

Abstract
Airway hyperresponsiveness is the excessive narrowing of the airway lumen caused by stimuli that would cause little or no narrowing in the normal individual. It is one of the cardinal features of asthma, but its mechanisms remain unexplained. In asthma, the key end-effector of acute airway narrowing is contraction of the airway smooth muscle cell that is driven by myosin motors exerting their mechanical effects within an integrated cytoskeletal scaffolding. However, in just the past few years, our understanding of the rules that govern muscle biophysics has dramatically changed, as has their classical relationship to airway mechanics. It has become well established, for example, that muscle length is equilibrated dynamically rather than statically, and that in a dynamic setting non-classical features of muscle biophysics come to the forefront, including unanticipated interactions between the muscle and its time-varying load, as well as the ability of the muscle cell to adapt (remodel) its internal microstructure rapidly in response to its ever-changing mechanical environment. Here we consider some of these emerging concepts and, in particular, focus on structural remodeling of the airway smooth muscle cell as it relates to excessive airway narrowing in asthma.
Keywords: airway smooth muscle, plasticity, cytoskeleton, stiffness, malleability
Introduction
It was recognized quite early that the lung is an irritable organ and that stimulation of its contractile machinery in an animal with an open chest can cause an increase in lung recoil, air to be expelled, a rise in intratracheal pressure, and an increase in airways resistance (Colebatch et al., 1966; Colebatch and Mitchell, 1971; Dixon and Brodie, 1903; Mead, 1973; Otis, 1983). However, until the second half of the last century, smooth muscle embedded in the airways was not regarded as being a tissue of any particular significance in respiratory mechanics (Otis, 1983); airway smooth muscle was first described in 1804 by Franz Daniel Reisseisen (as related by Otis (1983)) and its functional properties considered by Einthoven (1892) and Dixon and Brodie (1903). A notable exception in that regard was Henry Hyde Salter, who, in 1859, was well aware of the “spastic” nature of this muscle tissue and its potential role in asthma (Salter, 1868). Airway smooth muscle is now recognized as being the major end-effector of acute airway narrowing in asthma (Lambert and Paré, 1997; Macklem, 1996). There is also widespread agreement that shortening of the airway smooth muscle cell is the proximal cause of excessive airway narrowing during an asthmatic attack (Dulin et al., 2003), with swelling of airway wall compartments and plugging by airway liquid or mucous being important amplifying factors (Homer and Elias, 2000; James et al., 1989; Lambert and Paré, 1997; Yager et al., 1989).
Nonetheless, it was many years ago that research on the asthmatic airways and research on the biophysics of airway smooth muscle had a parting of the ways (Seow and Fredberg, 2001). The study of smooth muscle biophysics took on a life of its own and pursued a deeply reductionist agenda, one that became focused to a large extent on myosin II and regulation of the acto-myosin cycling rate by myosin light chain kinase/phosphatase (Dillon et al., 1981; Hai and Murphy, 1988; Murphy, 1994; Stull et al., 1980). The study of airway biology pursued a reductionist agenda as well, but one that became focused less and less on contractile functions of muscle and instead emphasized immune responses, inflammatory cells and mediators, and, to the extent that smooth muscle remained of interest, that interest centered mainly on its synthetic, proliferative and migratory functions (Amrani and Panettieri, 2003; Black and Johnson, 1996; Black and Johnson, 2000; Black et al., 2001; Holgate et al., 2003; Kelleher et al., 1995; Zhu et al., 2001).
To better understand the impact of inflammatory remodeling processes upon muscle shortening and acute airway narrowing, computational models of ever increasing sophistications were formulated but, remarkably, the muscle compartment of these models remained at a relatively primitive level, being represented by nothing more than the classical relationship of active isometric force vs. muscle length (Lambert and Paré 1997; Lambert et al. 1993; Macklem 1987; Macklem 1989; Macklem 1990; Macklem 1996; Wiggs et al. 1992). As discussed below, this description is now considered to be problematic because the very existence of a well-defined static force-length relationship has of late been called into question, as has the classical notion that the muscle possesses a well-defined optimal length. Rather, other factors intrinsic to the muscle, especially muscle dynamics and mechanical plasticity, as well as unanticipated interactions between the muscle and its load, are now understood to be major factors affecting the ability of smooth muscle to narrow the airways (An et al., 2007; Fredberg, 2000a; Fredberg et al., 1999; Pratusevich et al., 1995; Seow and Fredberg, 2001; Seow and Stephens, 1986; Seow and Stephens, 1988; Seow et al., 2000).
The topics addressed in this review are intended to highlight recent discoveries that bring airway biology and smooth muscle biophysics together once again and to focus on the question that remains at the heart of both – Why in asthma do the airways narrow excessively? Here we do not provide an exhaustive review of the literature, but rather emphasize key biophysical properties of the airway smooth muscle as they relate to excessive airway narrowing. In the balance of this review, we draw first the classical picture of smooth muscle behavior and then go on to describe what we know about its non-classical behavior in a dynamic setting and, in particular, the remarkable ability of the muscle cell to adapt (remodel) its internal microstructure. Finally, we conclude by providing an emerging biophysical assay that seems to probe discrete molecular level remodeling dynamics of such underlying microstructure in the living cell.
Classical behavior of airway smooth muscle and the balance of static forces
The microstructure of striated muscle is highly ordered whereas there is abundant evidence in the literature demonstrating that the internal microstructure of smooth muscle is quite disordered (Small, 1995; Small and Gimona, 1998); it is, after all, its amorphous structure that gives ‘smooth’ muscle its name. Moreover, the airway smooth muscle cytoskeleton (CSK) – denoted here as the network of all major stress-bearing and stress-generating structures within the muscle cell – is in a continuous state of remodeling, a point to which we return below. Despite these differences, it has been widely presumed that to a first approximation Huxley’s sliding-filament model of muscle contraction (Huxley, 1957) describes the function of both smooth and striated muscle (Murphy, 1988; Murphy, 1994; Mijailovich et al., 2000). For many of the biophysical phenomena observed in airway smooth muscle, such as active force generation and shortening velocity, Huxley’s model represents a useful tool for thought (Huxley, 1957; Mijailovich et al., 2000), while for others, like mechanical plasticity, it does not.
As in the case of striated muscle contraction, the principal biophysical parameters that characterize smooth muscle contraction include the maximum active isometric force (or stress, which is simply the force carried per unit area), the length at which the muscle can attain that maximal force (i.e., the optimum length, Lo), and the shortening capacity of the muscle. The sliding-filament model of Huxley is the starting point for understanding each of these phenomena. As described by Huxley (1957), isometric force as well as muscle stiffness are proportional to the number of acto-myosin cross links per unit volume. This is true because, assuming rigid filaments, all bridges within a given contractile unit must act mechanically in parallel, with their displacements being identical and their forces being additive. The optimal length (Lo) is then attributed to the extent of overlap between the myosin filament and the actin filament, with Lo corresponding to a maximum number of myosin heads finding themselves within the striking distance of an available binding sites on the actin filament: the maximum active stress supported by smooth vs. striated muscle is approximately the same and is of the order 105 Pascal. Interestingly, however, whereas unloaded striated muscle can shorten perhaps 20% from its optimum length, unloaded smooth muscle can shorten as much as 70% (Stephens, 1987; Stephens and Seow, 1993; Uvelius, 1976).
In striated muscle, this maximum shortening capacity is limited by the collision of the myosin filament end with the z-disc. Smooth muscle possesses no structure comparable to the z-disc; however, several physical factors may come into play to limit its capacity for unloaded shortening. For example, Small (1995) has shown that actin filaments of the smooth muscle contractile unit connect to the cytoskeleton at cytoplasmic dense bodies and with the longitudinal rib-like arrays of dense plaques of the membrane skeleton that couple to the extracellular matrix. These structural organizations, as well as the polarity of actin filaments inserted in the dense bodies/plaques (Bond and Somlyo, 1982) and the side-polar configuration of myosin filaments (Tonino et al., 2002; Xu et al., 1996) may come into play in limiting smooth muscle shortening. Still other factors likely to be involved include length-dependent activation (An and Hai, 1999; An and Hai, 2000; Mehta et al., 1996; Youn et al., 1998) and rearrangement (Pratusevich et al., 1995; Gunst et al., 1995) of the contractile unit and its associated scaffold, as well as length-dependent change in the internal load (Seow and Stephens, 1986; Seow and Stephens, 1988; Stephens and Kromer, 1971; Stephens and Seow, 1993; Warshaw et al., 1988).
What are the extramuscular factors that act to limit airway smooth muscle shortening? The basic notion, of course, is that muscle shortening stops when the total force generated by the muscle comes into a static balance with the load against which the muscle has shortened, both of which vary with muscle length. The factors setting the load include the elasticity of the airway wall, elastic tethering forces conferred by the surrounding lung parenchyma, active tethering forces conferred by contractile cells in the lung parenchyma (Nagase et al., 1994; Romero and Ludwig, 1991), mechanical coupling of the airway to the parenchyma by the peribronchial adventitia, and buckling of the airway epithelium and submucosa (Ding et al., 1987; Robatto et al., 1992; Wiggs et al., 1997). In addition, the airway smooth muscle itself is a syncytium comprised mostly of smooth muscle cells, aligned roughly along the axis of muscle shortening, and held together by an intercellular connective tissue network. In order to conserve volume, as the muscle shortens it must also thicken. And as the muscle shortens and thickens, the intercellular connective tissue network must distort accordingly. Meiss (1999) has shown evidence to suggest that at the extremes of muscle shortening it may be the loads associated with radial expansion (relative to the axis of muscle shortening) of the intercellular connective tissue network that limits the ability of the muscle to shorten further.
In the healthy intact dog, airway smooth muscle possesses sufficient force generating capacity to close all airways (Brown and Mitzner, 1998; Warner and Gunst, 1992). This fact may at first seem to be unremarkable, but it is not easily reconciled with the observation that when healthy animals or humans are challenged with inhaled contractile agonists in concentrations thought to be sufficient to activate the muscle maximally, resulting airway narrowing is limited in extent, and that limit falls far short of airway closure (Moore et al., 1997; Moore et al., 1998). Breathing remains unaccountably easy. Indeed, it is this lightness of breathing in the healthy challenged lung, rather than the labored breathing that is characteristic of the asthmatic lung, that in many ways presents the greater challenge to our understanding of the determinants of airway hyperresponsiveness (Fredberg and Shore, 1999; Sterk and Bel, 1989; Woolcock and Peat, 1989). Brown and Mitzner (1998) have suggested that such limitation of airway closure in the healthy challenged lung – the appearance of a plateau in dose-response curves – may reflect uneven or limited aerosol delivery to the airways. Still another possibility, however, is that there are precise mechanism(s) that act to limit the extent of muscle shortening in the healthy breathing lung and that these mechanism(s) become compromised in the asthmatic lung. It has been suspected that the impairment of those salutary mechanism(s), if they could only be understood, might help to unlock some of the secrets surrounding this excessive narrowing of the asthmatic airways, as well as the morbidity and mortality associated with asthma (Woolcock and Peat, 1989; Fish et al., 1981; Lim et al., 1987; Nadel and Tierney, 1961; Skloot et al., 1995). This brings us to muscle dynamics and to consider several plausible mechanisms that could account for airway hyperresponsiveness in asthma.
Shortening velocity and other manifestations of muscle dynamics
The oldest and certainly the simplest explanation of airway hyperresponsiveness would be that muscle from the asthmatic airways is stronger than muscle from the healthy airways, but evidence in support of that hypothesis remains equivocal (Black and Johnson, 1996; Black and Johnson, 2000; De Jongste et al., 1987; Solway and Fredberg, 1997). Indeed, a number of earlier studies, in which tissues were obtained post-mortem or surgically, have reported normal (Bai, 1990; Bjorck et al., 1992) and even hypo-contractility (Goldie et al., 1986; Whicker et al., 1988) of muscle from the asthmatic airways. Accordingly, studies from the laboratory of Stephens and colleagues (Antonissen et al., 1979; Fan et al., 1997; Jiang et al., 1992; Ma et al., 2002; Stephens et al., 1986) have emphasized that the force generation capacity of allergen-sensitized airway smooth muscle of the dog, or asthmatic muscle from the human, is no different from that of control muscle. As a result, the search for an explanation turned to other factors, and several alternative hypotheses have been advanced. These fall into three broad classes, each of which is consistent with remodeling events induced by the inflammatory microenvironment, and include an increase of muscle mass (Johnson et al., 2001; Lambert et al., 1993; Thomson et al., 1996; Wiggs et al., 1992), a decrease of the static load, possibly by the uncoupling of the airway wall to the surrounding lung parenchyma, against which the muscle shortens (Ding et al., 1987; James et al., 1989; Macklem, 1996; Wiggs et al., 1992; Wiggs et al., 1997), and a decrease of the fluctuating load that perturbs myosin binding during breathing (Fredberg, 1998; Fredberg, 2000a; Fredberg, 2000b; Fredberg et al., 1999; Mijailovich et al., 2000). Together, these hypotheses are attractive because they suggest a variety of mechanisms by which airway smooth muscle can shorten excessively even while the isometric force generating capacity of the muscle remains essentially unchanged.
Aside from changes in the static load and/or the dynamic load, however, a consistent association has been noted between airway hyperresponsiveness and unloaded shortening velocity of the muscle (Antonissen et al., 1979; Fan et al., 1997; Ma et al., 2002; Duguet et al., 2000; Stephens et al., 1986; Wang et al., 1997). This association suggests the possibility that the problem with airway smooth muscle in asthma may be that it is too fast rather than too strong. But how shortening velocity – a dynamic property of the muscle – might cause excessive airway narrowing – a parameter that was thought to be determined by a balance of static forces – remains unclear. To account for increased shortening capacity of unloaded cells, Stephens and colleagues have reasoned that upon activation virtually all muscle shortening is completed within the first few seconds (Ma et al., 2002; Seow and Stephens, 1986; Stephens et al., 1986). As such, the faster the muscle can shorten within this limited time window, the more it will shorten. However, in isotonic loading conditions at physiological levels of load, muscle shortening is indeed most rapid at the very beginning of the contraction, but appreciable shortening continues for at least 10 min after the onset of the contractile stimulus (Fredberg et al., 1999). An alternative hypothesis to explain why intrinsically faster muscle might shorten more comes from consideration of the temporal fluctuations of the muscle load that are attributable to the action of spontaneous breathing (Fredberg et al., 1997; Fredberg et al., 1999; Solway and Fredberg, 1997). Load fluctuations that are attendant to spontaneous breathing are the most potent of all known bronchodilating agencies (Gump et al., 2001; Shen et al., 1997). Among many possible effects, these load fluctuations perturb the binding of myosin to actin, causing the myosin head to detach from actin much sooner than it would have during an isometric contraction (Fredberg et al., 1997; Fredberg et al., 1999; Mijailovich et al., 2000). But the faster the myosin cycling (i.e., the faster the muscle), the more difficult it is for imposed load fluctuations to perturb the acto-myosin interaction. This is because the faster the intrinsic rate of cycling, the faster will a bridge, once becoming detached, reattach and contribute once again to active force and stiffness.
Why is muscle from the allergen-sensitized animal or asthmatic subject faster? For technical reasons, in their study on the single airway smooth muscle cell freshly isolated from bronchial biopsies obtained from an asthmatic subject, Ma and his colleagues (2002) did not measure protein expression levels of myosin light chain kinase (MLCK), but their finding of increased content of message strongly implicates MLCK. Although regulation of myosin phosphorylation is a complex process with multiple kinases and phosphatases, this finding also substantially narrows the search for the culprit that may account for the mechanical changes observed in these cells. Furthermore, this study seems to rule out changes in the distribution of myosin heavy chain isoforms; content and isoform distributions of message from asthmatic cells showed the presence of smooth muscle myosin heavy chain A (SM-A) but not SM-B (Ma et al., 2002), the latter of which contains a seven-amino acid insert that is typical of phasic rather than tonic smooth muscle, and is by far the faster of the two isoforms (Lauzon et al., 1998; Murphy et al., 1997).
Using laser capture microdissection of airway smooth muscle from bronchial biopsies obtained from normal vs. mild-to-moderate asthmatics, Woodruff et al. (2004) also found no differences in the expressions profile of a panel of genes that are often considered markers of hypercontractile phenotype (including MLCK, however) but detected nearly twofold increase in the number of airway smooth muscle cells in the asthmatic airways. Although the source of the increased cell number (increased proliferation, decreased apoptosis, and/or increased migration) remains unclear (Hirst et al., 2004; Johnson et al., 2001; Lazaar and Panettieri, 2005; Madison, 2003; Woodruff et al., 2004; Zacour and Martin, 1996), increased muscle mass alone is sufficient to predispose toward airway hyperresponsiveness (James et al., 1989; Lambert et al., 1993; Moreno et al., 1986). The question of whether muscle mass (quantity) and muscle contractility (quality) might covary remains to be elucidated, however. For example, it is likely that cells in the proliferative/synthetic/maturational state might be less contractile than similar airway smooth muscle cells differentiated into fully contractile state – an effect that would be compensatory – but no mechanical data are available to support that possibility.
Biophysical characterization of airway smooth muscle: bronchospam in culture?
With recent technological advances, such as atomic force microscopy (Alcaraz et al., 2003; Smith et al., 2005), two-point and laser-tracking microrheology (Van Citters et al., 2006; Yamada et al., 2000), magnetic tweezers (Bausch et al., 1998; Bausch et al., 1999), and traction microscopy (Butler et al., 2002; Tolic-Norrelykke et al., 2002), a single living cell in culture can be now characterized biophysically. While using cultured cells has certain limitations, they do offer the advantage that, when passaged in culture, serum-deprived airway smooth muscle cells retain functional responses to a wide panel of agonists and signaling pathways that are implicated in asthma (Halayko et al., 1999; Hubmayr et al., 1996; Panettieri et al., 1989; Shore et al., 1997; Tao et al., 1999; Tao et al., 2003; Tolloczko et al., 1995). To probe deeper into mechanical properties of the airway smooth muscle cell, in our laboratories, we use a technology that has its roots in an early contribution of Francis H.C. Crick.
Before his well known work on the double helical structure of deoxyribonucleic acid (DNA) (Watson and Crick, 1953a; Watson and Crick, 1953b), Crick measured the viscosity and elasticity of the medium inside cells by observing internalized magnetic particles and how these particles rotate in reaction to an applied magnetic field (Crick and Hughes, 1950). Extending this approach further, Valberg and his colleagues studied populations of particles internalized into populations of cells and measured induced bead rotations by remote sensing, namely, by means of changes in the horizontal projection of the remanent magnetic field produced by the magnetized particles as they rotate (Valberg, 1984; Valberg and Feldman, 1987). In a major step forward, we subsequently adapted this technique still further (Wang et al., 1993; Fabry et al., 2001) by using ligand-coated ferrimagnetic microbeads – not internalized as before – but rather bound to the cytoskeleton (CSK) via membrane-spanning integrin receptors. Recently, we showed that changes in cell stiffness measured in this way correlate well with stiffness changes in the same cells measured by atomic force microscopy (Alcaraz et al., 2003) and with force changes measured with traction microscopy (Wang et al., 2002). This method is now known as magnetic twisting cytometry (MTC) and has evolved into a useful tool to probe the mechanical properties of a variety of cell types, both cultured and freshly isolated, through different receptor systems, and with a variety of experimental interventions (Fabry et al., 2001; Puig-de-Morales et al., 2004; Laudadio et al., 2005; Deng et al., 2005; Trepat et al., 2007).
The principle of magnetic twisting cytometry is straightforward (Figure 1). A ferrimagnetic microbead (4.5 μm in diameter) is coated with a synthetic peptide containing the sequence Arg-Gly-Asp (RGD) and is then allowed to bind to the cell. Such RGD-coated bead binds avidly to cell surface integrin receptors (Wang et al., 1993), forms focal adhesions (Matthews et al., 2004), and becomes well-integrated into the cytoskeletal scaffold (Maksym et al., 2000): it also displays tight functional coupling to stress-bearing cytoskeletal structures and the contractile apparatus (An et al., 2002; Hu et al., 2003). By imposing a uniform magnetic field upon the magnetized bead, a small torque is applied and resulting bead motions deform structures deep in the cell interior (Hu et al., 2003). Such forced bead motions are impeded by mechanical stresses developed within the cell body, and the ratio of specific torque to lateral bead displacements – now measured optically instead of remote sensing – is taken as a measure of cell stiffness (Figure 1).
Figure 1. Magnetic twisting cytometry with optical detection.
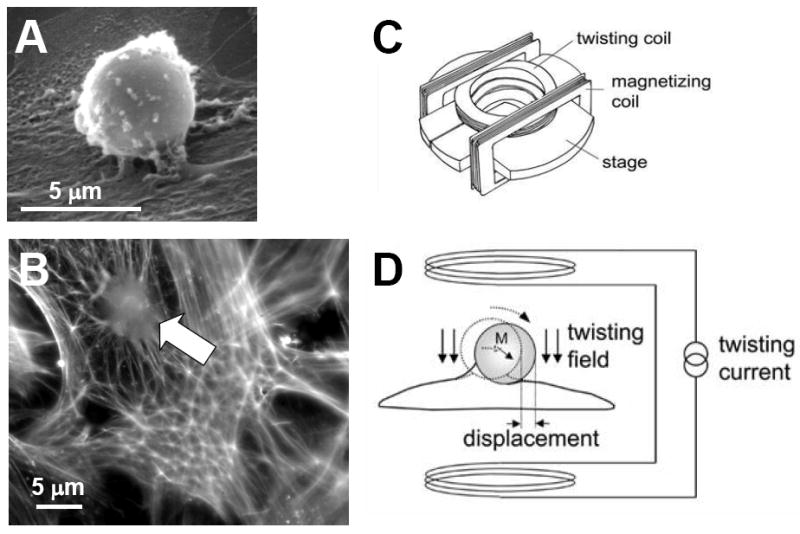
A. An RGD-coated bead (4.5 μm in diameter) binds to the surface of the adherent cell. B. Such bead (white arrow) becomes well-integrated into underlying actin lattice (phalloidin staining). C. The bead is magnetized horizontally (parallel to the surface on which cells are plated) and then twisted in a vertically aligned homogenous magnetic field that is varying sinusoidally in time. D. This sinusoidal twisting field causes both a rotation and a pivoting displacement of the bead. As the bead moves, the cell develops internal stresses which in turn resist bead motions. Here the ratio of specific torque to lateral bead displacement is computed and is expressed as cell stiffness in Pa/nm. [Reproduced by permission of J. Appl. Physiol., Vol. 91, pp. 986-994 (2001), © The American Physiological Society and of Phys. Rev. Lett., Vol. 87, 14 (2001), © The American Physical Society]
Using this technique, it has been previously demonstrated that airway smooth muscle cells in culture exhibit pharmacomechanical coupling to a wide panel of contractile and relaxing agonists (An et al., 2002; Hubmayr et al., 1996). For example, cell stiffness increases in response to agonists reported to increase intracellular Ca2+ concentration ([Ca2+]i) or inositol 1,4,5-trisphosphate (IP3) formation and decreases in response to agonists that are known to increase intracellular cAMP or cGMP levels (An et al., 2002; Hubmayr et al., 1996; Shore et al., 1997). Although stiffness is an indirect measure of contractility (Fredberg et al., 1997), changes in cell stiffness range appreciably from maximally relaxed to maximally activated states (Fabry et al., 2001), and such stiffening responses require, as in intact tissues, actin polymerization as well as myosin activation (An et al., 2002; Mehta and Gunst, 1999). Indeed, active stresses within individual airway smooth muscle cells as measured by traction microscopy span a similarly wide range (Figure 2) and closely track changes in cell stiffness as measured by magnetic twisting cytometry (Wang et al., 2002). Taken together, mechanical responsiveness of airway smooth muscle cells measured at the level of the single cell in vitro is consistent with physiological responses measured at tissue and organ levels (Fredberg et al., 1996; Mehta and Gunst, 1999).
Figure 2. Airway smooth muscle cell exerts traction upon an elastic substrate.
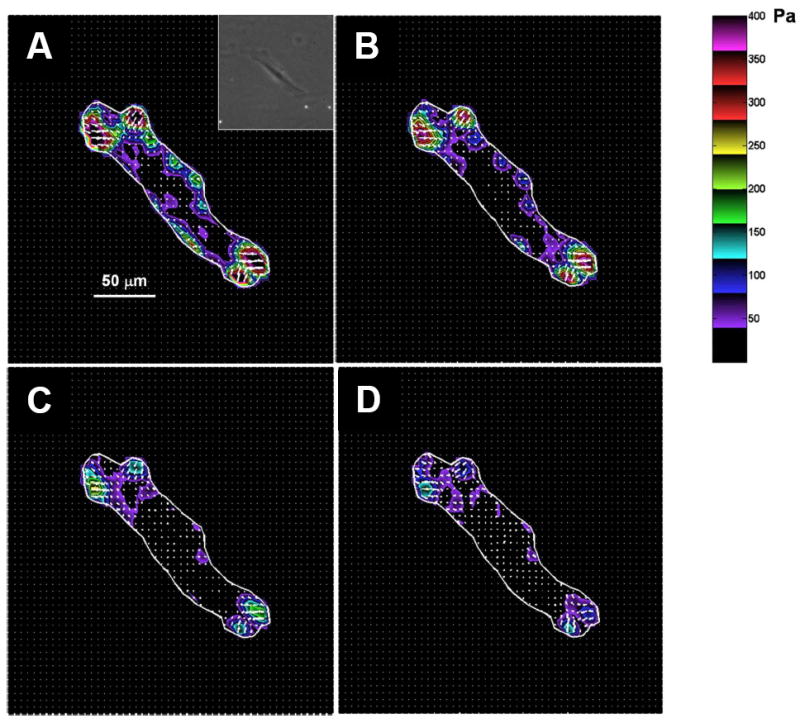
A representative changes in traction field of a single airway smooth muscle cell in response to isoproterenol (A, 0 μM; B, 0.1 μM; C, 1 μM; D, 10 μM). The traction field was computed from the displacement field using Fourier transform traction cytometry (FTTC) (Butler et al. 2002; Tolic-Norrelykke et al. 2002; Wang et al. 2002). The cell boundary is shown by the white line. Colors show the magnitude of the tractions in Pascal (Pa) (see color scale). Arrows show the direction and relative magnitude of the tractions. In general, the greatest tractions are at the cell periphery and directed centripetally. Inset: A phase-contrast image of the respective airway smooth muscle cell. Scale bar, 50 μm.
Does mechanical responsiveness of the airway smooth muscle cell predict airway hyperresponsiveness? To address this question, we have recently contrasted biophysical properties of the airway smooth muscle cell isolated from the relatively hyporesponsive Lewis rat vs. the relatively hyperresponsive Fisher rat (An et al., 2006b); these strains represent an attractive model of airway hyperresponsiveness. In agreement with biochemical changes that have been previously reported in these cells (Tao et al., 1999; Tao et al., 2003; Tolloczko et al., 1995), compared with cells isolated from Lewis rat, those isolated from Fisher rat demonstrate in turn greater extent of the stiffening response to a panel of contractile agonists that are known to increase [Ca2+]i or IP3 formation. Fisher airway smooth muscle cells stiffen fast and also stiffen more (Figure 3). Furthermore, consistent with these changes in cell stiffness, airway smooth muscle cells isolated from the relatively hyperresponsive Fisher rat also exert bigger contractile forces and exhibit greater scope of these forces (An et al., 2006b). Taken together, these findings firmly establish that comprehensive biophysical characterization of bronchospasm in culture is a reality, and these characterizations at the level of the single cell show mechanical responses that are consistent with phenotypic differences in airway responsiveness measured at tissue and organ levels (Dandurand et al., 1993a; Dandurand et al., 1993b; Eidelman et al., 1991; Jia et al., 1995; Tao et al., 1999).
Figure 3. Fisher airway smooth muscle cells stiffen fast and also stiffen more.
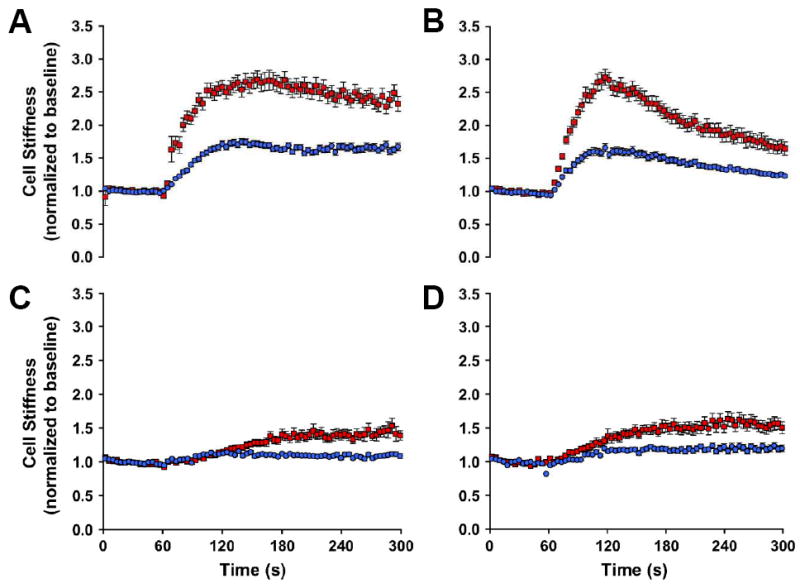
Airway smooth muscle cells isolated from the relatively hyporesponsive Lewis rat (blue closed circles) and the relatively hyperresponsive Fisher rat (red closed squares) were maximally stimulated with a panel of contractile agonists: A, 5-HT [1 μM]; B, bradykinin [1 μM]; C, acetylcholine [1 μM]; D, carbachol [100 μM]. For each agonist, changes in cell stiffness were normalized to the baseline stiffness of each individual cell before stimulation. [Reproduced by permission of Am. J. Respir. Cell Mole. Biol., Vol. 35, pp. 55-64 (2006), © The American Thoracic Society]
Like human asthmatic airways (Johnson et al., 2001; Woodruff et al., 2004), the relatively hyperresponsive Fisher rat has more smooth muscle cells in its airways (Eidelman et al., 1991), and these cells show greater capacity to proliferate in culture (Zacour and Martin, 1996). Although these features, together with greater muscle contraction (shortening velocity as well as contractile force), may account for enhanced airway responsiveness of Fisher rat, to what extent this animal model recapitulates the pathophysiology associated with human disease is still ill-defined: the precise role of human airway smooth muscle cell in the pathogenesis of airway hyperresponsiveness in asthma remains unclear. To this end, in our laboratories, efforts are now being focused on characterizing biophysical properties of the human airway smooth muscle cell isolated from bronchial biopsies obtained from clinically well-characterized non-asthmatic and asthmatic volunteers, who have been sub-phenotyped according to the extent of airway hyperresponsiveness vs. fixed airflow obstruction (Haitchi et al., 2005; An et al., 2006a). We now turn to some of the newly discovered muscle biophysics in a dynamic setting and, in particular, focus on the ability of the airway smooth muscle cell to remodel its internal microstructure rapidly in response its ever-changing mechanical microenvironment, as well as an emerging biophysical assay that probes such remodeling in a living cell.
Mechanical plasticity: a non-classical feature of airway smooth muscle
When activated muscle in the muscle bath is subjected to progressively increasing load fluctuations approaching the magnitude and frequency expected during normal breathing, the muscle lengthens appreciably in response (Fredberg et al., 1999). But when load fluctuations are progressively reduced, the muscle re-shortens somewhat but fails to return to its original length. This incomplete re-shortening is not account for by muscle injury: the original operating length can be recovered simply by removing the contractile agonist and allowing the muscle a short interval to relax before contracting again. Neither can incomplete re-shortening be accounted for by myosin dynamics; myosin dynamics by themselves predict complete re-shortening when the load fluctuations are removed (Fredberg et al., 1999). Thus, the failure of activated muscle to re-shorten completely is evidence of a plasticity of the contractile response. During a sustained contraction, the operational length of the muscle for a given loading, or the force at a given length, can be reset by loading and the history of that loading (Ford et al., 1994; Fredberg et al., 1997; Fredberg et al., 1999; Gunst and Wu, 2001; Gunst et al., 1993; Pratusevich et al., 1995; Wang et al., 2001). In healthy individuals this plasticity seems to work in a favorable direction, allowing activated muscle to be reset to a longer length. The asthmatic, it has been argued, never manages to melt the contractile domain in the airway smooth muscle and, as such, the benefits of this plastic response are not attained (Fredberg, 200b).
It is now firmly established that airway smooth muscle can somehow adapt its contractile machinery, as well as the cytoskeletal scaffolding on which that machinery operates, in such a way that the muscle can maintain the same high force over an extraordinary range of muscle length (An et al., 2007; Ford et al., 1994; Fredberg, 1998; Gunst and Wu, 2001; Gunst et al., 1993; Gunst et al., 1995; Kuo et al., 2001; Kuo et al., 2003; Naghshin et al., 2003; Pratusevich et al., 1995; Qi et al., 2002; Seow and Fredberg, 2001; Seow et al., 2000; Wang et al., 2001). Accordingly, the airway smooth muscle cell is now characterized by its ability to disassemble its contractile apparatus when an appropriate stimulus is given, and its ability to reassemble that apparatus when accommodated at a fixed length. When exposed to contractile agonists, for example, airway smooth muscle cells in culture reorganize cytoskeletal biopolymers, especially actin (Hirshman and Emala, 1999), and become stiffer (An et al., 2002). Although cell stiffening is attributable largely to activation of the contractile machinery, an intact actin lattice has been shown to be necessary but not sufficient to account for the stiffening response (An et al., 2002).
Malleability of the airway smooth muscle cell: a novel tool for thought
Malleability of the cell and its mechanical consequences have been called by various authors mechanical plasticity, remodeling, accommodation or adaptation. Even though the force generating capacity varies little with length in the fully adapted muscle, the unloaded shortening velocity and the muscle compliance vary with muscle length in such a way as to suggest that the muscle cell adapts by adding or subtracting contractile units that are mechanically in series (Figure 4). The mechanisms by which these changes come about and the factors that control the rate of plastic adaptation are unknown, however.
Figure 4. Mechanical plasticity of the airway smooth muscle.
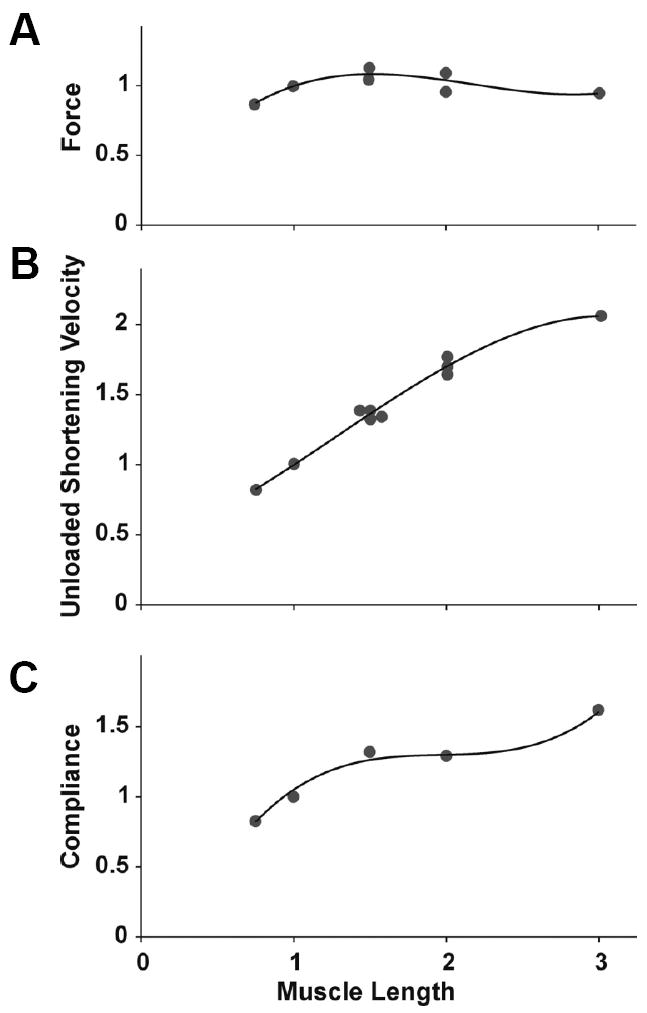
Isometric force (F), unloaded shortening velocity (V), and compliance (C) of canine tracheal smooth muscle activated over a range of muscle lengths. Circles represent data modified from Pratusevich et al. (1995) and Kuo et al. (2003), as compiled by Lambert et al. (2004); solid lines are 3rd-order polynominal functions adjusted to the original data (Silveira and Fredberg, 2005). [Reproduced by permission of Can. J. Physiol. Pharmacol. Vol. 83, pp. 923-931 (2005), © NRC Canada]
Several hypotheses have been advanced to explain smooth muscle plasticity. Ford and colleagues have suggested that the architecture of the myosin fibers themselves may change (Ford et al., 1994; Kuo et al., 2001; Kuo et al., 2003; Pratusevich et al., 1995; Seow et al., 2000), while Gunst and colleagues (Gunst and Wu, 2001; Gunst et al., 1993; Gunst et al., 1995) have argued that it is the connection of the actin filament to the focal adhesion plaque at the cell boundary that is influenced by loading history. Alternatively, another notion is that secondary but important molecules stabilize the cytoskeleton, and as the contractile domain melts under the influence of imposed load fluctuations, those loads must be borne increasingly by the scaffolding itself, and thus reflects malleability of the cytoskeletal domain (Fredberg, 2000a; Halayko and Solway, 2001; Gunst et al., 1995; Wang and Bitar, 1998). In that connection a role for the Rho-A pathway has been suggested (Halayko and Solway, 2001; Mehta et al., 2000) and some evidence now suggests that the p38 MAP kinase pathway may be involved (Lakser et al., 2002). For example, airway smooth muscle incubated with an inhibitor of the p38 MAP kinase pathway demonstrates a greater degree of fluctuation-driven muscle lengthening than does a control muscle, and upon removal of the force fluctuations it remains at a greater length. Moreover, force fluctuations themselves activate the p38 MAP kinase pathway. It is noteworthy in that connection that heat shock protein 27 (HSP27), a downstream target of Rho and p38, has been implicated as an essential element in cytoskeletal remodeling of the airway smooth muscle cell (Gerthoffer and Pohl, 1994; Hedges et al., 1998; Hedges et al., 1999; Hedges et al., 2000; Yamboliev et al., 2000). These findings are consistent with the hypothesis that stress response pathways may somehow stabilize the airway smooth muscle cytoskeleton and, thereby limit the bronchodilating effects of deep inspirations.
Recently, we have made a series of observations in a number of different cell types and reported a functional assay that probes discrete molecular level remodeling dynamics of the cytoskeleton (An et al., 2004; An et al., 2005; Bursac et al., 2005; Bursac et al., 2007). This assay is based on spontaneous nano-scale movements of an individual RGD-coated microbead tightly anchored to the cytoskeleton of the living cell (Figure 5): we reasoned that the bead can move spontaneously only if microstructure to which it is attached rearranges (remodels), and quantified these motions by calculating its mean square displacement (MSDb),
| (1) |
where r(t) is the bead position at time t, Δt is the time lag (Δt = 1/12s), and brackets indicate an average over many starting times (Bursac et al., 2005; Bursac et al., 2007). As shown below, MSDb of most beads increased with time according to a power law relationship; ensemble average of all MSDb (MSD) increased faster than linearly with time (~ t1.6), exhibiting superdiffusive motions (Figure 5). Such anomalous motions were also observed on cells seeded on a micropatterned substrate on which a cell could adhere but not crawl (Bursac et al., 2007; Parker et al., 2002). Accordingly, unlike a simple diffusive thermal Brownian motion that increases its MSD linearly with time (Kubo, 1986), spontaneous motions of an individual RGD-coated bead were non-thermal in nature and, instead consistent with the notion that these anomalous motions report ongoing molecular level reorganization (remodeling) of the underlying cytoskeleton in the living cell (An et al., 2004; An et al., 2005; Bursac et al., 2005; Bursac et al., 2007). Using this method, we have also demonstrated that the rate of cytoskeletal remodeling is appreciably different between airway smooth muscle cells isolated from the relatively hyporesponsive Lewis rat vs. the relatively hyperresponsive Fisher rat: Fisher cells exhibit faster remodeling dynamics (An et al., 2006b). Furthermore, such remodeling event in the living cell is dependent on the levels of intracellular ATP content (An et al., 2006b; Bursac et al., 2005) and becomes progressively slow with phosphorylation of HSP27 (An et al., 2004; An et al., 2005).
Figure 5. Cytoskeleton remodeling of the airway smooth muscle cell.
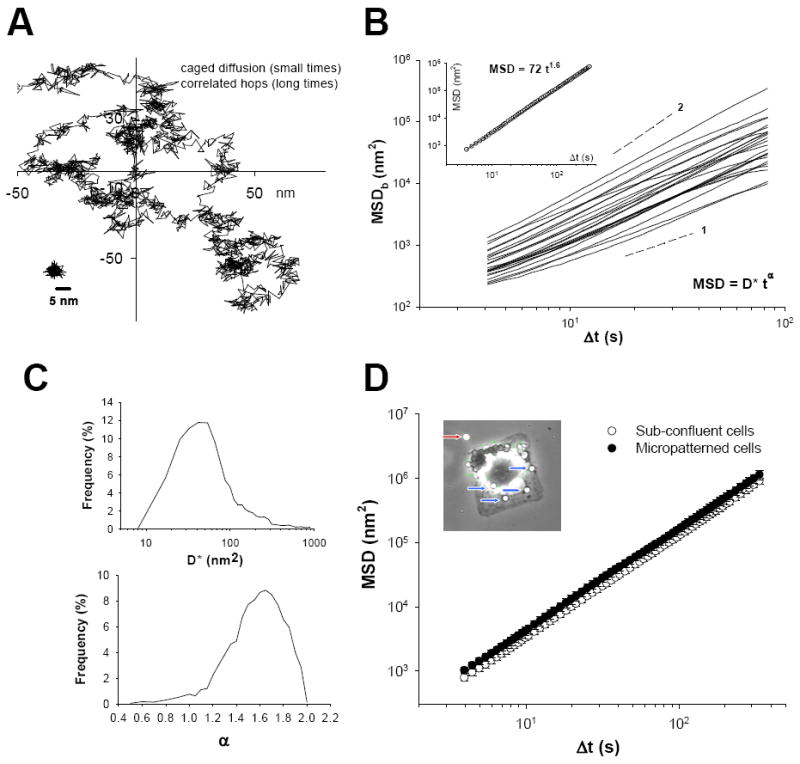
A. Spontaneous motions of a representative bead show intermittent dynamics, with periods of confinement alternating with hopping; a bead glued to the coverslip is shown at the bottom left and taken to represent the upper limit of measurement noise. B. MSDb calculated from Equation 1 is shown for representative beads. Inset:Ensemble average of all MSDb (MSD) increased with time as ~ t1.6. C. The histograms of diffusion coefficient D* and exponent α estimated from a least-square fits of a power-law (Equation 2) to the MSDb data. D. Ensemble average of all MSDb (MSD) increased faster than linearly with time (~ t1.6); beads attached to a cell seeded on a micropatterned substrate (50 μm × 50 μm), on which it could adhere but not crawl, exhibited the same anomalous motions. [Reproduced by permission of Biochem. Biophys. Res. Comm., Vol. 355, pp. 324-330 (2007), © Elsevier Inc.]
Indeed, evidence supporting the notion of a highly malleable cell is accumulating rapidly, but a molecular basis to explain this malleability is only beginning to emerge. Most recently, we have reported that, in response to a transient stretch-unstretch maneuver with zero residual macroscale strain, the airway smooth muscle cell promptly fluidizes and then slowly re-solidifies (Trepat et al., 2007). At the same time, the rate of spontaneous nano-scale structural rearrangements promptly accelerates and then slowly decays in a scale-free manner (Trepat et al., 2007). These findings are consistent with the notion that fluidization provides freedom of the cell to reorganize contractile units, stress fibers and focal adhesions in response to mechanical stress (Trepat et al., 2007) and, regardless of the specific molecules and mechanisms invoked to explain the plasticity of the contractile responses, therefore, the melting of the contractile domain would appear to be a necessary (or permissive) event (An et al., 2004; An et al., 2006; An et al., 2007; Bursac et al., 2005; Bursac et al., 2007; Deng et al., 2006; Fabry et al., 2001; Fabry and Fredberg, 2003; Gunst and Fredberg, 2003; Fredberg, 2000b; Trepat et al., 2007). How these molecular changes and malleability of the airway smooth muscle cell, in turn, correlate with the progression of asthma pathophysiology are currently under investigation in our laboratories.
Future directions
To understand the multifaceted problem of airway hyperresponsiveness in asthma, an integrative understanding that brings together a diversity of factors will be essential. We have outlined here an emerging picture of smooth muscle biophysics as it relates to excessive airway narrowing in asthma, but we need to keep in mind that asthma is a chronic inflammatory disorder and, therefore, understanding the impact of inflammatory remodeling of the airway wall and the airway smooth muscle cell on disease presentation will be vital. Fortunately, with recent technological advances, we are now equipped with both biochemical and biophysical tools to address nagging questions that have often separated the fields of airway biology and smooth muscle biophysics.
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute grants HL-59682 and HL-33009, and by National Institute of Environmental Health Sciences grant P30-ES03819-11.
References
- Alcaraz J, Buscemi L, Grabulosa M, Trepat X, Fabry B, Farre R, Navajas D. Microrheology of human lung epithelial cells measured by atomic force microscopy. Biophys J. 2003;84:2071–2079. doi: 10.1016/S0006-3495(03)75014-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani Y, Panettieri RA. Airway smooth muscle: contraction and beyond. Internatl J Biochem Cell Biol. 2003;35:272–276. doi: 10.1016/s1357-2725(02)00259-5. [DOI] [PubMed] [Google Scholar]
- An SS, Hai CM. Mechanical strain modulates maximal phosphatidylinositol turnover in airway smooth muscle. Am J Physiol. 1999;277:L968–L974. doi: 10.1152/ajplung.1999.277.5.L968. [DOI] [PubMed] [Google Scholar]
- An SS, Hai CM. Mechanical signals and mechanosensitive modulation of intracellular [Ca2+] in smooth muscle. Am J Physiol. 2000;279:C1375–C1384. doi: 10.1152/ajpcell.2000.279.5.C1375. [DOI] [PubMed] [Google Scholar]
- An SS, Laudadio RE, Lai J, Rogers RA, Fredberg JJ. Stiffness changes in cultured airway smooth muscle cells. Am J Physiol. 2002;283:C792–C801. doi: 10.1152/ajpcell.00425.2001. [DOI] [PubMed] [Google Scholar]
- An SS, Fabry B, Mellema M, Bursac P, Gerthoffer WT, Kayyali US, Gaestel M, Shore SA, Fredberg JJ. Role of heat shock protein 27 in cytoskeletal remodeling of the airway smooth muscle cell. J Appl Physiol. 2004;96:1707–1713. doi: 10.1152/japplphysiol.01129.2003. [DOI] [PubMed] [Google Scholar]
- An SS, Pennella CM, Gonnabathula A, Chen J, Wang N, Gaestel M, Hassoun PM, Fredberg JJ, Kayyali US. Hypoxia alters biophysical properties of endothelial cells via p38 MAPK- and Rho kinase-dependent pathways. Am J Physiol. 2005;289:C521–C530. doi: 10.1152/ajpcell.00429.2004. [DOI] [PubMed] [Google Scholar]
- An SS, Andrews AL, Deng L, Fredberg JJ, Holgate ST, Davies DE. Biophysical properties of fibroblasts from normal versus asthmatic airways. Proc Am Thorac Soc. 2006a;3:A264. [Google Scholar]
- An SS, Fabry B, Trepat X, Wang N, Fredberg JJ. Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? Am J Respir Cell Mole Biol. 2006b;35:55–64. doi: 10.1165/rcmb.2005-0453OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An SS, Bai TR, Bates JHT, Black JL, Brown RH, Brusasco V, Chitano P, Deng L, Dowell M, Eidelman DH, Fabry B, Ford LE, Fredberg JJ, Gerthoffer WT, Gilbert SH, Gunst SJ, Halayko AJ, Ingram RH, Irvin CG, James AL, Janssen LJ, King GG, Knight DA, Lauzon AM, Lakser OJ, Ludwig MS, Lutchen KR, Maksym GN, Martin JG, Mauad T, McParland BE, Mijailovich SM, Mitchell HW, Mitchell RW, Mitzner W, Murphy TM, Paré PD, Pellegrino R, Seow CY, Smith PG, Solway J, Schellenberg RR, Silveira PS, Stephens NL, Sterk PJ, Stewart AG, Tang DD, Tepper RS, Wang L. Airway smooth muscle dynamics: a final common pathway of airway obstruction in asthma. Eur Respir J. 2007;29:834–860. doi: 10.1183/09031936.00112606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonissen LA, Mitchell RW, Kroeger EA, Krepon W, Tse KS, Stephens NL. Mechanical alterations of airway smooth muscle in a canine asthmatic model. J Appl Physiol. 1979;46:681–687. doi: 10.1152/jappl.1979.46.4.681. [DOI] [PubMed] [Google Scholar]
- Bai TR. Abnormalities in airway smooth muscle in fatal asthma. Am Rev Respir Dis. 1990;141:552–557. doi: 10.1164/ajrccm/141.3.552. [DOI] [PubMed] [Google Scholar]
- Bausch AR, Ziemann F, Boulbitch AA, Jacobson K, Sackmann E. Local measurements of viscoelastic parameters of adherent cell surfaces by magnetic bead microrheology. Biophys J. 1998;75:2038–2049. doi: 10.1016/S0006-3495(98)77646-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch AR, Moller W, Sackmann E. Measurement of local viscoelasticity and forces in living cells by magnetic tweezers. Biophys J. 1999;76:573–579. doi: 10.1016/S0006-3495(99)77225-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorck T, Gustafsson LE, Dahlen SE. Isolated bronchi from asthmatics are hyperresponsive to adenosine, which apparently acts indirectly by liberation of leukotrienes and histamine. Am Rev Respir Dis. 1992;145:1087–1091. doi: 10.1164/ajrccm/145.5.1087. [DOI] [PubMed] [Google Scholar]
- Black JL, Johnson PR. Airway smooth muscle in asthma. Respir. 1996;1:153–158. doi: 10.1111/j.1440-1843.1996.tb00026.x. [DOI] [PubMed] [Google Scholar]
- Black JL, Johnson PR. What determines asthma phenotype? Is it the interaction between allergy and the smooth muscle? Am J Respir Crit Care Med. 2000;161:S207–S210. doi: 10.1164/ajrccm.161.supplement_2.a1q4-12. [DOI] [PubMed] [Google Scholar]
- Black JL, Johnson PR, Armour CL. Factors controlling transduction signaling and proliferation of airway smooth muscle. Curr. Allergy Asthma Rep. 2001;1:116–121. doi: 10.1007/s11882-001-0078-3. [DOI] [PubMed] [Google Scholar]
- Bond M, Somlyo AV. Dense bodies and actin polarity in vertebrate smooth muscle. J Cell Biol. 1982;95:403–413. doi: 10.1083/jcb.95.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RH, Mitzner W. The myth of maximal airway responsiveness in vivo. J Appl Physiol. 1998;85:2012–2017. doi: 10.1152/jappl.1998.85.6.2012. [DOI] [PubMed] [Google Scholar]
- Bursac P, Lenormand G, Fabry B, Oliver M, Weitz DA, Viasnoff V, Butler JP, Fredberg JJ. Cytoskeletal remodelling and slow dynamics in the living cell. Nature Mater. 2005;4:557–561. doi: 10.1038/nmat1404. [DOI] [PubMed] [Google Scholar]
- Bursac P, Fabry B, Trepat X, Lenormand G, Butler JP, Wang N, Fredberg JJ, An SS. Cytoskeleton dynamics: fluctuations within the network. Biochem Biophys Res Comm. 2007;355:324–330. doi: 10.1016/j.bbrc.2007.01.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JP, Tolic-Norrelykke IM, Fabry B, Fredberg JJ. Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol. 2002;282:C595–C605. doi: 10.1152/ajpcell.00270.2001. [DOI] [PubMed] [Google Scholar]
- Colebatch HJH, Mitchell CA. Constriction of isolated living liquid-filled dog and cat lungs with histamine. J Appl Physiol. 1971;30:691–702. doi: 10.1152/jappl.1971.30.5.691. [DOI] [PubMed] [Google Scholar]
- Colebatch HJH, Olsen CR, Nadel JA. Effect of histamine, serotonin, and acetylcholine on the peripheral airways. J Appl Physiol. 1966;21:217–226. doi: 10.1152/jappl.1966.21.1.217. [DOI] [PubMed] [Google Scholar]
- Crick FHC, Hughes AFW. The physical properties of cytoplasm: a study by means of the magnetic particle method. Exp Cell Res. 1949;1:37–80. [Google Scholar]
- Dandurand RJ, Wang CG, Phillips NC, Eidelman DH. Responsiveness of individual airways to methacholine in adult rat lung explants. J Appl Physiol. 1993a;75:364–372. doi: 10.1152/jappl.1993.75.1.364. [DOI] [PubMed] [Google Scholar]
- Dandurand RJ, Xu LJ, Martin JG, Eidelman DH. Airway-parenchymal interdependence and bronchial responsiveness in two highly inbred rat strains. J Appl Physiol. 1993b;74:538–544. doi: 10.1152/jappl.1993.74.2.538. [DOI] [PubMed] [Google Scholar]
- De Jongste JC, Mons H, Bonta IL, Kerrebijin KF. In vitro responses of airways from an asthmatic patient. Eur J Respir Dis. 1987;71:23–29. [PubMed] [Google Scholar]
- Deng L, Trepat X, Butler JP, Millet E, Morgan KG, Weitz DA, Fredberg JJ. Fast and slow dynamics of the cytoskeleton. Nature Mater. 2006;5:636–640. doi: 10.1038/nmat1685. [DOI] [PubMed] [Google Scholar]
- Dillon PF, Aksoy MO, Driska SP, Murphy RA. Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science. 1981;211:495–497. doi: 10.1126/science.6893872. [DOI] [PubMed] [Google Scholar]
- Ding DJ, Martin JG, Macklem PT. Effects of lung volume on maximal methacholine-induced bronchoconstriction in normal humans. J Appl Physiol. 1987;62:1324–1330. doi: 10.1152/jappl.1987.62.3.1324. [DOI] [PubMed] [Google Scholar]
- Dixon WE, Brodie TG. Contributions to the physiology of the lungs. Part I. The bronchial muscles, their innervation, and the action of drugs upon them. J Physiol (London) 1903;29:97–173. doi: 10.1113/jphysiol.1903.sp000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguet A, Biyah K, Minshall E, Gomes R, Wang CG, Taoudi-Benchekroun M, Bates JH, Eidelman DH. Bronchial responsiveness among inbred mouse strains. Role of airway smooth-muscle shortening velocity. Am J Respir Crit Care Med. 2000;161:839–848. doi: 10.1164/ajrccm.161.3.9906054. [DOI] [PubMed] [Google Scholar]
- Dulin NO, Fernandes DJ, Dowell M, Bellam S, McConville J, Lakser O, Mitchell R, Camoretti-Mercado B, Kogut P, Solway J. What evidence implicates airway smooth muscle in the cause of BHR? Clin Rev Allergy Immunol. 2003;24:73–84. doi: 10.1385/CRIAI:24:1:73. [DOI] [PubMed] [Google Scholar]
- Eidelman DH, Dimaria GU, Bellofiore S, Wang NS, Guttmann RD, Martin JG. Strain-related differences in airway smooth muscle and airway responsiveness in the rat. Am Rev Respir Dis. 1991;144:792–796. doi: 10.1164/ajrccm/144.4.792. [DOI] [PubMed] [Google Scholar]
- Einthoven W. Ueber die Wirkung der Bronchialmuskeln, nach einer neuen Methode untersucht, und ueber Asthma nervosum. Pfluegers Arch. 1892;51:367–444. [Google Scholar]
- Fabry B, Fredberg JJ. Remodeling of the airway smooth muscle cell: are we built of glass? Respir Physiol Neurobiol. 2003;137:109–124. doi: 10.1016/s1569-9048(03)00141-1. [DOI] [PubMed] [Google Scholar]
- Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the mircrorheology of living cells. Phys Rev Let. 2001;87:1481021. doi: 10.1103/PhysRevLett.87.148102. [DOI] [PubMed] [Google Scholar]
- Fan T, Yang M, Halayko A, Mohapatra SS, Stephens NL. Airway responsiveness in two inbred strains of mouse disparate in IgE and IL-4 production. Am J Respir Cell Mole Biol. 1997;17:156–163. doi: 10.1165/ajrcmb.17.2.2628. [DOI] [PubMed] [Google Scholar]
- Fish JE, Ankin MG, Kelly JF, Peterman VI. Regulation of bronchomotor tone by lung inflation in asthmatic and nonasthmatic subjects. J Appl Physiol. 1981;50:1079–1086. doi: 10.1152/jappl.1981.50.5.1079. [DOI] [PubMed] [Google Scholar]
- Ford LE, Seow CY, Pratusevich VR. Plasticity in smooth muscle, a hypothesis. Can J Physiol Pharm. 1994;72:1320–1324. doi: 10.1139/y94-190. [DOI] [PubMed] [Google Scholar]
- Fredberg JJ. Airway smooth muscle in asthma: flirting with disaster. Eur Respir J. 1998;12:1252–1256. doi: 10.1183/09031936.98.12061252. [DOI] [PubMed] [Google Scholar]
- Fredberg JJ. Airway smooth muscle in asthma. Perturbed equilibria of myosin binding. Am J Respir Crit Care Med. 2000a;161:S158–S160. doi: 10.1164/ajrccm.161.supplement_2.a1q4-1. [DOI] [PubMed] [Google Scholar]
- Fredberg JJ. Frozen objects: small airways, big breaths, and asthma. J Allergy Clin Immunol. 2000b;106:615–624. doi: 10.1067/mai.2000.109429. [DOI] [PubMed] [Google Scholar]
- Fredberg JJ, Shore SA. The unbearable lightness of breathing. J Appl Physiol. 1999;86:3–4. doi: 10.1152/jappl.1999.86.1.3. [DOI] [PubMed] [Google Scholar]
- Fredberg JJ, Jones KA, Nathan M, Raboudi S, Prakash YS, Shore SA, Butler JP, Sieck GC. Friction in airway smooth muscle: mechanism, latch and implications in asthma. J Appl Physiol. 1996;81:2703–2712. doi: 10.1152/jappl.1996.81.6.2703. [DOI] [PubMed] [Google Scholar]
- Fredberg JJ, Inouye D, Miller B, Nathan M, Jafari S, Raboudi SH, Butler JP, Shore SA. Airway smooth muscle, tidal stretches, and dynamically determined contractile states. Am J Respir Crit Care Med. 1997;156:1752–1759. doi: 10.1164/ajrccm.156.6.9611016. [DOI] [PubMed] [Google Scholar]
- Fredberg JJ, Inouye DS, Mijailovich SM, Butler JP. Perturbed equilibrium of myosin binding in airway smooth muscle and its implications in bronchospasm. Am J Respir Crit Care Med. 1999;159:959–967. doi: 10.1164/ajrccm.159.3.9804060. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT, Pohl J. Caldesmon and calponin phosphorylation in regulation of smooth muscle contraction. Can J Physiol Pharm. 1994;72:1410–1414. doi: 10.1139/y94-203. [DOI] [PubMed] [Google Scholar]
- Goldie RG, Spina D, Henry PJ, Lulich KM, Paterson JW. In vitro responsiveness of human asthmatic bronchus to carbachol, histamine, beta-adrenoceptor agonists and theophylline. Brit J Clin Pharm. 1986;22:669–676. doi: 10.1111/j.1365-2125.1986.tb02956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gump A, Haughney L, Fredberg J. Relaxation of activated airway smooth muscle: relative potency of isoproterenol vs. tidal stretch. J Appl Physiol. 2001;90:2306–2310. doi: 10.1152/jappl.2001.90.6.2306. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Fredberg JJ. The first three minutes: smooth muscle contraction, cytoskeletal events, and soft glasses. J Appl Physiol. 2003;95:413–425. doi: 10.1152/japplphysiol.00277.2003. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Wu MF. Plasticity of airway smooth muscle stiffness and extensibility: role of length-adaptive mechanisms. J Appl Physiol. 2001;90:741–749. doi: 10.1152/jappl.2001.90.2.741. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Wu MF, Smith DD. Contraction history modulates isotonic shortening velocity in smooth muscle. Am J Physiol. 1993;265:C467–C476. doi: 10.1152/ajpcell.1993.265.2.C467. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Meiss RA, Wu MF, Rowe M. Mechanisms for the mechanical placticity of tracheal smooth muscle. Am J Physiol. 1995;268:C1267–C1276. doi: 10.1152/ajpcell.1995.268.5.C1267. [DOI] [PubMed] [Google Scholar]
- Hai CM, Murphy RA. Regulation of shortening velocity by cross-bridge phosphorylation in smooth muscle. Am J Physiol. 1988;255:C86–C94. doi: 10.1152/ajpcell.1988.255.1.C86. [DOI] [PubMed] [Google Scholar]
- Haitchi HM, Powell RM, Shaw TJ, Howarth PH, Wilson SJ, Wilson DI, Holgate ST, Davies DE. ADAM33 expression in asthmatic airways and human embryonic lungs. Am J Respir Crit Care Med. 2005;171:958–965. doi: 10.1164/rccm.200409-1251OC. [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol. 2001;90:358–368. doi: 10.1152/jappl.2001.90.1.358. [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Camoretti-Mercado B, Forsythe SM, Vieira JE, Mitchell RW, Wylam ME, Hershenson MB, Solway J. Divergent differentiation paths in airway smooth muscle culture: induction of functionally contractile myocytes. Am J Physiol. 1999;276:L197–L206. doi: 10.1152/ajplung.1999.276.1.L197. [DOI] [PubMed] [Google Scholar]
- Hedges JC, Yamboliev IA, Ngo M, Horowitz B, Adam LP, Gerthoffer WT. p38 mitogen-activated protein kinase expression and activation in smooth muscle. Am J Physiol. 1998;275:C527–C534. doi: 10.1152/ajpcell.1998.275.2.C527. [DOI] [PubMed] [Google Scholar]
- Hedges JC, Dechert MA, Yamboliev IA, Martin JL, Hickey E, Weber LA, Gerthoffer WT. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J Biol Chem. 1999;274:24211–24219. doi: 10.1074/jbc.274.34.24211. [DOI] [PubMed] [Google Scholar]
- Hedges JC, Oxhorn BC, Carty M, Adam LP, Yamboliev IA, Gerthoffer WT. Phosphorylation of caldesmon by ERK MAP kinases in smooth muscle. American J Physiol. 2000;278:C718–C726. doi: 10.1152/ajpcell.2000.278.4.C718. [DOI] [PubMed] [Google Scholar]
- Hirshman CA, Emala CW. Actin reorganization in airway smooth muscle cells involves Gq and Gi-2 activation of Rho. Am J Physiol. 1999;277:L653–L661. doi: 10.1152/ajplung.1999.277.3.L653. [DOI] [PubMed] [Google Scholar]
- Hirst SJ, Martin JG, Bonacci JV, Chan V, Fixman ED, Hamid QA, Herszberg B, Lavoie JP, McVicker CG, Moir LM, Nguyen TT, Peng Q, Ramos-Barbon D, Stewart AG. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol. 2004;114:S2–S17. doi: 10.1016/j.jaci.2004.04.039. [DOI] [PubMed] [Google Scholar]
- Homer RJ, Elias JA. Consequences of long-term inflammation. Airway remodeling. Clin Chest Med. 2000;21:331–343. doi: 10.1016/s0272-5231(05)70270-7. [DOI] [PubMed] [Google Scholar]
- Hu S, Chen J, Fabry B, Numaguchi Y, Gouldstone A, Ingber DE, Fredberg JJ, Butler JP, Wang N. Intracellular stress tomography reveals stress focusing and structural anisotropy in the cytoskeleton of living cells. Am J Physiol. 2003;285:C1082–C1090. doi: 10.1152/ajpcell.00159.2003. [DOI] [PubMed] [Google Scholar]
- Hubmayr RD, Shore SA, Fredberg JJ, Planus E, Panettieri RA, Moller W, Heyder J, Wang N. Pharmacological activation changes stiffness of cultured human airway smooth muscle cells. Am J Physiol. 1996;271:C1660–C1668. doi: 10.1152/ajpcell.1996.271.5.C1660. [DOI] [PubMed] [Google Scholar]
- Huxley AF. Muscle structure and theories of contraction. Prog Biophys Biophys Chem. 1957;7:255–318. [PubMed] [Google Scholar]
- James AL, Paré PD, Hogg JC. The mechanics of airway narrowing in asthma. Am Rev Respir Dis. 1989;139:242–246. doi: 10.1164/ajrccm/139.1.242. [DOI] [PubMed] [Google Scholar]
- Jia Y, Xu L, Heisler S, Martin JG. Airways of a hyperresponsive rat strain show decreased relaxant response to sodium nitroprusside. Am J Physiol. 1995;269:L85–L91. doi: 10.1152/ajplung.1995.269.1.L85. [DOI] [PubMed] [Google Scholar]
- Jiang H, Rao K, Halayko AJ, Liu X, Stephens NL. Ragweed sensitization-induced increase of myosin light chain kinase content in canine airway smooth muscle. Am J Respir Cell Mole Biol. 1992;7:567–573. doi: 10.1165/ajrcmb/7.6.567. [DOI] [PubMed] [Google Scholar]
- Johnson PR, Roth M, Tamm M, Hughes M, Ge Q, King G, Burgess JK, Black JL. Airway smooth muscle cell proliferation is increased in asthma. Am J Respir Crit Care Med. 2001;164:474–477. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- Kelleher MD, Abe MK, Chao TS, Jain M, Green JM, Solway J, Rosner MR, Hershenson MB. Role of MAP kinase activation in bovine tracheal smooth muscle mitogenesis. Am J Physiol. 1995;268:L894–L901. doi: 10.1152/ajplung.1995.268.6.L894. [DOI] [PubMed] [Google Scholar]
- Kubo R. Brownian motion and nonequilibrium statistical mechanics. Science. 1986;233:330–334. doi: 10.1126/science.233.4761.330. [DOI] [PubMed] [Google Scholar]
- Kuo KH, Wang L, Paré PD, Ford LE, Seow CY. Myosin thick filament lability induced by mechanical strain in airway smooth muscle. J Appl Physiol. 2001;90:1811–1816. doi: 10.1152/jappl.2001.90.5.1811. [DOI] [PubMed] [Google Scholar]
- Kuo KH, Herrera AM, Wang L, Paré PD, Ford LE, Stephens NL, Seow CY. Structure-function correlation in airway smooth muscle adapted to different lengths. Am J Physiol. 2003;285:C384–C390. doi: 10.1152/ajpcell.00095.2003. [DOI] [PubMed] [Google Scholar]
- Lakser OJ, Lindeman RP, Fredberg JJ. Inhibition of the p38 MAP kinase pathway destabilizes smooth muscle length during physiological loading. Am J Physiol. 2002;282:L1117–L1121. doi: 10.1152/ajplung.00230.2000. [DOI] [PubMed] [Google Scholar]
- Lambert RK, Paré PD. Lung parenchymal shear modulus, airway wall remodeling, and bronchial hyperresponsiveness. J Appl Physiol. 1997;83:140–147. doi: 10.1152/jappl.1997.83.1.140. [DOI] [PubMed] [Google Scholar]
- Lambert RK, Wiggs BR, Kuwano K, Hogg JC, Paré PD. Functional significance of increased airway smooth muscle in asthma and COPD. J Appl Physiol. 1993;74:2771–2781. doi: 10.1152/jappl.1993.74.6.2771. [DOI] [PubMed] [Google Scholar]
- Laudadio RE, Millet E, Fabry B, An SS, Butler JP, Fredberg JJ. Rat airway smooth muscle cell during actin modulation: rheology and glassy dynamics. Am J Physiol. 2005;289:C1388–C1395. doi: 10.1152/ajpcell.00060.2005. [DOI] [PubMed] [Google Scholar]
- Lauzon AM, Tyska MJ, Rovner AS, Freyzon Y, Warshaw DM, Trybus KM. A 7-amino-acid insert in the heavy chain nucleotide binding loop alters the kinetics of smooth muscle myosin in the laser trap. J Muscle Res Cell Mot. 1998;19:825–837. doi: 10.1023/a:1005489501357. [DOI] [PubMed] [Google Scholar]
- Lazaar AL, Panettieri RA. Airway smooth muscle: a modulator of airway remodeling in asthma. J Allergy Clin Immunol. 2005;116:488–495. doi: 10.1016/j.jaci.2005.06.030. [DOI] [PubMed] [Google Scholar]
- Lim TK, Pride NB, Ingram RH. Effects of volume history during spontaneous and acutely induced air-flow obstruction in asthma. Am Rev Respir Dis. 1987;135:591–596. doi: 10.1164/arrd.1987.135.3.591. [DOI] [PubMed] [Google Scholar]
- Ma X, Cheng Z, Kong H, Wang Y, Unruth H, Stephens NL, Laviolette M. Changes in biophysical and biochemical properties of single bronchial smooth muscle cells from asthmatic subjects. Am J Physiol. 2002;283:L1181–L1189. doi: 10.1152/ajplung.00389.2001. [DOI] [PubMed] [Google Scholar]
- Macklem PT. Bronchial hyperresponsiveness. Chest. 1987;91:189S–191S. doi: 10.1378/chest.91.6_supplement.189s. [DOI] [PubMed] [Google Scholar]
- Macklem PT. Mechanical factors determining maximum bronchoconstriction. Eur Respir J. 1989;2:516S–519S. [PubMed] [Google Scholar]
- Macklem PT. A hypothesis linking bronchial hyperreactivity and airway inflammation: implications for therapy. Ann Allergy. 1990;64:113–116. [PubMed] [Google Scholar]
- Macklem PT. A theoretical analysis of the effect of airway smooth muscle load on airway narrowing. Am J Respir Crit Care Med. 1996;153:83–89. doi: 10.1164/ajrccm.153.1.8542167. [DOI] [PubMed] [Google Scholar]
- Madison JM. Migration of airway smooth muscle cells. Am J Respir Cell Mole Biol. 2003;29:8–11. doi: 10.1165/rcmb.F272. [DOI] [PubMed] [Google Scholar]
- Maksym GN, Fabry B, Butler JP, Navajas D, Tschumperlin DJ, Laporte JD, Fredberg JJ. Mechanical properties of cultured human airway smooth muscle cells from 0.05 to 0.4 Hz. J Appl Physiol. 2000;89:1619–1632. doi: 10.1152/jappl.2000.89.4.1619. [DOI] [PubMed] [Google Scholar]
- Matthews BD, Overby DR, Alenghat FJ, Karavitis J, Numaguchi Y, Allen PG, Ingber DE. Mechanical properties of individual focal adhesions probed with a magnetic microneedle. Biochem Biophys Res Comm. 2004;313:758–764. doi: 10.1016/j.bbrc.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Mead J. Respiration: pulmonary mechanics. Ann Rev Physiol. 1973;35:169–192. doi: 10.1146/annurev.ph.35.030173.001125. [DOI] [PubMed] [Google Scholar]
- Mehta D, Gunst SJ. Actin polymerization stimulated by contractile activation regulates force development in canine tracheal smooth muscle. J Physiol. 1999;519:829–840. doi: 10.1111/j.1469-7793.1999.0829n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Wu MF, Gunst SJ. Role of contractile protein activation in the length-dependent modulation of tracheal smooth muscle force. Am J Physiol. 1996;270:C243–C252. doi: 10.1152/ajpcell.1996.270.1.C243. [DOI] [PubMed] [Google Scholar]
- Mehta D, Tang DD, Wu MF, Atkinson S, Gunst SJ. Role of Rho in Ca2+-insensitive contraction and paxillin tyrosine phosphorylation in smooth muscle. Am J Physiol. 2000;279:C308–C318. doi: 10.1152/ajpcell.2000.279.2.C308. [DOI] [PubMed] [Google Scholar]
- Meiss RA. Influence of intercellular tissue connections on airway muscle mechanics. J Appl Physiol. 1999;86:3–4. doi: 10.1152/jappl.1999.86.1.5. [DOI] [PubMed] [Google Scholar]
- Mijailovich SM, Butler JP, Fredberg JJ. Perturbed equilibria of myosin binding in airway smooth muscle: bond-length distributions, mechanics, and ATP metabolism. Biophys J. 2000;79:2667–2681. doi: 10.1016/S0006-3495(00)76505-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BJ, Verburgt LM, King GG, Paré PD. Effect of deep inspiration on methacholine dose-response curves in normal subjects. Am J Respir Crit Care Med. 1997;156:1278–1281. doi: 10.1164/ajrccm.156.4.96-11082. [DOI] [PubMed] [Google Scholar]
- Moore BJ, King GG, D’Yachkova Y, Ahmad HR, Paré PD. Mechanism of methacholine dose-response plateaus in normal subjects. Am J Respir Crit Care Med. 1998;158:666–669. doi: 10.1164/ajrccm.158.2.9709048. [DOI] [PubMed] [Google Scholar]
- Moreno R, Hogg JC, Paré PD. Mechanics of airway narrowing. Am Rev Respir Dis. 1986;133:1171–1180. doi: 10.1164/arrd.1986.133.6.1171. [DOI] [PubMed] [Google Scholar]
- Murphy RA. Muscle cells of hollow organs. News Physiol Sci. 1988;3:124–128. [Google Scholar]
- Murphy RA. What is special about smooth muscle? The significance of covalent crossbridge regulation. FASEB J. 1994;8:311–318. doi: 10.1096/fasebj.8.3.8143937. [DOI] [PubMed] [Google Scholar]
- Murphy RA, Walker JS, Strauss JD. Myosin isoforms and functional diversity in vertebrate smooth muscle. Comp Biochem Physiol. 1997;117B:51–60. doi: 10.1016/s0305-0491(96)00314-8. [DOI] [PubMed] [Google Scholar]
- Nadel JA, Tierney DF. Effect of a previous deep inspiration on airway resistance in man. J Appl Physiol. 1961;16:717–719. doi: 10.1152/jappl.1961.16.4.717. [DOI] [PubMed] [Google Scholar]
- Nagase T, Moretto A, Ludwig MS. Airway and tissue behavior during induced constriction in rats: intravenous vs. aerosol administration. J Appl Physiol. 1994;76:830–838. doi: 10.1152/jappl.1994.76.2.830. [DOI] [PubMed] [Google Scholar]
- Naghshin J, Wang L, Paré PD, Seow CY. Adaptation to chronic length change in explanted airway smooth muscle. J Appl Physiol. 2003;95:448–453. doi: 10.1152/japplphysiol.01180.2002. [DOI] [PubMed] [Google Scholar]
- Otis AB. A perspective of respiratory mechanics. J Appl Physiol. 1983;54:1183–1187. doi: 10.1152/jappl.1983.54.5.1183. [DOI] [PubMed] [Google Scholar]
- Panettieri RA, Murray RK, DePalo LR, Yadvish PA, Kotlikoff MI. A human airway smooth muscle cell line that retains physiological responsiveness. Am J Physiol. 1989;256:C329–C335. doi: 10.1152/ajpcell.1989.256.2.C329. [DOI] [PubMed] [Google Scholar]
- Parker KK, Brock AL, Brangwynne C, Mannix RJ, Wang N, Ostuni E, Geisse NA, Adams JC, Whitesides GM, Ingber DE. Directional control of lamellipodia extension by constraining cell shape and orienting cell tractional forces. FASEB J. 2002;16:1195–1204. doi: 10.1096/fj.02-0038com. [DOI] [PubMed] [Google Scholar]
- Pratusevich VR, Seow CY, Ford LE. Plasticity in canine airway smooth muscle. J Gen Physiol. 1995;105:73–94. doi: 10.1085/jgp.105.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-de-Morales M, Millet E, Fabry B, Navajas D, Wang N, Butler JP, Fredberg JJ. Cytoskeletal mechanics in adherent human airway smooth muscle cells: probe specificity and scaling of protein-protein dynamics. Am J Physiol. 2004;287:C643–C654. doi: 10.1152/ajpcell.00070.2004. [DOI] [PubMed] [Google Scholar]
- Qi D, Mitchell RW, Burdyga T, Ford LE, Kuo KH, Seow CY. Myosin light chain phosphorylation facilitates in vivo myosin filament reassembly after mechanical perturbation. Am J Physiol. 2002;282:C1298–C1305. doi: 10.1152/ajpcell.00554.2001. [DOI] [PubMed] [Google Scholar]
- Robatto FM, Simard S, Orana H, Macklem PT, Ludwig MS. Effect of lung volume on plateau response of airways and tissue to methacholine in dogs. J Appl Physiol. 1992;73:1908–1913. doi: 10.1152/jappl.1992.73.5.1908. [DOI] [PubMed] [Google Scholar]
- Romero P, Ludwig MS. Maximal methacholine-induced constriction in rabbit lungs: interactions between airways and tissues? J Appl Physiol. 1991;70:1044–1050. doi: 10.1152/jappl.1991.70.3.1044. [DOI] [PubMed] [Google Scholar]
- Salter HH. On asthma: its pathology and treatment. John Churchill & Sons, Science Press Limited; London: 1868. pp. 24–60. [Google Scholar]
- Seow CY, Fredberg JJ. Historical perspective on airway smooth muscle: the saga of a frustrated cell. J Appl Physiol. 2001;91:938–952. doi: 10.1152/jappl.2001.91.2.938. [DOI] [PubMed] [Google Scholar]
- Seow CY, Stephens NL. Force-velocity curves for smooth muscle: analysis of internal factors reducing velocity. Am J Physiol. 1986;251:C362–C368. doi: 10.1152/ajpcell.1986.251.3.C362. [DOI] [PubMed] [Google Scholar]
- Seow CY, Stephens NL. Velocity-length-time relations in canine tracheal smooth muscle. J Appl Physiol. 1988;54:2053–2057. doi: 10.1152/jappl.1988.64.5.2053. [DOI] [PubMed] [Google Scholar]
- Seow CY, Pratusevich VR, Ford LE. Series-to-parallel transition in the filament lattice of airway smooth muscle. J Appl Physiol. 2000;89:869–876. doi: 10.1152/jappl.2000.89.3.869. [DOI] [PubMed] [Google Scholar]
- Shen X, Gunst SJ, Tepper RS. Effect of tidal volume and frequency on airway responsiveness in mechanically ventilated rabbits. J Appl Physiol. 1997;83:1202–1208. doi: 10.1152/jappl.1997.83.4.1202. [DOI] [PubMed] [Google Scholar]
- Shore SA, Laporte J, Hall I, Hardy E, Panettieri RA. Effect of IL-1β on responses of cultured human airway smooth muscle cells to bronchodilator agonists. Am J Respir Cell Mole Biol. 1997;16:702–712. doi: 10.1165/ajrcmb.16.6.9191472. [DOI] [PubMed] [Google Scholar]
- Silveira PSP, Fredberg JJ. Smooth muscle length adaptation and actin filament length: a network model of the cytoskeletal dysregulation. Can J Physiol Pharmacol. 2005;83:923–931. doi: 10.1139/y05-092. [DOI] [PubMed] [Google Scholar]
- Skloot G, Permutt S, Togias A. Airway hyperresponsiveness in asthma: a problem of limited smooth muscle relaxation with inspiration. J Clin Invest. 1995;96:2393–2403. doi: 10.1172/JCI118296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small JV. Structure-function relationships in smooth muscle: the missing links. Bioessays. 1995;17:785–792. doi: 10.1002/bies.950170908. [DOI] [PubMed] [Google Scholar]
- Small JV, Gimona M. The cytoskeleton of the vertebrate smooth muscle cell. Acta Physiol Scand. 1998;164:341–348. doi: 10.1046/j.1365-201X.1998.00441.x. [DOI] [PubMed] [Google Scholar]
- Smith BA, Tolloczko B, Martin JG, Grutter P. Probing the viscoelastic behavior of cultured airway smooth muscle cells with atomic force microscopy: stiffening induced by contractile agonist. Biophys J. 2005;88:2994–3007. doi: 10.1529/biophysj.104.046649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solway J, Fredberg JJ. Perhaps airway smooth muscle dysfunction does contribute to bronchial hyperresponsiveness afer all. Am J Respir Cell Mole Biol. 1997;17:144–146. doi: 10.1165/ajrcmb.17.2.f137. [DOI] [PubMed] [Google Scholar]
- Stephens NL. Airway smooth muscle. Am Rev Respir Dis. 1987;135:960–975. doi: 10.1164/arrd.1987.135.4.960. [DOI] [PubMed] [Google Scholar]
- Stephens NL, Kromer U. Series elastic component of tracheal smooth muscle. Am J Physiol. 1971;220:1890–1895. doi: 10.1152/ajplegacy.1971.220.6.1890. [DOI] [PubMed] [Google Scholar]
- Stephens NL, Seow CY. Airway smooth muscle: physiology, bronchomotor tone, pharmacology, and relation to asthma. In: Weiss EB, Stein M, editors. Bronchial Asthma. Little, Brown; Boston, MA: 1993. pp. 314–332. [Google Scholar]
- Stephens NL, Morgan G, Kepron W, Seow CY. Changes in cross-bridge properties of sensitized airway smooth muscle. J Appl Physiol. 1986;61:1492–1498. doi: 10.1152/jappl.1986.61.4.1492. [DOI] [PubMed] [Google Scholar]
- Sterk PJ, Bel EH. Bronchial hyperresponsiveness: the need to distinguish between hypersensitivity and excessive airway narrowing. Eur Respir J. 1989;2:267–274. [PubMed] [Google Scholar]
- Stull JT, Blumenthal DK, Miller JR, DiSalvo J. Regulation of myosin phosphorylation. J Mol Cell Cardiol. 1980;3:105–110. doi: 10.1016/0022-2828(82)90137-7. [DOI] [PubMed] [Google Scholar]
- Tao FC, Tolloczko B, Eidelman DH, Martin JG. Enhanced Ca2+ mobilization in airway smooth muscle contributes to airway hyperresponsiveness in an inbred strain of rat. Am J Respir Crit Care Med. 1999;160:446–453. doi: 10.1164/ajrccm.160.2.9811098. [DOI] [PubMed] [Google Scholar]
- Tao FC, Shah S, Pradhan AA, Tolloczko B, Martin JG. Enhanced calcium signaling to bradykinin in airway smooth muscle from hyperresponsive inbred rats. Am J Physiol. 2003;284:L90–L99. doi: 10.1152/ajplung.00023.2002. [DOI] [PubMed] [Google Scholar]
- Thomson RJ, Bramley AM, Schellenberg RR. Airway muscle stereology: implications for increased shortening in asthma. Am J Respir Crit Care Med. 1996;154:749–757. doi: 10.1164/ajrccm.154.3.8810615. [DOI] [PubMed] [Google Scholar]
- Tolic-Norrelykke IM, Butler JP, Chen J, Wang N. Spatial and temporal traction response in human airway smooth muscle cells. Am J Physiol. 2002;283:C1254–C1266. doi: 10.1152/ajpcell.00169.2002. [DOI] [PubMed] [Google Scholar]
- Tolloczko B, Jia YL, Martin JG. Serotonin-evoked calcium transients in airway smooth muscle cells. Am J Physiol. 1995;269:L234–L240. doi: 10.1152/ajplung.1995.269.2.L234. [DOI] [PubMed] [Google Scholar]
- Tonino P, Simon M, Craig R. Mass determination of native smooth muscle myosin filaments by scanning transmission electron microscopy. J Mole Biol. 2002;318:999–1007. doi: 10.1016/S0022-2836(02)00191-2. [DOI] [PubMed] [Google Scholar]
- Trepat X, Deng L, An SS, Navajas D, Tschumperlin DJ, Gerthoffer WT, Butler JP, Fredberg JJ. Universal physical responses to stretch in the living cell. Nature. 447:592–595. doi: 10.1038/nature05824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uvelius B. Isometric and isotonic length-tension relations and variations in cell length in longitudinal smooth muscle from rabbit urinary bladder. Acta Physiol Scand. 1976;97:1–12. doi: 10.1111/j.1748-1716.1976.tb10230.x. [DOI] [PubMed] [Google Scholar]
- Valberg PA. Magnetometry of ingested particles in pulmonary macrophages. Science. 1984;224:513–516. doi: 10.1126/science.6710153. [DOI] [PubMed] [Google Scholar]
- Valberg PA, Feldman HA. Magnetic particle motions within living cells. Biophys J. 1987;52:551–561. doi: 10.1016/S0006-3495(87)83244-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Citters KM, Hoffman BD, Massiera G, Crocker JC. The role of F-actin and myosin in epithelial cell rheology. Biophys J. 2006;91:3946–3956. doi: 10.1529/biophysj.106.091264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CG, Almirall JJ, Dolman CS, Dandurand RJ, Eidelman DH. In vitro bronchial responsiveness in two highly inbred rat strains. J Appl Physiol. 1997;82:1445–1452. doi: 10.1152/jappl.1997.82.5.1445. [DOI] [PubMed] [Google Scholar]
- Wang L, Paré PD, Seow CY. Effect of chronic passive length change on airway smooth muscle length-tension relationship. J Appl Physiol. 2001;90:734–740. doi: 10.1152/jappl.2001.90.2.734. [DOI] [PubMed] [Google Scholar]
- Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- Wang N, Tolic-Norrelykke IM, Chen J, Mijailovich SM, Butler JP, Fredberg JJ, Stamenovic D. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am J Physiol. 2002;282:C606–C616. doi: 10.1152/ajpcell.00269.2001. [DOI] [PubMed] [Google Scholar]
- Wang P, Bitar KN. Rho A regulates sustained smooth muscle contraction through cytoskeletal reorganization of HSP27. Am J Physiol. 1998;275:G1454–G1462. doi: 10.1152/ajpgi.1998.275.6.G1454. [DOI] [PubMed] [Google Scholar]
- Warner DO, Gunst SJ. Limitation of maximal bronchoconstriction in living dogs. Am Rev Respir Dis. 1992;145:553–560. doi: 10.1164/ajrccm/145.3.553. [DOI] [PubMed] [Google Scholar]
- Warshaw DM, Rees DD, Fay FS. Characterization of cross-bridge elasticity and kinetics of cross-bridge cycling force development in single smooth muscle cells. J Gen Physiol. 1988;91:761–779. doi: 10.1085/jgp.91.6.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson JD, Crick FHC. Molecular structure of nucleic acids. Nature. 1953a;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- Watson JD, Crick FHC. Genetic implications of the structure of deoxyribonucleic acid. Nature. 1953b;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- Whicker SD, Armour CL, Black JL. Responsiveness of bronchial smooth muscle from asthmatic patients to relaxant and contractile agonists. Pulm Pharm. 1988;1:25–31. doi: 10.1016/0952-0600(88)90007-5. [DOI] [PubMed] [Google Scholar]
- Wiggs BR, Bosken C, Paré PD, James A, Hogg JC. A model of airway narrowing in asthma and in chronic obstructive pulmonary disease. Am Rev Respir Dis. 1992;145:1251–1258. doi: 10.1164/ajrccm/145.6.1251. [DOI] [PubMed] [Google Scholar]
- Wiggs BR, Hrousis CA, Drazen JM, Kamm RD. On the mechanism of mucosal folding in normal and asthmatic airways. J Appl Physiol. 1997;83:1814–1821. doi: 10.1152/jappl.1997.83.6.1814. [DOI] [PubMed] [Google Scholar]
- Woodruff PG, Dolganov GM, Ferrando RE, Donnelly S, Hays SR, Solberg OD, Carter R, Wong HH, Cadbury PS, Fahy JV. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med. 2004;169:1001–1006. doi: 10.1164/rccm.200311-1529OC. [DOI] [PubMed] [Google Scholar]
- Woolcock AJ, Peat JK. Epidemiology of bronchial hyperresponsiveness. Clin Rev Allergy. 1989;7:245–256. doi: 10.1007/BF02914477. [DOI] [PubMed] [Google Scholar]
- Xu JQ, Harder BA, Uman P, Craig R. Myosin filament structure in vertebrate smooth muscle. J Cell Biol. 1996;134:53–66. doi: 10.1083/jcb.134.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager D, Butler JP, Bastacky J, Israel E, Smith G, Drazen JM. Amplification of airway constriction due to liquid filling of airway interstices. J Appl Physiol. 1989;66:2873–2884. doi: 10.1152/jappl.1989.66.6.2873. [DOI] [PubMed] [Google Scholar]
- Yamada S, Wirtz D, Kuo SC. Mechanics of living cells measured by laser tracking microrheology. Biophys J. 2000;78:1736–1747. doi: 10.1016/S0006-3495(00)76725-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamboliev IA, Hedges JC, Mutnick JL, Adam LP, Gerthoffer WT. Evidence for modulation of smooth muscle force by the p38 MAP kinase/HSP27 pathway. Am J Physiol. 2000;278:H1899–H1907. doi: 10.1152/ajpheart.2000.278.6.H1899. [DOI] [PubMed] [Google Scholar]
- Youn T, Kim SA, Hai CM. Length-dependent modulation of smooth muscle activation: effects of agonist, cytochalasin, and temperature. Am J Physiol. 1998;274:C1601–C1607. doi: 10.1152/ajpcell.1998.274.6.C1601. [DOI] [PubMed] [Google Scholar]
- Zacour ME, Martin JG. Enhanced growth response of airway smooth muscle in inbred rats with airway hyperresponsiveness. Am J Respir Cell Mole Biol. 1996;15:590–599. doi: 10.1165/ajrcmb.15.5.8918366. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Lee CG, Zheng T, Chupp G, Wang J, Homer RJ, Noble PW, Hamid Q, Elias JA. Airway inflammation and remodeling in asthma. Lessons from interleukin 11 and interleukin 13 transgenic mice. Am J Respir Crit Care Med. 2001;164:S67–S70. doi: 10.1164/ajrccm.164.supplement_2.2106070. [DOI] [PubMed] [Google Scholar]