

Abstract
Adoptive transfer of T lymphocytes equipped with tumor-antigen specific T-cell receptors (TCRs) represents a promising strategy in cancer immunotherapy, but the approach remains technically demanding. Using influenza virus (Flu)-specific T-cell responses as a model system we compared different methods for the generation of T-cell clones and isolation of antigen-specific TCRs. Altogether, we generated 12 CD8+ T-cell clones reacting to the Flu matrix protein (Flu-M) and 6 CD4+ T-cell clones reacting to the Flu nucleoprotein (Flu-NP) from 4 healthy donors. IFN-γ-secretion-based enrichment of antigen-specific cells, optionally combined with tetramer staining, was the most efficient way for generating T-cell clones. In contrast, the commonly used limiting dilution approach was least efficient. TCR genes were isolated from T-cell clones and cloned into both a previously used gammaretroviral LTR-vector, MP91 and the novel lentiviral self-inactivating vector LeGO-MP that contains MP91-derived promotor and regulatory elements. To directly compare their functional efficiencies, we in parallel transduced T-cell lines and primary T cells with the two vectors encoding identical TCRs. Transduction efficiencies were approximately twice higher with the gammaretroviral vector. Secretion of high amounts of IFN-γ, IL-2 and TNF-α by transduced cells after exposure to the respective influenza target epitope proved efficient specificity transfer of the isolated TCRs to primary T-cells for both vectors, at the same time indicating superior functionality of MP91-transduced cells. In conclusion, we have developed optimized strategies to obtain and transfer antigen-specific TCRs as well as designed a novel lentiviral vector for TCR-gene transfer. Our data may help to improve adoptive T-cell therapies.
Keywords: T-cell receptor (TCR), gene transfer, influenza antigen, adoptive immunotherapy, TCR gene therapy, lentiviral vectors
Introduction
Adoptive immunotherapy with autologous antigen-specific T cells has been shown to be an efficient approach in fighting life-threatening infections1-3 and even malignant diseases.4-7 However, obtaining sufficient numbers of antigen-directed T cells in a reasonable period of time is often very difficult and expensive. Moreover, expanded tumor-specific T-cell clones have often turned out to be inactive in patients.4,8 One way to overcome this limitation may be the generation of large numbers of tumor-specific T cells by directly transferring the T-cell receptor (TCR) and thus the specificity of highly active T cells. To this end, sequences of the respective TCR need to be identified and transferred into T cells using appropriate vector systems.9-12 A number of recent clinical studies using TCR gene transfer have provided very promising results.4,13
Generally, the production of TCR-transduced T cells for adoptive immunotherapy comprises several critical steps, which are all technically demanding, time-consuming and often error-prone. First, antigen-specific and fully functional effector T-cell clones have to be identified. Second, the respective TCRs need to be isolated and cloned. Finally, efficient ways for the transfer and stable expression of the TCR in primary T cells need to be developed. Here, we have compared different techniques within stages one to three in order to optimize the production of TCR-transduced T cells for adoptive immunotherapy. So far, influenza virus (Flu)-specific T cells have not yet been generated applying TCR transduction. Using Flu, which naturally induces strong polyclonal T-cell responses in vast parts of the population,14 as a target antigen we aimed at developing an optimized technique for the generation of CD4+ and CD8+ T-cell clones independently from the nature of the target epitope and the respective HLA restriction pattern. We isolated the respective TCRs of the generated Flu-specific T-cell clones and cloned them into lentiviral as well as gammaretroviral vectors with similar configurations. After transferring the Flu-specific TCRs into the T-cell line J76 as well as primary T cells, we were able to compare the two different vector systems and show, for the first time, the successful generation of active anti-Flu effector T cells by TCR transduction.
Results
Generation of influenza-specific CD4+ and CD8+ T-cells
To generate influenza-specific CD4+ and CD8+ T cells, PBMCs of 4 healthy HLA-A2-positive donors (BC-126, BC-142, BC-143 and BC-144) were used. T cells were presensitized with the influenza matrix protein (Flu-M) for CD8+ and the influenza nucleoprotein (Flu-NP) for CD4+ cells. To assess the specificity of presensitized T cells three different assays were performed: an IFN-γ ELISPOT, the flow cytometry-based IFN-γ-secretion assay and tetramer staining. We successfully generated Flu-specific T cells from all 4 donors. As evidenced by the three types of read-out assays, the CD4+ and CD8+ T cells showed strong reactivity against Flu-M- and Flu-NP-peptides, respectively, but no reactivity against the control peptide SSX-2 (Fig. 1A and B).

Figure 1. Chronological order of experiments for the identification of Flu-specific clones for BC-143. (A) T-cell responses measured by IFN-γ ELISPOT assay: CD8+ T-cell responses to Flu-M- and SSX-2-peptides (left) as well as CD4+ T-cell response to Flu-NP- and SSX-2-peptides (right). Five × 104 (upper part) and 1 × 104 (lower part) cells per well were used. (B) T-cell responses measured by IFN-γ-secretion assay: CD4+ T-cell responses to Flu-NP and SSX-2 peptides by IFN-γ secretion (upper part) and CD8+ T-cell responses to Flu-M- and SSX-2-peptides measured by a combination of IFN-γ secretion and tetramer staining (lower part). (C) Clone specificity analysis: Analysis of CD8+ T-cell clone specificity via IFN-γ ELISPOT (upper part) and tetramer-FACS analysis (lower part).
Generation of influenza-specific T-cell clones
In the next step we aimed at optimizing the generation of influenza antigen-specific T-cell clones from T cells obtained by presensitization of memory precursors. To this end, we compared different cloning procedures.
For healthy donor BC-126 we used a limiting dilution approach seeding cells from CD4+ T cells evidencing Flu-NP-specific CD4+ T cells directly into cell culture plates in serial dilutions of 10, 1 and 0.3 cells/well, respectively. We were able to generate 35 CD4+ T-cell clones from 384 cells seeded (9.1%). However, only one of the CD4+ T-cell clones (0.3%) was found to be specific for Flu-NP in the ELISPOT, a frequency which would be in agreement with the percentage of Flu-NP-specific CD4+ T cells within the original T-cell bulk culture after presensitization.
Based on these suboptimal results we chose to apply a different technique for healthy donor BC-142. In this case, we decided to first enrich for influenza-specific T cells before seeding cells on a single-cell level. To this end, we applied FACS sorting of live cells based on IFN-γ secretion or based on TCR specificity using tetramer staining, respectively. Using tetramer-based sorting of cells from Flu-M-specific CD8+ T cells we were able to generate two (1.4% of 144 cells seeded) T-cell clones recognizing Flu-M. Using the cytokine-secretion assay, which allows for staining of IFN-γ on the surface of living cells, we were able to obtain three Flu-NP-specific CD4+ T-cell clones (0.6% of 480 cells seeded) from T cells which had contained 1.6% Flu-NP-specific cells after presensitization.
In the case of healthy donor BC-143 we compared all different approaches with regard to their potential to obtain CD4+ and CD8+ T-cell clones: the IFN-γ-secretion assay, tetramer staining, tetramer staining combined with IFN-γ secretion as well as limiting dilution. With the IFN-γ-secretion method alone we were able to obtain two Flu-NP-specific CD4+ T-cell clones out of 480 cells seeded (0.4%) from T cells that had originally contained 2.46% Flu-NP-specific cells. Using limiting dilution with the same T cells we were able to generate only one positive clone (0.3%). Using CD8+ T cells from the same donor, which had originally contained 1.07% of Flu-M-specific cells, not a single clone was obtained using tetramer-based FACS sorting. In contrast, combining Flu-tetramer staining and IFN-γ-secretion assay we were able to obtain a total of 8 Flu-M-specific CD8+ T-cell clones (1.19%) derived from 672 cells seeded from the same T cells (Fig. 1C).
For healthy donor BC-144 no specific T-cell clone was obtained from cells containing 0.3% and 0.24% Flu-M- and Flu-NP-specific T cells, respectively, irrespective of the approach used for cloning.
To summarize these data, the IFN-γ-secretion assay was the most efficient way for generating CD4+ T-cell clones from T cells containing Flu-NP-specific precursors. For Flu-M-specific CD8+ T cells, tetramer staining and the IFN-γ-secretion assay both were efficient. On the other hand, results obtained by directly seeding cells from influenza-specific T cells using limited dilution conditions were disappointing.
Isolation of T-cell receptor genes and genetic modifications of TCR chains
We isolated TCR genes from 14 of the generated CD8+ and from two CD4+ T-cell clones via RLM-Race-PCR. Integrity of TCR genes was confirmed by DNA sequencing. Analysis of acquired TCR-gene sequences against the IMGT-Database confirmed clonal identity of T cells from individual clones. Eleven out of 14 of all identified CD8+ T-cell clones specific for the Flu-M peptide expressed the same TCR genes: TRAV17*01, TRAJ42*01, TRBV19*01, TRBJ2–7*01, TRBD2*02. For the two CD4+ T-cell clones specific for the Flu-NP peptide two different TCR genes were identified: TRAV8–1*01, TRAJ43*01, TRBV6–1*01, TRBJ2–4*01, TRBD1*01 and TRAV8–1*01, TRAJ30*01, TRBV6–5*01, TRBJ2–7*01, TRBD2*01.
In initial experiments we were not able to detect Flu-M-specific cells by tetramer staining despite successful transduction of PBMCs with the native CD8+ Flu-M specific TCR chains. Therefore, we modified the cloned TCR genes by replacing the human constant regions by their murine counterparts in order to reduce TCR mispairing with the endogenous TCR chains. Additionally, the TCR chains were codon-optimized to enhance expression levels. These modifications were exemplarily performed for the identified CD8+ T-cell-receptor genes TRAV17*01, TRAJ42*01, TRBV19*01, TRBJ2–7*01, TRBD2*02. Optimized TCR sequences were subsequently cloned into lentiviral and gammaretroviral vectors.
Vectors used for TCR gene transfer
To efficiently transfer the identified Flu-specific T-cell receptor genes into T-cell lines and primary T cells we used different viral vectors. We aimed at directly comparing suitability of lentiviral and gammaretroviral constructs for TCR-gene transfer. To do so, we used both well-established gammaretroviral and lentiviral vectors. Similar promotors and expression cassettes were used in both vectors for better comparability (Fig. 2). The third generation lentiviral vector LeGO-MP, is based on the LeGO-iG2 vector.15 For stronger expression in T cells we exchanged the internal SSFV-promotor by an internal MPSV-promotor, followed by a leader sequence derived from the murine embryonic stem cell virus. A very similar promotor configuration is present in the gammaretroviral vector MP91.16 In both constructs, the two genes for the influenza-specific TCR chains were linked via a 2A-sequence from the porcine teschovirus-1 (P2A) for equimolar co-expression. Also, both vectors contained a Woodchuck hepatitis virus post-transcriptional regulatory element (wPRE) and an IRES-eGFP sequence. As stated above, TCR genes were genetically modified by codon optimization and by replacing the human constant regions with the murine constant regions. The modified TCR sequences were cloned into the two vectors and the vector constructs are hereinafter termed “LeGO-MP-muFlu” (Fig. 2A) and “MP91-muFlu” (Fig. 2B), respectively.
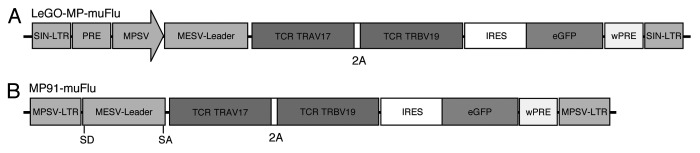
Figure 2. Schematic representation of vector constructs used in this study. The lentiviral vector (A) “Lego-MP-muFlu” and the gammaretroviral vector (B) “MP91-muFlu” are displayed [not to scale]. MPSV, myeloproliferative sarcoma virus; LTR, long-terminal repeat; MESV, murine embryonic stem cell virus; IRES, internal ribosome entry site; wPRE, Woodchuck hepatitis virus post-transcriptional regulatory element; P2A, 2A sequence of porcine teschovirus-1; eGFP, enhanced green fluorescent protein; SD, splice donor; SA, splice acceptor.
Viral transfer of Flu-specific TCRs into the αβ-TCR deficient T-cell line J76
To test functionality and specificity of the constructed TCR-chains, we first used the αβ-TCR deficient T-cell line J76. J76 cells lack expression of endogenous TCR chains, but possess all necessary CD3 molecules, which are only recruited to the surface in the presence of a functional T-cell receptor. Therefore, functional expression of introduced TCR transgenes is possible without interference with endogenous TCRs. In fact, high-level functional expression of introduced TCR genes has been shown in J76 cells.17 Conveniently, expression of introduced TCR genes can easily be detected using anti-TCR and/or anti-CD3 antibodies.
We produced VSV-pseudotyped viral supernatants for both vector constructs. The obtained infectious titers were in the range of 1 to 2 × 107 viral particles/ml. The J76 cell line was transduced with the viral supernatants of LeGO-MP-muFlu, MP91-muFlu and LeGO-iG2, as a mock control, at a multiplicity of infection (MOI) of 1 as estimated on 293T cells. J76 cells were successfully transduced with all vector constructs. Based on eGFP expression we detected comparable transduction rates of 73.6% for MP91-muFlu and 66.5% for LeGO-MP-muFlu (Fig. 3A). Additionally we assessed expression of the Flu-specific TCR by staining the cells with monoclonal antibodies against murine TCR (mTCR) and CD3. The obtained data was in very good accordance with the percentages of eGFP+ cells (Fig. 3A). In line with that, almost all eGFP positive cells simultaneously showed expression of the transgenic TCR and CD3 molecules on their cell surface (Fig. 3B). In contrast, J76 cells transduced with the control vector LeGO-iG2 also showed high eGFP-expression (91.6%), but no TCR- or CD3-antibody staining was detectable by FACS (Fig. 3B), underlining the functionality of the chosen J76 cell-based approach.

Figure 3. Expression of the muFlu-vector constructs in J76 cells. J76 cells were transduced with LeGO-MP-muFlu, MP91-muFlu and LeGO-iG2, serving as a mock control. (A) 3 d post transduction eGFP-, CD3- and mTCR- expression were analyzed. Numbers indicate percentages of eGFP, mTCR and CD3 positive cells, respectively. (B) eGFP positive cells were double-stained with the mTCR- and CD3-specific antibodies. eGFP+ cells expressing both CD3 and the Flu-TCR are depicted in the upper right quadrant.
Cytokine secretion by TCR-transduced J76 cells upon specific stimulation
Next we assessed whether the cloned TCR showed antigen-specific effector functions upon contact with the cognate influenza matrix antigen (Flu-M). To address this question, Flu-TCR transduced J76 cells were co-cultured with T2-target cells either pulsed with Flu-M peptide or the irrelevant SSX-2 control peptide. LeGO-iG2 transduced J76 cells served as an additional control to investigate the target-effector interaction specificity. After 18–20 h of co-culture, supernatant was harvested and analyzed by ELISA for the presence of IL-2 and IFN-γ. As shown in Figure 4 Flu-TCR-transduced J76 T cells specifically secreted high amounts of IL-2 in response to T2 cells presenting the Flu-M peptide. In contrast, no IL-2 secretion was detected upon co-cultivation with the control peptide. Comparison between the LeGO-MP-muFlu and the MP91-muFlu indicates that both vectors were able to mediate high IL-2 secretion to a similar extent. Also, Flu-TCR transduced J76 cells secreted moderate amounts of IFN-γ upon contact with the Flu-M-, but not the control peptide (data not shown). LeGO-iG2 (mock control) transduced J76 cells showed no secretion upon co-cultivation with peptide-pulsed T2 cells. These results prove that the antigen-specificity of the influenza-specific T-cell clones was successfully transferred to the J76 T-cell line and that the transduced cells retained their effector functions.
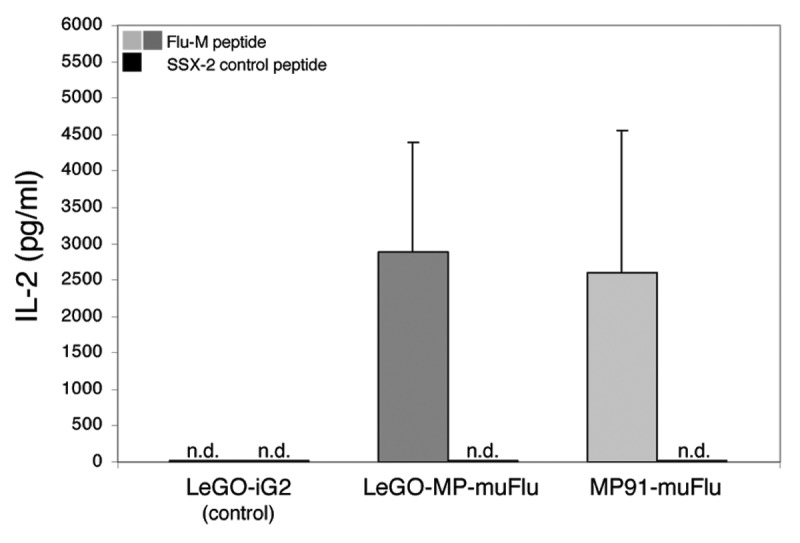
Figure 4. Specific IL-2 secretion of Flu-transduced J76 cells upon encounter with the Flu-M-peptide. J76 cells were stably transduced with the lentiviral construct LeGO-MP-muFlu and the gammaretroviral construct MP91-muFlu or mock-transduced with LeGO-iG2. Transduced cells were co-cultured with Flu- or SSX-2- peptide pulsed T2 cells, serving as APCs. After 18–20 h the supernatant was harvested. IL-2 secretion was analyzed by ELISA. Mean values and range of two independent experiments are shown. No IL-2 secretion was detectable for J76 cells stimulated with the irrelevant peptide SSX-2 and for mock-transduced J76 cells. n.d., not detectable.
Transfer of Flu-specific TCR constructs into primary T cells
We went on by investigating the possibility to transfer the influenza-specificity of the generated TCR constructs to αβ-harboring primary T cells. To this end, we transduced prestimulated T cells of two healthy donors (BCBBe3, BCBBe4) with GALV-pseudotyped viral vectors, since the GALV envelope was previously shown to mediate efficient gene transfer into primary T cells.18-20
Transduction rates for the two vectors were measured by FACS based on eGFP-expression. They were similar for both donors, but overall relatively low (as compared with J76 cells). Higher transduction efficiency was found for the gammaretroviral vector MP91-muFlu (around 20–30%), compared with the LeGO-MP-muFlu vector (10–18%).
The proportion of transduced GFP+ primary T cells expressing the influenza-specific TCR on their cell surface was measured by FACS after antibody-staining with the murine TCR-antibody and tetramer staining with the influenza-specific tetramer. We observed that about 40% - 50% of the GFP+ cells were labeled by the mTCR antibody and the tetramer and hence expressed the introduced influenza-TCR at sufficiently high levels. This percentage was consistent for both vector constructs. Figure 5 shows representative FACS plots of one transduction for donor BCBBe3 with LeGO-MP-muFlu. More than half of the 17.3% GFP- positive cells (9.7%) expressed detectable levels of the influenza-specific TCR. Three quarters of the TCR-positive cells (7.2% of all) were stained by the Flu-specific tetramer. In agreement with that, approximately one third of all GFP-positive cells (5.8% of total cells) were double stained for mTCR and tetramer. Mock-transduced T cells showed no staining with the mTCR-antibody and tetramer (data not shown).

Figure 5. Transduction of peripheral blood T cells with the LeGO-MP-muFlu-vector construct. Peripheral blood T cells of buffy coat BCBBe3 were transduced with LeGO-MP-muFlu 72 h after activation with CD3/CD28-Dynabeads. Transduction efficiencies were measured 3 d post transduction via FACS, looking at eGFP-, mTCR- and Flu-tetramer-expression and staining, respectively. Numbers indicate percentages of eGFP, mTCR and Flu-tetramer positive cells.
Release of effector cytokines after influenza antigen-specific stimulation by transduced primary cells
Finally we analyzed whether the transduced and Flu-TCR-expressing primary T cells, which made up 2–6% (mTCR and tetramer double positives) of the total culture were able to react to their cognate antigen. Five days after transduction non-selected T cells were co-cultured with T2-target cells pulsed with the specific Flu-M peptide or with the irrelevant peptide SSX-2 (negative control). Antigen recognition was assessed using IL-2, IFN-γ and TNF-α cytokine release assays. As shown in Figure 6 the lentiviral transduced T-cell culture, as well as the one transduced with the gammaretroviral Flu-vector were able to secrete significant amounts of IFN-γ when stimulated with T2 cells loaded with the Flu-M peptide, but not with the negative control SSX-2 peptide (Fig. 6A). The cells also secreted high amounts of IL-2 (Fig. 6B) and moderate amounts of TNF-α (Fig. 6C) when co-cultured with Flu-pulsed T2 cells. These results indicate that the T-cell receptor isolated from a Flu-specific clone obtained from healthy donor PBMCs and introduced into primary T cells was still reactive to the cognate influenza antigen, independently of the vector (lentiviral or gammaretroviral) used for transduction of primary T cells. Comparing the two vector constructs, T cells transduced with the gammaretroviral vector MP91-muFlu secreted greater amounts of IL-2, IFN-γ and TNF-α when compared with T cells transduced at the same MOI but using the lentiviral vector LeGO-MP-muFlu. To exclude an impact of the higher transduction efficiencies seen with the gammaretroviral vector on functionality of TCR-modified T lymphocytes we next used T cells transduced to equal amounts (as assessed by Flu-tetramer staining) for functional analysis. Notably, in the cytokine-secretion ELISA cells transduced with the gammaretroviral vector still secreted more IFN-γ (Fig. 6D).

Figure 6. IL-2-, IFN-γ and TNF-α- secretion by transduced primary human T lymphocytes upon co-culture with the Flu-M vs. SSX-2- peptide. Primary T cells were transduced with LeGO-MP-muFlu or MP91-muFlu 72h after activation of PBMCs with CD3/CD28-Dynabeads. Five days after transduction cells were co-cultured with peptide (Flu-M, SSX-2) pulsed T2 cells and the secretion of different cytokines was analyzed by ELISA. (A–C) Mean values and ranges for two to three independent experiments with two buffy coats (BCBBe3, 4) are shown. (A) IFN-γ, (B) IL-2, (C) TNF-α. (D) IFN-γ cytokine secretion of transduced primary cells showing the same amount of Flu-tetramer positive cells were compared by ELISA. Mean values of 3 different buffy coats and two experiments are shown. n.d., not detectable.
Discussion
In this study, we aimed at establishing a straightforward protocol for the identification of antigen-specific T-cell clones and for the generation of TCR-transduced T cells by comparatively evaluating different strategies and vector systems. It has previously been shown that the adoptive transfer of antigen-specific T cells can be an efficient strategy in fighting viral infections1-3 and malignant diseases.4-7 However, the identification of antigen-specific T-cell clones and the subsequent steps - identification and cloning of the specific TCR, efficient transfer/expression of the TCR in primary T cells using suitable vectors - remain technically challenging.
As a model, we cloned and transferred TCRs specific for influenza A virus antigens. We chose the Flu matrix protein (Flu-M) and the Flu nucleoprotein (Flu-NP) because a large proportion of the human population shows strong T cell-mediated immune responses against them.14 Moreover, the Flu-NP peptide used in this study shows a promiscuous behavior being presented by a large variety of HLA class II molecules21 allowing for the expansion of specific CD4+ memory T cells from the majority of donors included. After we had identified influenza-specific CD4+ and CD8+ T cells we compared different approaches with regard to their efficiency in the isolation of specific T-cell clones. Interestingly, we found that the commonly used limiting dilution assay was least efficient. Thus, we would not recommend to use this as a single approach for the generation of CD4+ and CD8+ T-cell clones from pre-existing antigens-specific T cells. In contrast, we found the cytokine-secretion assay, either alone or in combination with tetramer staining, to show the highest yield of T-cell clones derived from presensitized bulk cultures. This approach also has the advantage that it allows for the generation of T-cell clones independently of the antigen used and the individual HLA context. Hence, we would suggest to preferably applying the cytokine-secretion assay to obtain antigen-specific T-cell clones from T cells produced by presensitization of memory precursors, in particular when the precursor frequency is low and a variety of HLA haplotypes need to be taken into account.
Functional testing of cloned TCRs may become a bottleneck of the strategy. J76 cells have been proposed for testing TCR-construct functionality and specificity. J76 cells lack endogenous TCR chains, which helps to avoid interference problems in this model cell line.22 Consequently, expression of the genetically introduced TCR on the surface of these cells can easily be detected via anti-TCR and anti-CD3 antibodies. Indeed, transduction of J76 cells with our gammaretroviral and lentiviral constructs resulted in high levels of CD3 and TCR expression readily detectable by flow cytometry. Importantly, J76 cells retained the capability to function as effector T cells.23 In line with that we found high-level IL-2, and also IFN-γ secretion by J76 cells transduced with Flu-specific TCRs in the presence of APCs presenting the cognate influenza antigen. Notably, IL-2 secretion was comparable for both vector types (lentiviral and gammaretroviral constructs) used for TCR gene transfer. This finding indicates that at least in J76 cells both gammaretroviral and lentiviral vectors are equally effective. It also confirms that J76 cells are a very convenient tool for initial testing of TCR functionality and specificity.
To become practically useful, cloned TCRs also need to prove their functionality in primary T cells harboring an endogenous T-cell receptor. Introducing a second T-cell receptor is associated with a number of important challenges. First, the presence of four TCR chains may result in a so-called mispairing, i.e., the pairing of an introduced α/β chain with the complementary endogenous TCR α/β chain. Such mispairing can have several consequences, e.g., an impairment of surface expression of the introduced TCR, which can be the cause of insufficient effector function.24,25 Also, TCR mispairing can lead to the generation of TCRs with unknown specificities. In a worst-case scenario the latter could be autoreactive and lead to a lethal graft-vs.-host disease.26 Another problem is the competition between the endogenous TCR and the introduced TCR for the CD3 molecules.27 TCR cell-surface expression requires the CD3 molecules to form a complex with the TCR, which then migrates to the cell surface. T-cell receptors that do not assemble properly with the CD3 molecules are degraded.25 Availability of CD3 molecules is thus a critical point, because the competition may lead to reduced surface expression of the introduced TCR resulting in impairment of effector functions.27 Indeed, we have seen these effects with our Flu-M-specific TCR in preliminary experiments. Transduction of the T-cell line J76 with the genetically unmodified Flu-M-specific TCR resulted in its strong expression and functional transfer of specificity (s. above). On the contrary, after transduction of primary T cells with the same TCR, we did not detect its expression in transduced cells by Flu-M-tetramer staining. Moreover, we did also not observe any IL-2, IFN-γ or TNF-α production of transduced primary T cells in cytokine-secretion assays after stimulation with APCs pulsed with the relevant influenza-specific peptide.
Several strategies have been proposed to maximize cell surface expression of the introduced T-cell receptor including codon optimization of the transgene-cassette or addition of a second disulfide bond.28,29 It was also observed that murine TCRs when transduced into human primary T cells often replaced the endogenous TCR on the cell surface.30,31 One underlying mechanism is the reduction of mispaired TCR dimers, because murine TCR preferentially pair with their murine counterparts.30 In addition, more efficient interaction of the murine constant regions with human CD3 molecules compared with the human constant regions has been observed. Therefore, TCRs containing murine constant regions are more efficient in the competition for the CD3 molecules.22,30
Based on the observations outlined above we modified the influenza-specific T-cell receptor accordingly. We performed codon optimization and substituted the human TCR constant region with the murine counterpart. These modifications led to dramatic changes. While the unmodified TCR had not been detectable in primary T cells, the optimized TCR was readily stained with the Flu-M-specific tetramer. Also, staining with the murine TCR-antibody resulted in efficient detection of the introduced TCR. Double staining with Flu-tetramer and mTCR-antibody indicated that at least 30–40% of transduced T cells expressed a functional TCR (as normalized for eGFP expression). Taken together, these data indicate that the applied genetic modifications indeed facilitated and strongly improved expression of the introduced TCR. For clinical use, a humanized murine constant TCR region has been developed. To do so, the amino acids responsible for the more efficient CD3 recruitment by murine TCRs were defined and introduced into the human constant region, resulting in only 4 amino acid changes in the TCRα C region and 5 amino acid changes in the TCRβ C region.22
Notably, we found that a large proportion of transduced GFP+ primary T cells did apparently not express the optimized TCR. This could reflect the fact that T-cell receptors need a certain threshold to be transported to the cells surface and to become activated.28 Based on the used co-expression strategy it is very likely that cells expressing only moderate amounts of eGFP also express only moderate amounts of the TCR chains and thus cannot be detected by the mTCR antibody and Flu-tetramer. Another explanation is based on the observation made by Heemskerk and colleagues,32 who showed that the expression levels of the introduced TCR are influenced by the strength of the endogenous TCR, which is obviously different in individual T cells. In regard to the strength of the vector constructs one need to take into account that the IRES-eGFP sequences included in the vector for convenient analysis, probably lead to lower efficiencies in transducing T cells. Moreover the IRES-eGFP sequences are certainly not applicable for use in potential future clinical trials.
Among the different vector systems used for TCR gene delivery, integrating retroviral vectors represent the most popular delivery system today, because these vectors have high gene transfer efficiency and usually mediate high and stable expression of the transgene.33 In the next part of this study we directly compared the efficiency of two different retroviral vectors and their functionality for T-cell receptor gene transfer. The usefulness of lentiviral and gammaretroviral vectors for TCR gene transfer was previously compared, indicating superior performance of the used lentiviral vector.34 However, vectors used in that study were not optimized for T-cell gene transfer. In this study we used an optimized MPSV-based gammaretroviral backbone35 previously shown to facilitate efficient TCR gene transfer.36-39 This vector was directly compared with the novel lentiviral vector LeGO-MP designed for this study. In both our vector constructs, the two genes for the influenza-specific TCR chains were linked via a 2A-sequence from the porcine teschovirus-1. Additionally both vectors contain a similar promotor configuration and a woodchuck hepatitis virus post-transcriptional regulatory element (wPRE) and are therefore well comparable with each other. The comparison of the lentiviral vector LeGO-MP-muFlu and the gammaretroviral vector MP91-muFlu in T-cell line J76 showed that both vectors were equally good in transducing T cells, as well as in regard to the strength of the effector functions. In primary T cells we detected that the gammaretroviral vector was able to transduce on average twice as many T cells as the LeGO-MP-muFlu construct. However, both the gammaretroviral and the lentiviral vector constructs were functional in primary T cells and were able to recognize their influenza specific peptide presented by APCs. This recognition resulted in secretion of high amounts of IFN-γ, IL-2 and TNF-α. We also observed that the gammaretroviral vector MP91 mediated stronger secretion of cytokines in comparison to the lentiviral vector LeGO-MP. To investigate whether these differences in cytokine secretion just reflected different transduction rates, we performed additional experiments using different MOIs for the two vectors. Based thereon we were able to directly compare functionality of cells with equalized TCR expression levels as assessed by tetramer-staining. As previously, the gammaretroviral vector was still able to mediate higher cytokine secretion compared with the lentiviral vector. This outcome probably reflects different capabilities of the two vectors to express functional levels of T-cell receptor molecules in primary T cells, indicating some inherent advantage of the gammaretroviral vector. The latter is probably due to higher expression strength mediated by the LTR configuration.
An advantage of lentivirus-based vector systems over gammaretrovirus-based systems is the potentially lower insertional genotoxicity, especially in regard to gene-therapy trials with human stem cells. Several cases of T-cell leukemia in children, which were caused by insertional mutagenesis, were reported.40,41 Gammaretroviral LTR vectors can activate genes by promotor activation from the long-terminal repeats (LTRs), whereas third-generation lentiviral vectors are self-inactivating, thereby reducing this risk. Currently it is believed that more mature cells, like fully differentiated T cells, which are the natural target of T-cell receptor gene therapy approaches, are less susceptible to transformation by insertional mutagenesis.16 However it was also shown that gammaretroviral insertional mutagenesis can happen in mature T lymphocytes even if the chances are relatively low.35 In summary, in respect to the comparison of the two vector constructs, we have shown that the novel lentiviral vector LeGO-MP mediates similar effector functions as the already frequently used gammaretroviral vector MP91. So far, the MP91 vector is more effective in transducing primary T cells and is also easier and less expensive to produce for clinical trials. If the production challenges can be overcome, the future will show if the lower insertional mutagenesis frequencies of lentiviral vectors, as observed in hematopoietic stem cells, will also be beneficial for gene therapy of mature T cells. This would potentially make a change to lentiviral vectors in T-cell receptor gene therapy beneficial.
In summary, based on the systematic evaluation of different, widely used techniques we established an optimized, straight-forward protocol for the generation of antigen-specific CD4+ and CD8+ T-cell clones, which will be beneficial for different strategies of adoptive immunotherapy. Using this protocol we, for the first time, successfully transferred T-cell receptors highly specific against broadly recognized influenza epitopes. While clinical application of influenza-directed TCRs cannot be expected, this model system might be very useful for testing novel techniques or vectors as demonstrated here. Finally, in this work we directly compared the performance of an MPSV-based gammaretroviral LTR vector frequently employed for T-cell gene transfer with a novel third-generation LeGO-MP vector equipped with identical gene expression cassettes. This enabled us to show that the LeGO-MP vector is principally useful for the transfer of T-cell specificity, but the classical gammaretroviral LTR vector was superior with regard to transduction and expression levels, particularly in primary T cells.
In conclusion, we believe that the approach outlined herein will facilitate the production of TCR-transduced T cells for the adoptive transfer of virus- and also tumor-specific T cells.
Materials and Methods
Peripheral blood mononuclear cells (PBMCs) and cell lines
PBMCs were isolated by Ficoll gradient centrifugation from blood (buffy coats) donated by healthy individuals after informed consent. The cell line T2 (ATCC CRL-1992) is an HLA-A*0201-expressing lymphoblastoid line that is deficient in transporter associated with antigen processing (TAP) function and is, therefore, unable to present peptides derived from endogenous cytosolic proteins. T2 cells can thus be loaded with exogenous peptide to serve as antigen-presenting cells (APCs). The cell line J76 is a αβ-TCR-deficient T-cell line, which was kindly provided by W. Uckert (Max-Delbrück-Center, Berlin, Germany). J76 cells are very convenient for testing TCR-construct functionality because of the absence interfering endogenous TCR chains. T2 and J76 cells were cultured in RPMI 1640, GlutamaxTM (Life Technologies, Darmstadt, Germany), supplemented with 10% FCS, 2% L-Glutamine and 1% Sodium Pyruvate (Life Technologies). The cell line 293T (ATCC CRL-11268) used for virus production and titration of viral supernatants was cultured in DMEM, GlutamaxTM (Life Technologies), supplemented with 10% FCS. Cell-culture material was purchased from Corning Inc. (Corning, NY), Greiner Bio One (Frickenhausen, Germany) and Sarstedt (Nürnbrecht, Germany).
Peptides, proteins and antibodies used for flow cytometry
In this study the influenza matrix protein (Flu-M) peptide 58–66 (GIL GFV FTL) produced by Bio-Synthesis (Lewisville, TX), the influenza A nucleoprotein (Flu-NP) peptide 206–229 (FWRGENGRKTRIAYERMCNILKGK) and the control SSX-2 peptide 101–120 (FGRLQGISPKIMPKKPAEEG), both synthesized by Iris Biotech (Marktredwitz, Germany) were used. The HLA-A2 tetramer assembled with influenza matrix peptide 58–66 was provided by the Ludwig Institute for Cancer Research (Lausanne, Switzerland). We used the following mouse anti-human antibodies: anti-TCRβ-APC (eBioscience, San Diego, CA), anti-CD3-APC and anti-TCRα/β-PE (Miltenyi Biotec, Bergisch Gladbach, Germany).
Identification of Flu-specific CD4+ and CD8+ T-cell clones
CD4+ and CD8+ T cells were sequentially purified from PBMCs of 4 healthy donors (HLA-A2 serotype) using bead-based magnetic separation (CD4+/CD8+ Isolation Kit; Life Technologies). Read-out assays were performed after a single cycle of in-vitro presensitization, as described previously.21 Briefly, remaining CD4- CD8- cell populations were used as antigen-presenting cells (APC). Initially, T cells were stimulated once with irradiated APCs pulsed with either 20 µM influenza-specific peptide or the irrelevant control peptide. After 10–20 d of culture in RPMI 1640 (Life Technologies) supplemented with 10% human AB serum, 1% penicillin and streptomycin (Life Technologies), human IL-2 (10 U/ml, Roche Diagnostics, Mannheim, Germany) and human IL-7 (20 ng/ml, R&D Systems, Minneapolis, MN, USA), CD8+ and CD4+ cells were analyzed for their specificity in IFN-γ ELISPOT assay, tetramer staining and IFN-γ secretion assay prior to clonal separation.
The ELISPOT was performed according to manufacturer’s instructions (IFN-γ ELISPOT kit, Mabtech) using nitrocellulose-bottom ELISPOT plates (MAHA S4510, Millipore). A fraction of initially separated and subsequently activated with 2 µg/ml phytohemagglutinin (PHA, Roche) autologous CD4+ cells served as T-APCs. T-APCs were pulsed in serum-free X-VIVO 15 medium (Lonza, Wuppertal, Germany) overnight with cognate influenza-derived peptides or the control peptide of the SSX-2 protein as described above. Five × 104 or 1 × 104 presensitized effector T cells together with 1 × 105 target T-APCs were co-cultivated 16 h on ELISPOT plates prior to detection. Ten times less cells (e:t ratio) were taken when analyzing T-cell clones. Spot-forming units (SFU) were counted using an ELISPOT reader and ELISPOT software version 4.0 (Autoimmun Diagnostika, Strassberg, Germany). A positive response was defined as > 10 spots/well and > three times the number of spots counted for the irrelevant control target. Results were representative of at least 2 repeated experiments.
Three different approaches were compared with regard to their efficiency of generating influenza-specific T-cell clones from specific T cells. The first procedure used limiting-dilution conditions and T cells were seeded into U-bottom 96-well cell-culture plates in serial dilutions of 10, 1 and 0.3 cells/well and further analyzed by ELISPOT and Flu-specific tetramer staining.
The second approach took advantage of the cytokine-secretion potential of activated T cells upon co-cultivation with pulsed T-APCs. Vital IFN-γ secreting antigen-specific T cells were detected and further separated on single-cell level by FACS-based sorting using the IFN-γ Secretion-Assay-Detection-Kit (Miltenyi Biotec) according to manufacturer’s instructions with some modifications. Briefly, 1 × 105 target T-APCs were pulsed overnight with a Flu-specific peptide or an irrelevant control peptide of the SSX-2 protein. Cells were washed once in X-VIVO 15 medium and stained with 0.33 µM CFSE (Sigma-Aldrich) for 10 min at 37°C. After thoroughly washing with cold X-VIVO 15 medium, presensitized effector T cells were co-cultivated in an effector to target ratio of 1:1 in 100 µl X-VIVO 15 medium for 2.5 h at 37°C. Cells were washed in cold assay buffer and secreted IFN-γ was bound for 45 min at 37°C under rotating conditions using a volume of 10 ml X-VIVO 15 medium. Cells were stained with the detection antibody anti-IFN-γ-APC and the co-staining antibodies CD4-PerCP or CD8-PerCP (BD Biosciences, Heidelberg, Germany). Samples were measured using a FACSAriaIIIu (Becton Dickinson, Heidelberg, Germany, equipped with 405-/488-/561- and 640-nm lasers) with FACSDiva software (BD Biosciences) and analyzed with FlowJo software (Tree Star Inc.). IFN-γ+ Flu-specific effector T cells were equalized against the corresponding negative control of effector T cells pulsed with the irrelevant SSX-2 peptide. For obtaining single and viable IFN-γ-producing effector T cells, IFN-γ+ cells were sorted into U-bottom 96-well cell-culture plates and cultivated as described.
The third approach was based on tetramer staining to detect and sort CD8+ T cells specific for the HLA-A2-restricted Flu-M peptide on a single cell level. Five × 105 CD8+ T cells were incubated with 1 µl Flu-tetramer in 50 µl PBS with 1% bovine serum albumin (BSA) at 37°C for 15 min prior sorting. Tetramer-positive cells were sorted by FACS into U-bottom 96-well cell culture plates.
T-cell clones derived by the described methods were cultivated in T-cell medium with 1 × 105 feeder cells per well and 1 µg/µl PHA. Feeder cells were obtained by pooling irradiated (60 Gy) PBMCs from buffy coats of 3 healthy donors. Every two weeks, new irradiated feeder cells and PHA were added. T-cell clone specificity was confirmed by ELISPOT analysis before the clones were subjected to TCR isolation.
Isolation of T-cell receptor genes by RLM-Race-PCR
For the isolation of influenza-specific TCR genes, mRNA of individual clones was isolated using the μMACS™ mRNA Isolation Kit (Miltenyi Biotec) following manufacturer’s instructions. The mRNA was amplified using a 5′ Race protocol based on the First Choice RLM-Race Kit (Ambion, Austin, TX, USA) with the provided universal 5′ primers and 3′-specific primers for the TCR-α constant region or the two TCR-β constant regions, respectively. PCR products were subcloned into PJet1.2/blunt (CloneJET™ PCR Cloning Kit, Thermo Scientific, St. Leon-Rot, Germany) and sequenced (Seqlab, Göttingen, Germany). V, D, J usage of the sequenced chains was checked with the IMGT-Database (http://www.imgt.org).
Cloning of lentiviral and gammaretroviral vector constructs
The identified α and β chains were linked via a 2A sequence by overlapping polymerase chain reactions in the orientation Beta-2A-Alpha as previously described.42 TCR sequences were codon optimized and the human constant regions were replaced by the murine constant regions, as previously reported.22 Codon optimization was done by GeneArt (Life Technologies). The construct was ligated into the lentiviral vector LeGO-iG2 (ref. 15) and the gammaretroviral vector MP91.35 For stronger expression in T cells we exchanged the internal SSFV-promotor in the LeGO-iG2 for an internal MPSV-promotor,43,44 followed by a leader sequence derived from the murine embryonic stem cell virus. This new vector was named LeGO-MP. Besides the TCR genes both vectors additionally contained IRES-eGFP-sequences for easier tracing of transduced cells.
Generation of lentiviral and gammaretroviral particles
Viral supernatants of the lentiviral and gammaretroviral constructs were generated by transient transfection of 293T packaging cells as previously described.15,45 Viruses were pseudotyped with the vesicular stomatitis virus (VSV) glycoprotein for transduction of T-cell lines or with gibbon ape leukemia virus (GALV) envelope protein for transduction of PBMCs. Viral supernatants were titrated on 293T cells.
Lentiviral and gammaretroviral TCR gene transfer
T-cell lines were transduced in their respective growth medium by spin-infection. Briefly 1–2 × 105 target cells were incubated with viral supernatant in the presence of 8 µg/ml polybrene (Sigma-Aldrich) and centrifuged at 1,000 × g for 1 h at 22°C. The T-cell line was transduced at MOIs of 1–2 to obtain transduction rates of 60–80%.
Before transduction of primary T cells, PBMCs were activated for 72 h with Dynabeads® Human T-Activator CD3/CD28 (Life Technologies) according to the manufacturer’s instructions in X-VIVO 15, supplemented with 8% autologous serum and 100U/ml IL-2.46 For transduction, non-tissue culture-treated 24-well-plates were coated with 20 µg RetroNectin (Takara Bio Inc., Otsu, Shiga, Japan) for 2 h at RT or overnight at 4°C.
Viral supernatant (MOI: 5 – 20) was loaded onto the RetroNectin-coated plates by four consecutive centrifugation rounds at 940 × g and 4°C for 30 min. Subsequently, approximately 2 × 105 activated PBMCs per well were added and incubated at 37°C and 5% CO2.
Flow cytometric analysis of transduced T-cell lines and PBMCs was conducted 3 d post transduction on an LSRFortessaTM cytometer (Becton Dickinson), equipped with 405-/488-/561- and 640-nm lasers.
Determination of antigen specificity and effector function of TCR-transduced T cells
Antigen specificity and effector functions of TCR-transduced T cells were determined by enzyme-linked immunosorbent (ELISA) assays. In the ELISA assays we evaluated the secretion of different cytokines by transduced effector cells (J76 and PBMCs) in response to their cognate antigen. T2 cells, serving as APCs, were pulsed with 20 µM Flu-M peptide or SSX-2 peptide in cell-culture medium for 2 h at 37°C. One × 105 target T2 cells and 5 × 105 transduced J76 or 3 × 105 transduced PBMCs, were then cocultured in 96-well round bottom plates in 250 µl culture medium. After 18–20 h the supernatant was harvested and analyzed for IFN-γ, interleukin-2 (IL-2) and tumor necrosis factor-α (TNF-α) secretion using the appropriate Ready-Set-Go!® ELISA-Kit (eBioscience) following the manufacturer’s instructions. Absorbance was measured at 450 nm (minus 570 nm for wavelength correction) using the Sunrise Basic Tecan ELISA Analyzer (Tecan Austria GmbH, Grödig, Austria). Data was analyzed using MyAssays Software (MyAssays Ltd, http://www.myassays.com) and a four parameter logistic fit curve.
Acknowledgments
This work is part of the PhD thesis of Belinda Berdien at the University of Hamburg. It was supported by a grant from the Deutsche Krebshilfe. We wish to thank the FACS Core unit of the UMC Hamburg-Eppendorf, the Z2 project of SFB841 and the Dept. of Transfusion Medicine of the University Medical Center Hamburg-Eppendorf for kind support.
Glossary
Abbreviations:
- TCR
T-cell receptor
- Flu
influenza
- Flu-NP
influenza nucleoprotein
- Flu-M
influenza matrix protein
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/vaccines/article/24051
References
- 1.Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119:2644–56. doi: 10.1182/blood-2011-08-371971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peggs KS, Verfuerth S, Pizzey A, Khan N, Guiver M, Moss PA, et al. Adoptive cellular therapy for early cytomegalovirus infection after allogeneic stem-cell transplantation with virus-specific T-cell lines. Lancet. 2003;362:1375–7. doi: 10.1016/S0140-6736(03)14634-X. [DOI] [PubMed] [Google Scholar]
- 3.Gustafsson Å, Levitsky V, Zou JZ, Frisan T, Dalianis T, Ljungman P, et al. Epstein-Barr virus (EBV) load in bone marrow transplant recipients at risk to develop posttransplant lymphoproliferative disease: prophylactic infusion of EBV-specific cytotoxic T cells. Blood. 2000;95:807–14. [PubMed] [Google Scholar]
- 4.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg SA. Raising the bar: the curative potential of human cancer immunotherapy. Sci Transl Med. 2012;4:ps8. doi: 10.1126/scitranslmed.3003634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bobisse S, Zanovello P, Rosato A. T-cell receptor gene transfer by lentiviral vectors in adoptive cell therapy. Expert Opin Biol Ther. 2007;7:893–906. doi: 10.1517/14712598.7.6.893. [DOI] [PubMed] [Google Scholar]
- 10.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 11.Stripecke R, Kasahara N. Lentiviral and Retroviral Vector Systems. Cancer Drug Discovery: Gene Therapy for Cancer. 2009:39–71. [Google Scholar]
- 12.Schambach A, Swaney WP, van der Loo JCM. Design and production of retro- and lentiviral vectors for gene expression in hematopoietic cells. Methods Mol Biol. 2009;506:191–205. doi: 10.1007/978-1-59745-409-4_14. [DOI] [PubMed] [Google Scholar]
- 13.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naumov YN, Naumova EN, Yassai MB, Kota K, Welsh RM, Selin LK. Multiple glycines in TCR alpha-chains determine clonally diverse nature of human T cell memory to influenza A virus. J Immunol. 2008;181:7407–19. doi: 10.4049/jimmunol.181.10.7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weber K, Bartsch U, Stocking C, Fehse B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Mol Ther. 2008;16:698–706. doi: 10.1038/mt.2008.6. [DOI] [PubMed] [Google Scholar]
- 16.Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, et al. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278–86. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- 17.Sommermeyer D, Neudorfer J, Weinhold M, Leisegang M, Engels B, Noessner E, et al. Designer T cells by T cell receptor replacement. Eur J Immunol. 2006;36:3052–9. doi: 10.1002/eji.200636539. [DOI] [PubMed] [Google Scholar]
- 18.Bunnell BA, Muul LM, Donahue RE, Blaese RM, Morgan RA. High-efficiency retroviral-mediated gene transfer into human and nonhuman primate peripheral blood lymphocytes. Proc Natl Acad Sci U S A. 1995;92:7739–43. doi: 10.1073/pnas.92.17.7739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam JS, Reeves ME, Cowherd R, Rosenberg SA, Hwu P. Improved gene transfer into human lymphocytes using retroviruses with the gibbon ape leukemia virus envelope. Hum Gene Ther. 1996;7:1415–22. doi: 10.1089/hum.1996.7.12-1415. [DOI] [PubMed] [Google Scholar]
- 20.Ayuk F, Li Z, Kühlcke K, Lindemann C, Schade U, Eckert HG, et al. Establishment of an optimised gene transfer protocol for human primary T lymphocytes according to clinical requirements. Gene Ther. 1999;6:1788–92. doi: 10.1038/sj.gt.3300999. [DOI] [PubMed] [Google Scholar]
- 21.Atanackovic D, Matsuo M, Ritter E, Mazzara G, Ritter G, Jäger E, et al. Monitoring CD4+ T cell responses against viral and tumor antigens using T cells as novel target APC. J Immunol Methods. 2003;278:57–66. doi: 10.1016/S0022-1759(03)00209-6. [DOI] [PubMed] [Google Scholar]
- 22.Sommermeyer D, Uckert W. Minimal amino acid exchange in human TCR constant regions fosters improved function of TCR gene-modified T cells. J Immunol. 2010;184:6223–31. doi: 10.4049/jimmunol.0902055. [DOI] [PubMed] [Google Scholar]
- 23.Schub A, Schuster IG, Hammerschmidt W, Moosmann A. CMV-specific TCR-transgenic T cells for immunotherapy. J Immunol. 2009;183:6819–30. doi: 10.4049/jimmunol.0902233. [DOI] [PubMed] [Google Scholar]
- 24.Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas S, Stauss HJ, Morris EC. Molecular immunology lessons from therapeutic T-cell receptor gene transfer. Immunology. 2010;129:170–7. doi: 10.1111/j.1365-2567.2009.03227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bendle GM, Linnemann C, Hooijkaas AI, Bies L, de Witte MA, Jorritsma A, et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat Med. 2010;16:565–70, 1p, 570. doi: 10.1038/nm.2128. [DOI] [PubMed] [Google Scholar]
- 27.Ahmadi M, King JW, Xue S-A, Voisine C, Holler A, Wright GP, et al. CD3 limits the efficacy of TCR gene therapy in vivo. Blood. 2011;118:3528–37. doi: 10.1182/blood-2011-04-346338. [DOI] [PubMed] [Google Scholar]
- 28.Govers C, Sebestyén Z, Coccoris M, Willemsen RA, Debets R. T cell receptor gene therapy: strategies for optimizing transgenic TCR pairing. Trends Mol Med. 2010;16:77–87. doi: 10.1016/j.molmed.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 29.Kuball J, Dossett ML, Wolfl M, Ho WY, Voss R-H, Fowler C, et al. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood. 2007;109:2331–8. doi: 10.1182/blood-2006-05-023069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–86. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bialer G, Horovitz-Fried M, Ya’acobi S, Morgan RA, Cohen CJ. Selected murine residues endow human TCR with enhanced tumor recognition. J Immunol. 2010;184:6232–41. doi: 10.4049/jimmunol.0902047. [DOI] [PubMed] [Google Scholar]
- 32.Heemskerk MHM, Hagedoorn RS, van der Hoorn MAWG, van der Veken LT, Hoogeboom M, Kester MGD, et al. Efficiency of T-cell receptor expression in dual-specific T cells is controlled by the intrinsic qualities of the TCR chains within the TCR-CD3 complex. Blood. 2007;109:235–43. doi: 10.1182/blood-2006-03-013318. [DOI] [PubMed] [Google Scholar]
- 33.Walther W, Stein U. Viral vectors for gene transfer: a review of their use in the treatment of human diseases. Drugs. 2000;60:249–71. doi: 10.2165/00003495-200060020-00002. [DOI] [PubMed] [Google Scholar]
- 34.Bobisse S, Rondina M, Merlo A, Tisato V, Mandruzzato S, Amendola M, et al. Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer Res. 2009;69:9385–94. doi: 10.1158/0008-5472.CAN-09-0494. [DOI] [PubMed] [Google Scholar]
- 35.Newrzela S, Cornils K, Heinrich T, Schläger J, Yi J-H, Lysenko O, et al. Retroviral insertional mutagenesis can contribute to immortalization of mature T lymphocytes. Mol Med. 2011;17:1223–32. doi: 10.2119/molmed.2010.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Engels B, Cam H, Schüler T, Indraccolo S, Gladow M, Baum C, et al. Retroviral vectors for high-level transgene expression in T lymphocytes. Hum Gene Ther. 2003;14:1155–68. doi: 10.1089/104303403322167993. [DOI] [PubMed] [Google Scholar]
- 37.Leisegang M, Engels B, Meyerhuber P, Kieback E, Sommermeyer D, Xue S-A, et al. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J Mol Med (Berl) 2008;86:573–83. doi: 10.1007/s00109-008-0317-3. [DOI] [PubMed] [Google Scholar]
- 38.Thomas S, Xue SA, Cesco-Gaspere M, San José E, Hart DP, Wong V, et al. Targeting the Wilms tumor antigen 1 by TCR gene transfer: TCR variants improve tetramer binding but not the function of gene modified human T cells. J Immunol. 2007;179:5803–10. doi: 10.4049/jimmunol.179.9.5803. [DOI] [PubMed] [Google Scholar]
- 39.Engels B, Noessner E, Frankenberger B, Blankenstein T, Schendel DJ, Uckert W. Redirecting human T lymphocytes toward renal cell carcinoma specificity by retroviral transfer of T cell receptor genes. Hum Gene Ther. 2005;16:799–810. doi: 10.1089/hum.2005.16.799. [DOI] [PubMed] [Google Scholar]
- 40.Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–42. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–50. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, Vignali DAA. Generation of T-cell receptor retrogenic mice. Nat Protoc. 2006;1:406–17. doi: 10.1038/nprot.2006.61. [DOI] [PubMed] [Google Scholar]
- 43.Baum C, Hegewisch-Becker S, Eckert HG, Stocking C, Ostertag W. Novel retroviral vectors for efficient expression of the multidrug resistance (mdr-1) gene in early hematopoietic cells. J Virol. 1995;69:7541–7. doi: 10.1128/jvi.69.12.7541-7547.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hildinger M, Abel KL, Ostertag W, Baum C. Design of 5′ untranslated sequences in retroviral vectors developed for medical use. J Virol. 1999;73:4083–9. doi: 10.1128/jvi.73.5.4083-4089.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weber K, Thomaschewski M, Benten D, Fehse B. RGB marking with lentiviral vectors for multicolor clonal cell tracking. Nat Protoc. 2012;7:839–49. doi: 10.1038/nprot.2012.026. [DOI] [PubMed] [Google Scholar]
- 46.Fehse B, Schade UM, Li Z, Uhde A, Koch S, Goller B, et al. Highly-efficient gene transfer with retroviral vectors into human T lymphocytes on fibronectin. Br J Haematol. 1998;102:566–74. doi: 10.1046/j.1365-2141.1998.00785.x. [DOI] [PubMed] [Google Scholar]