Arduino C., Mancini C., Freudenthal B., Stanescu H. C., Zdebik A. A., Chaganti R. K., Nussbaum R. L., Kleta R., Gahl W. A., Boehm M. (2011) NT5E mutations and arterial calcifications. N. Engl. J. Med. 364, 432–442 [DOI] [PMC free article] [PubMed] [Google Scholar]