

Abstract
Targeting of drug carriers to cell-surface receptors involved in endocytosis is commonly used for intracellular drug delivery. However, most endocytic receptors mediate uptake via clathrin or caveolar pathways associated with ≤200-nm vesicles, restricting carrier design. We recently showed that endocytosis mediated by intercellular adhesion molecule 1 (ICAM-1), which differs from clathrin- and caveolar-mediated pathways, allows uptake of nano- and micro-carriers in cell culture and in vivo due to recruitment of cellular sphingomyelinases to the plasmalemma. This leads to ceramide generation at carrier binding sites and formation of actin stress-fibers, enabling engulfment and uptake of a wide size-range of carriers. Here we adapted this paradigm to enhance uptake of drug carriers targeted to receptors associated with size-restricted pathways. We coated sphingomyelinase onto model (polystyrene) submicro- and micro-carriers targeted to clathrin-associated mannose-6-phosphate receptor. In endothelial cells, this provided ceramide enrichment at the cell surface and actin stress-fiber formation, modifying the uptake pathway and enhancing carrier endocytosis without affecting targeting, endosomal transport, cell-associated degradation, or cell viability. This improvement depended on the carrier size and enzyme dose, and similar results were observed for other receptors (transferrin receptor) and cell types (epithelial cells). This phenomenon also enhanced tissue accumulation of carriers after intravenous injection in mice. Hence, it is possible to maintain targeting toward a selected receptor while bypassing natural size-restrictions of its associated endocytic route by functionalization of drug carriers with biological elements mimicking the ICAM-1 pathway. This strategy holds considerable promise to enhance flexibility of design of targeted drug delivery systems.
Keywords: Intracellular drug delivery, receptor-mediated endocytosis, carrier size, ICAM-1 pathway, clathrin-associated receptors, sphingomyelinase, ceramide
Our increased ability to fabricate functional materials at the nano- and micro-scales has resulted in the design of numerous drug carrier platforms capable of improving the therapeutic potential of pharmaceutical agents, including enhanced control of solubility, circulation, degradation, and release.1–3 In addition, surface functionalization of drug delivery systems with antibodies, peptides, aptamers, and other affinity moieties offers the opportunity to target treatments more precisely to sites in the body where their action is required.4–6 In order to further enhance therapeutic efficacy, cell-surface receptors involved in endocytic pathways are often targeted.7 This allows transport of drugs intracellularly and/or across cellular linings, improving access to their therapeutic targets.7 Hence, selecting appropriate endocytic receptors is key to achieving selectivity toward the tissue or organ to be targeted, and as a portal into or across its cells.
It has also become well recognized that the precise pathway by which cells mobilize drug carriers plays a crucial role with regard to carrier fate and therapeutic activity. Most receptors relate to one of four canonical endocytic pathways, including those dependent on clathrin, caveolae, phagocytosis, or macropinocytosis.8,9 Macropinocytosis is a non-selective pathway and phagocytosis is mostly present in immune cells, limiting the range of applications derived from targeting these pathways.10–11 Also, phagocytosis and macropinocytosis typically sort materials to vacuolar or lysosomal compartments for degradation, in detriment of prolonged therapeutic activity.7,12 In contrast, clathrin- and caveolae-mediated endocytosis are used by most cells in the body.7,12 While some receptors associated with these pathways result in lysosomal degradation, others avoid this compartment and provide recycling to the cell surface, transcytosis across cells, or routing to alternative subcellular compartments.8 This, along with availability of numerous clathrin- or caveolae-associated receptors and ample biological knowledge of their function and regulation, has positioned these pathways at the focus of research on intracellular drug delivery.
A limitation to exploiting endocytic transport concerns the size of carriers which can be internalized by these natural routes.7,13 Numerous studies have focused on the importance of this parameter, demonstrating the role of carrier size on binding, internalization, intracellular transport, and fate of therapeutics (apart from other aspects).14–17 Macropinocytosis and phagocytosis can internalize micron-sized objects, but they represent less desirable pathways, as described above.7,10,11 In turn, while clathrin- and caveolae-mediated endocytosis offer numerous advantages, they pose size limitations to efficient uptake of drug carriers, e.g., ~50–100-nm in the case of caveolae and ≤200-nm for clathrin-mediated endocytosis.7,13 These factors greatly limit design and application of drug delivery strategies.
Interestingly, a few cell-surface markers have been associated with independent, far less understood pathways of endocytosis.18 Of these, cell adhesion molecule (CAM)-mediated endocytosis, induced by binding to intercellular adhesion molecule 1 (ICAM-1), is the best characterized in the context of drug targeting, as well as intra- and trans-cellular delivery of drug carriers.19–27 ICAM-1 is a transmembrane glycoprotein overexpressed on endothelial and other cell types in the body affected by numerous pathological factors.28,29 It serves as a co-receptor for β2 integrins expressed on activated leukocytes.28,29 Hence, its natural ligand represents a multivalent micrometer-size “object”, in contrast to most canonical endocytic receptors of small molecular ligands. In accord to this, endocytosis of ICAM-1-targeted carriers is efficient under a wide range of carrier sizes (from 200-nm to several μm) both in cell culture and in vivo.7,20,30
We recently described that this ability of the CAM-mediated pathway is, at least in part, attributed to its link with sphingomyelin/ceramide signaling.31 Binding of ICAM-1-targeted carriers to ICAM-1 induces cellular secretion of acid sphingomyelinase. This enzyme, which is not involved in the initial binding event, contributes to the subsequent signaling cascade conducive to carrier engulfment and internalization.31 Carrier-engaged ICAM-1 also interacts with the Na+/H+ exchanger protein NHE1,32 which is known to extrude H+ extracellularly and may provide the local acidic environment required for acid sphingomyelinase activity.31 This enzyme can then hydrolyze the phospholipid sphingomyelin into ceramide, which becomes enriched at carrier-binding sites on the plasmalemma.31 Ceramide is known to form lipid microdomains, which helps clustering of receptors and acts as a second messenger for cytoskeletal reorganization.33–36 Ceramide enrichment alters the biophysical properties of the plasma membrane, facilitating formation of engulfment structures and vesicles, and causes fusion of smaller vesicles into larger endocytic compartments.37,38 Genetic or pharmacological disruption of acid sphingomyelinase inhibits ceramide enrichment, actin stress-fiber formation, and endocytosis via the CAM pathway.31 A similar phenomenon has also been associated with cell invasion of micrometer-sized pathogens such as Escherichia coli, Staphylococcus aureus, and Neisseria gonorrhoeae.39,40
Therefore, it seemed plausible that surface-functionalization of carriers with sphingomyelinases may exogenously provide a similar ability to enrich ceramide at the plasma membrane even when carriers are targeted to receptors naturally associated to other endocytic pathways. Using model polymer (polystyrene) carriers targeted to the clathrin-associated mannose-6-phosphate receptor41 or transferrin receptor,42 we demonstrate that this hypothesis is viable. This strategy may enhance intracellular transport of a wider range of carrier sizes in cases where the receptor targeted does not induce such function endogenously.
RESULTS AND DISCUSSION
Comparative Binding of Carriers Targeted to CAM (ICAM-1) vs. Clathrin (M6PR) Pathways
The goal of this study was to explore whether surface functionalization of drug carriers with biological elements of the CAM-mediated pathway (namely, sphingomyelinases) can facilitate intracellular transport for carriers targeted to receptors of more size-restricted pathways. To prove this concept, we selected mannose-6-phosphate receptor (M6PR) as a target. M6PR is a bifunctional receptor that mediates binding and endocytosis of proteins containing M6P-residues and of insulin-like growth factor II.41,43 As for ICAM-1, M6PR is also expressed by numerous cell types in the body.44,45 Contrarily to ICAM-1, M6PR mediates endocytosis via the clathrin-associated pathway, which is restricted to the size of clathrin-coated pits (≤200-nm diameter).43 M6PR is expressed in most tissues, but at a relatively higher level in the kidneys and lungs. Hence, this example is relevant to drug delivery for treatment of conditions affecting these organs.44–46 Indeed, several therapeutic strategies (both experimental and already in the clinics) are based on M6PR targeting.41,44–51
We first compared binding and uptake of carriers targeted to these different receptors, using model polystyrene particles in order to avoid potential confounding results of concomitant carrier degradation. We have previously shown that, after coating with antibodies, these particles provide similar targeting, endocytosis, and in vivo biodistribution as compared to biodegradable poly(lactic-co-glycolic acid) counterparts and, hence, represent a valid model.52,53 We used 1-μm diameter particles to accentuate the differences between targeting CAM- vs. clathrin-associated receptors. As per the cell model, we selected vascular endothelial cells (HUVECs) since this would be one of the first cell types in contact with carriers injected in circulation, and cells were activated with tumor necrosis factor α (TNFα) to mimic a pathological stimulation.54
Table 1 shows that carriers coated with anti-ICAM or anti-M6PR had similar density of antibodies on their surface, and also a comparable size and polydispersity. When incubated for 30 min with fixed cells to avoid concomitant endocytosis, anti-M6PR carriers showed 19-fold lower binding as compared to anti-ICAM carriers (Fig. 1A and Suppl. Fig. S1), and this difference was further enhanced over time (29-fold at 1 h and 34-fold at 3 h; Fig. 1B). Yet, this low level of binding was still specific over that of IgG carriers (e.g., 10-fold at 30 min; Fig. 1A). IgG carriers represent control counterparts, since IgG is a matching non-specific antibody compared to anti-ICAM or anti-M6PR. Indeed, IgG carriers did not bind to or internalize within cells (Figs. 1 and 2).
Table 1.
Characterization of carrier coat.
| Preparation | Surface Coverage | Molecules/μm2 of Carrier | Diameter (μm) | PDI | |
|---|---|---|---|---|---|
| Targeting Ab | IgG or NSM | ||||
| Parent Carriers | |||||
| Anti-ICAM | 100 | 25,734 | N/A | 1.198 | 0.22 |
| Anti-M6PR | 100 | 23,544 | N/A | 1.144 | 0.32 |
| Combined Coating | |||||
| Anti-M6PR/IgG | 80:20 | 19,608 | 1,632 | 1.156 | 0.37 |
| 50:50 | 12,616 | 4,328 | 1.095 | 0.22 | |
| Anti-M6PR/NSM | 80:20 | 16,052 | 2,532 | 1.051 | 0.26 |
| 50:50 | 11,498 | 5,658 | 1.142 | 0.30 | |
Ab = antibody; N/A = Not Applicable; PDI = Polydispersity index; Surface coverage = approximate fraction of carrier surface that a molecule type occupies with respect to the total coating observed for parent anti-M6PR NCs.
Figure 1. Comparative binding of carriers targeted to ICAM-1 vs. M6PR.

(A) Phase-contrast microscopy showing low, yet specific binding of 1-μm carriers coated with anti-M6PR compared to anti-ICAM carriers or control IgG carriers, after 30 min incubation at room temperature with fixed activated HUVECs. Dashed lines mark cell borders and arrowheads mark carriers. Scale bar = 10 μm. (B) Binding of these carriers to cells was similarly compared at 30 min, 1 h, and 3 h to reflect their relative binding kinetics. Mean ± standard error of the mean. *Compared to anti-ICAM carriers; #compared to control IgG carriers (p < 0.05).
Figure 2. Comparative endocytosis of carriers targeted to ICAM-1 vs. M6PR.

(A) Endocytosis of 1-μm carriers coated with anti-M6PR after incubation for 30 min at 37°C with activated HUVECs, compared to anti-ICAM carriers or control IgG carriers. Phase contrast (top panels) shows total carriers bound and internalized. Fluorescence images (bottom panels) show surface-bound, non-internalized carriers. Arrowheads mark non-internalized carriers and arrows mark endocytosed ones. Scale bar = 10 μm. (B) Low, although specific, internalization of anti-M6PR carriers compared to anti-ICAM carriers and control IgG carriers, quantified as in (A). Mean ± standard error of the mean. *Compared to anti-ICAM carriers; #compared to IgG carriers (p< 0.05).
Such poor binding of microcarriers to M6PR on the cell surface (compared to anti-ICAM carriers) may be due to different factors, such as suboptimal exposure of the receptor epitope targeted by the anti-M6PR antibody used. However, free non-coated anti-M6PR bound to cells only 3-fold below the level of binding of free anti-ICAM (not shown). This suggests that low binding of carriers to M6PR is rather due to steric hindrances posed by such large micrometer-range ligands as compared to natural ligands of this receptor (M6P-modified proteins or IGF-II).
Comparative Endocytosis of Carriers Targeted to CAM (ICAM-1) vs. Clathrin (M6PR) Pathways
We next determined the level of endocytosis of anti-M6PR carriers compared to that of anti-ICAM counterparts. As described above, endocytosis is a key factor determining the potential for intracellular drug delivery and needs to be studied independently from binding. This is because, although interdependent, internalization within cells is not granted by simply achieving carrier binding to cell receptors. Indeed, although binding of 1-μm anti-M6PR carriers to activated HUVECs was 10-fold elevated as compared to control IgG carriers (Fig. 1A), the endocytosis rate (internalized carriers/total cell-associated carriers) of these formulations was similarly ineffective (Fig. 2A and Suppl. Fig. S2): 22% for anti-M6PR carriers and 11% for IgG carriers at 30 min, much lower than that of anti-ICAM carriers (70% at this time point). This led to a very low number of carriers being internalized by cells in the case of targeting to M6PR compared to ICAM-1 (25-fold difference at 30 min), which was enhanced over time (46-fold at 1 h and 143-fold at 3h; Fig. 2B). The absolute level of anti-M6PR carriers internalized by cells still exceeded that of control IgG carriers (e.g., 7-fold at 30 min), validating the specificity of this system despite its poor efficiency.
It may be speculated that lower cell binding of anti-M6PR carriers compared to anti-ICAM carriers may account for this decreased uptake into cells. Although this can influence the result obtained, one must realize that binding is necessary but not always sufficient to specifically trigger endocytosis. Indeed, low internalization of anti-M6PR carriers was independent from the absolute number of carriers bound per cell: cells presenting up to 3-fold difference in the amount of bound carrier showed similarly low uptake (between 18% and ~20%; not shown). This phenomenon has been previously observed for other receptors: e.g., carriers targeted to certain epitopes of PECAM-1 (a molecule of the same family as ICAM-1) failed to induce endocytosis despite binding to cells more profusely than carriers targeted to “internalizable” PECAM-1 epitopes55. Also, the rate of internalization of anti-ICAM carriers was similarly high in control (low expression and binding) vs. pathological (high expression and binding) situations.21 Poor endocytosis of microcarriers targeted to M6PR vs. ICAM-1 was expected based on the size-dependency of the endocytic pathways naturally associated to these receptors (clathrin- vs. CAM-mediated).8,30
Binding Effects of Sphingomyelinase-Functionalized Carriers Targeted to the Clathrin (M6PR) Pathway
As described above, acid sphingomyelinase recruited to plasmalemma sites where anti-ICAM carriers bind to ICAM-1, along with putative NHE1 acidifying activity, seem to provide the means for the enrichment in ceramide required to facilitate endocytosis of micrometer size objects via the CAM pathway.31,32 This is opposite to the occurrence of clathrin-mediated endocytosis: ceramide has been shown to be excluded from clathrin-positive regions and clathrin-associated receptor clusters.56 This makes M6PR an appropriate example to determine whether signaling elements of the CAM-mediated pathway (e.g., sphingomyelinases) can be used to enhance uptake of drug carriers by cells. Since clathrin-mediated endocytosis (including that of M6PR) is independent from NHE1 activity (hence, precluding the use of the acid enzyme), we selected neutral sphingomyelinase (NSM) to demonstrate this paradigm.
We co-coated 1-μm model carriers with both anti-M6PR (to target M6PR, associated with size-restricted clathrin-mediated endocytosis) and NSM (to provide CAM-like signal transduction, facilitating endocytosis) at either 80:20 or 50:50 carrier surface-coverage ratios (see Table 1 for a definition). These represent NSM doses expected to be active based on our previous study on ICAM-1 endocytosis.31 As control carriers bearing similar targeting valency (anti-M6PR density) but not NSM, we co-coated particles with anti-M6PR and control IgG at similar ratios (see Methods). As shown in Table 1, these preparations displayed similar size, polydispersity, and relative coating with anti-M6PR between 70% and 83%, or between 49% and 54%, respectively, as compared to parent anti-M6PR carriers. Approximately 1.5-fold more molecules of NSM were present on the coat of anti-M6PR carriers as compared to IgG molecules, for each surface-coating ratio. This was expected given the smaller molecular weight of the enzyme. Yet, neither NSM nor IgG (but anti-M6PR) are expected to provide targeting; hence, this is a valid control.
As shown in Fig. 3, both anti-M6PR carrier preparations displaying 80% and 50% of the valency of parent anti-M6PR still had significantly higher binding to cells as compared to IgG carriers (dotted line in graph; Fig. 3B). Curiously, anti-M6PR carriers bearing 50% of maximal valency bound to cells significantly higher than parent anti-M6PR carriers (continuous line in graph; Fig. 3B) or 80% valency counterparts. This is in accord to negative cooperativity observed between the two M6P binding sites present on this dimeric receptor.57 Previous studies showed that saturating levels of M6PR targeting moieties on a carrier offer poor targeting compared to lower targeting densities.57 Similar outcomes have been reported for other targeted drug delivery strategies.58 Nevertheless, no significant difference was observed regarding binding of anti-M6PR/NSM carriers vs. that of anti-M6PR/IgG carriers for either targeting valency. Also, presence of anti-M6PR in the cell medium to compete for binding sites on the cell surface resulted in decreased binding of anti-M6PR/NSM carriers to cells, and anti-M6PR/IgG carrier binding was reduced to the same extent (Suppl. Fig. S3). This indicates that NSM presence on the carrier coat does not impact cell binding, which remains to reach the same level and occurs via M6PR.
Figure 3. Effect of surface functionalization with NSM on the binding of anti-M6PR carriers.

(A) Phase-contrast microscopy showing binding of 1-μm parent anti-M6PR carriers compared to 50:50 anti-M6PR/IgG carriers and anti-M6PR/NSM carriers, 3 h after incubation at room temperature with fixed activated HUVECs (dashed lines mark cell borders). Scale bar = 10 μm. (B) Binding of anti-M6PR/IgG carriers vs. anti-M6PR/NSM carriers (for both 80:20 and 50:50 formulations) was determined by phase-contrast microcopy as in Fig. 1A. Mean ± standard error of the mean. *Compared to parent anti-M6PR carriers (continuous line in graph); ^compared to control IgG carriers (dotted line in graph); +comparison between different surface-coverage ratios (80:20 vs. 50:50) of the same formulation type (p < 0.05). No difference was found comparing anti-M6PR/IgG vs. anti-M6PR/NSM carriers.
Effects on Endocytosis of Sphingomyelinase-Functionalized Carriers Targeted to the Clathrin (M6PR) Pathway
Despite lack of an effect on binding, both anti-M6PR/NSM carrier formulations showed a marked and significant increase in the number of carriers internalized by cells as compared to anti-M6PR carrier preparations lacking NSM (Fig. 4A and B), including parent anti-M6PR carriers and anti-M6PR/IgG counterparts. In contrast, anti-M6PR/IgG showed similarly poor internalization compared to IgG carriers and anti-M6PR carriers despite a greater binding efficacy (compare Fig. 4B to Fig. 3B). Hence, this confirms our hypothesis that carrier size, rather than absolute level of binding, represents an obstacle to endocytosis via this route and that surface functionalization with NSM can enhance carrier uptake even when targeting a receptor associated to a size-restrictive pathway. This is further shown in Fig. 4C: presence of NSM on the surface of anti-M6PR carriers enhanced the rate of endocytosis by ~2.5–3-fold and these formulations achieved up to ~60–65% the internalization efficacy of anti-ICAM carriers (dashed line in graph 4C).
Figure 4. Effect of surface functionalization with NSM on internalization of carriers.

(A) Endocytosis of 1-μm anti-M6PR/IgG carriers or anti-M6PR/NSM carriers (displaying either 80:20 or 50:50 surface-coverage ratios) incubated with live, activated HUVECs for 3 h at 37°C. Phase contrast shows total carriers bound and internalized. Fluorescence images show surface-bound, non-internalized carriers. Arrowheads mark non-internalized carriers and arrows mark endocytosed ones. Scale bar = 10 μm. (B) Total number of carriers internalized per cell. (C) Percent of carriers internalized out of the total numbers of carriers associated to cells (bound plus internalized). Dashed line in (C) represents anti-ICAM carriers. Mean ± standard error of the mean. *Compared to parent anti-M6PR carriers (continuous line in graph); ^compared to control IgG carriers (dotted line in graph); #compared to anti-M6PR/IgG carriers (p<0.05).
This improvement in endocytosis was also observed in cell types other than the primary endothelial cells used above, such as Caco-2 cells, an established cell line of epithelial colorectal adenocarcinoma origin (50% increase; p<0.05; Suppl. Fig. S4A). NSM functionalization also enhanced endocytosis of microcarriers targeted to receptors different from M6PR, such as the transferrin receptor (TfR; 30% increase; p<0.05; Suppl. Fig. S4B), naturally involved in iron transport and extensively used in the context of drug delivery.42
Although the optimal dose of NSM necessary for such effect is likely to vary depending on the particular physiological properties of each receptor, cell type, etc., these results suggests that sphingomyelinase-functionalization may enhance endocytosis of drug carriers in a generic manner, highlighting the potential for this strategy.
Effects of NSM Functionalization on Endocytosis as a Factor of Enzyme Dose and Carrier Size
The dependency on NSM dose to provide enough sphingomyelinase function for effective uptake should depend on the size of carriers. To examine this, we compared endocytosis of anti-M6PR/NSM carriers to that of their anti-M6PR/IgG control counterparts for carriers 200-nm, 1-μm, and 4.5-μm in size (Suppl. Fig. S5). We selected these sizes because carriers 200-nm in diameter are still close to the limit of clathrin-coated pits vs. microcarriers and, within the latter group, 1-μm is still somewhat amenable for a cell to internalize, while 4.5-μm is far from the range permissible in a non-immune cell.7 Increasing the size of anti-M6PR/IgG carriers decreased endocytosis (8 carriers/10 cells for 200-nm carriers, 3 carriers/10 cells for 1-μm carriers, 1 carrier/10 cells for 4.5-μm carriers), and functionalization with NSM resulted in enhanced uptake for all carrier sizes (54, 20, and 5 carriers/10 cells, respectively). Since absolute binding of objects of such different size greatly differs, we also examined the rate (%) of internalization for each bound carrier. This parameter also decreased with increasing carrier size (Fig. 5A), NSM functionalization enhanced uptake for all carriers tested (Δ > 1; Fig. 5B), and this improvement in uptake depended on carrier size (the improvement was greater for larger carriers; Fig. 5B). Additionally, a reduction in NSM from 50% to 20% of the carrier surface did not significantly reduce this improvement in the case of 200-nm or 1-μm carriers (~1–1.5-fold difference comparing 50% over 20% NSM; Fig. 5C), but it did for 4.5-μm carriers (by 3-fold). Therefore, the larger the carrier size, the greater the improvement provided by NSM and the greater the dependence on the enzyme dose.
Figure 5. Enhanced carrier endocytosis as a function of NSM dose and carrier size.

(A) Percent of anti-M6PR/IgG (50:50) carriers internalized out of the total number of carriers associated to cells (bound plus internalized) in activated HUVECs (3 h, 37°C). Carriers were 200-nm, 1-μm, or 4.5-μm in diameter. (B) Fold increase (Δ) in the percent internalization of anti-M6PR/NSM carriers over control anti-M6PR/IgG carriers (both 50:50) for each carrier size. (C) Comparative improvement in endocytosis (Δ in B) for anti-M6PR/NSM carriers bearing 50% NSM carrier-surface coverage (50:50) over that of carriers bearing 20% coverage (80:20). Mean ± standard error of the mean. *Compares microcarriers to 200-nm carriers; #compares anti-M6PR/NSM carriers to anti-M6PR/IgG carriers, ^compares anti-M6PR/NSM:anti-M6PR/IgG of 50:50 surface-coverage to that of 80:20 carriers for each given size (p<0.05).
Although spherical polymer carriers of such large size (~4–5-μm) do not seem viable for drug delivery due to concerns of mechanical entrapment in capillaries, tissue matrix, etc., the literature shows numerous examples of large, several micrometer-sized vehicles not only adequate for drug delivery but also offering advantages over submicrometer counterparts.14–16 This is the case for filomicelles and other elongated carriers with several micrometers in length, which align to the blood-flow direction, avoid fast uptake by macrophages, and sustain prolonged circulation compared to submicrometer spherical counterparts.16,30,59 Hence, our finding is expected to widen the range of applications for these formulations by lowering the size restrictions naturally associated with most target receptors and their native endocytic pathways.
Endocytic Pathway Induced by NSM-Functionalization of M6PR-Targeted Carriers
The mechanism underlying the role of acid sphingomyelinase on CAM-mediated endocytosis is through endogenous generation of ceramide and formation of actin stress-fibers, both enabling formation of large endocytic structures.31 We examined if this is also the case with M6PR-targeted carriers when functionalized to provide exogenous NSM signaling.
First, we looked at the colocalization of anti-M6PR/NSM carriers vs. control anti-M6PR/IgG carriers with ceramide at the plasma membrane (Suppl. Fig. S6 and Fig. 6A top panels show examples of colocalization). The level of carrier-ceramide colocalization was compared to that of anti-ICAM/IgG carriers of similar targeting valency. As shown in Fig. 6A (graph), anti-M6PR/IgG carriers presented ~50% the level of ceramide co-localization of anti-ICAM/IgG counterparts. Since ceramide is inherently present on the cell surface and micrometer-sized carriers occupy a considerable surface area of the plasmalemma, it is possible that this level of co-localization represents background passive co-localization or simply the threshold for this detection. In contrast, ceramide was significantly enriched at carrier binding-sites after NSM functionalization: anti- M6PR/NSM carriers reached ~90% the level of ceramide co-localization of anti-ICAM/IgG carriers. This may help engulfment and vesiculation, conducive to the enhanced endocytosis observed.31,34,35,37
Figure 6. Mechanism associated with NSM-functionalization of carriers.

(A) Enhanced colocalization of 1-μm or 4.5-μm anti-M6PR/NSM carriers (50:50 surface-coverage ratio) with ceramide at the surface of activated HUVECs after 30 min incubation at 37°C, compared to anti-M6PR/IgG carriers. An example of 1-μm anti-M6PR/NSM carriers (phase contrast) colocalizing with ceramide (green fluorescence) and the corresponding fluorescence surface-plot (upper right) is shown. Blue = DAPI-positive cell nuclei. Scale bars = 10 μm (left) and 2 μm (right). Mean ± standard error of the mean. *Compares anti-M6PR/NSM carriers to anti-M6PR/IgG counterparts (p < 0.05). (B) F-actin staining with fluorescent phalloidin shows stress fibers in activated HUVECs after incubation with control media or media containing 1-μm anti-M6PR/NSM carriers (50:50) for 30 min at 37°C. Anti-M6PR/IgG carriers are shown as negative controls, while anti-ICAM/IgG carriers are positive controls. Arrows indicate stress fibers and arrowheads indicate ruffling. Scale bar = 10 μm. (C) Percent endocytosis of 200-nm and 1-μm anti-M6PR/NSM carriers (50:50) was assessed using activated HUVECs incubated for 3 h at 37°C in the absence (control) vs. presence of pharmacological inhibitors of pathways mediated by clathrin (monodalsylcadaverine, MDC), caveolae (filipin, Fil), CAM and macropinocytosis (amiloride, Amil), or macropinocytosis (wortmannin, Wort). Mean ± standard error of the mean. *Compares inhibitor to control for each carrier size (p < 0.05).
Second, we examined the actin cytoskeleton of cells subjected to incubation with anti-M6PR carriers. As shown in Fig. 6B, in the absence of carriers endothelial cells presented typical cortical actin around the cell periphery. ICAM-1 targeting (using anti-ICAM/IgG carriers as a valency control) elicited formation of long, thick actin stress-fibers across the cell body (arrows), as previously reported.20,31,32 Binding of anti-M6PR/IgG carriers to cells resulted in small, thin actin filaments through the cell body and actin ruffles at the cell periphery (arrowheads). However, when cells were incubated with anti-M6PR/NSM a pattern of ruffles at the cell border and actin stress fibers through the cell body was observed. Membrane ruffles suggest contribution of macropinocytosis, but this was also observed in the absence of NSM. This is likely due to the size of these microparticles, reflecting an attempt of cells to uptake these via macropinocytosis. Formation of actin stress-fibers only upon NSM functionalization suggests the contribution of the CAM pathway, since this is a rather unique feature not observed for clathrin pits, caveolae, macropinocytosis, or phagocytosis.20 Actin is involved in clathrin- and caveolae-mediated endocytosis but rather presents as small fibers,60,61 as shown for anti-M6PR/IgG carriers (Fig. 6B). Phagocytosis also associates with formation of larger endocytic vesicles, quite dependent of cytoskeletal re-arrangements, yet these form actin cups surrounding phagosomes, not stress fibers.62 Interestingly, actin stress fibers are induced in endothelial cells upon binding of activated leukocytes to cell adhesion molecules, including ICAM-1.63 This is in accord to a common underlying mechanism of ICAM-1 signaling upon binding of its natural ligands or artificial ICAM-1-targeted carriers,64 and explains that this pathway is amenable to engulfment of micrometer-sized carriers.30 Hence, functionalizing the surface of carriers targeted to other receptors (M6PR) with NSM seems to mimic this function of the CAM pathway, by providing cells with similar signals.
Acquisition of CAM-like signaling properties by NSM functionalization of anti-M6PR carriers was further validated using inhibitors of endocytic pathways (Suppl. Fig. S7 and Fig. 6C) and carriers of different size (submicrometer vs. micrometer). Internalization of either anti-M6PR/NSM nano- or micro-carriers was not affected by filipin, which served as a negative control since this agent inhibits caveolar pathways. In agreement with signaling through M6PR associated to clathrin pits, uptake of 200-nm anti-M6PR/NSM carriers was inhibited by monodansylcadaverine (MDC). Also, amiloride (but not wortmannin) inhibited endocytosis of these carriers, suggesting involvement of the CAM pathway and validating the contribution of NSM to signaling induced by anti-M6PR/NSM carriers. Interestingly, no effect of MDC was found in the case of microcarriers (1-μm), indicating no contribution of the clathrin pathway to the uptake of such large carriers, as expected due to natural size restrictions of clathrin pits. Both amiloride and wortmannin reduced uptake, suggesting that CAM- and macropinocytic-like pathways were induced. This correlates well with the actin rearrangement observed for anti-M6PR/NSM microcarriers, showing CAM-like stress fibers and macropinocytic-like ruffles (Fig. 6B). Furthermore, endocytosis of anti-M6PR/NSM carriers was outcompeted by anti-M6PR in the cell medium but enhanced by presence of anti-ICAM (Suppl. Fig. S8). This verifies that binding of anti-M6PR carriers to M6PR is needed and, also, that inducing CAM-mediated endocytosis (e.g. by binding of anti-ICAM antibodies to ICAM-1) improves uptake of anti-M6PR/NSM carriers, which is in agreement with NSM inducing the CAM pathway. Altogether, these data indicate that exogenous NSM functionalization provides signaling additional to and independent from the receptor engaged, and this is conducive to enhanced endocytosis, to which CAM related pathways contribute.
Effect of NSM-Functionalization on the Fate of Clathrin (M6PR)-Targeted Carriers and Cell Viability
We next examined the role of NSM on the carrier coat in terms of intracellular trafficking and potential degradation of carriers targeted to M6PR. This receptor, as described above, is known to mediate binding and endocytosis of proteins containing M6P-residues and subsequently transports them through the endo-lysosomal route.41,43 Carriers targeted to this receptor would be expected to follow this path. Also, since endo-lysosomal compartments are characterized by presence of hydrolases, it would be expected that the protein coat of carriers would be subjected to degradation (polystyrene particles are not biodegradable and remain intact). Interestingly, this was not the case. As presented in Fig. 7A, colocalization of either anti-M6PR/NSM microcarriers or control anti-M6PR/IgG counterparts with an early endosomal marker, EEA-1, was minimal during a 5 h period. Accordingly, the protein coat (the antibody counterpart) remained immunodetectable during this time (Fig. 7B). These results were unexpected based on the biological function of M6PR and illustrate the fact that engaging endocytic receptors with targeted carriers often results in outcomes different from those elicited by their natural ligands. Since no differences were observed in the absence or presence of NSM functionalization, it is possible that this outcome is due to the micrometer-range size of these carriers. Poor trafficking through the endolysosomal route has been reported for other receptors upon targeting with micrometer-sized carriers vs. submicrometer couterparts,30 although the opposite effect has also been observed,7,13,14 highlighting the unpredictability of these events.
Figure 7. Fate of NSM-functionalization carriers.

(A) Activated HUVECs were incubated with 1-μm anti-M6PR/NSM carriers or anti-M6PR/IgG carriers (50:50 surface-coverage) at 37°C for either 3 h or 5 h. After this, cells were permeabilized and immunostained to label early endosomal marker EEA-1 in Texas Red (arrows). (B) The stability of the biodegradable component of carriers (the protein coat; e.g., the antibody component) was additionally assessed after cell permeabilization, by immunostaining in Texas Red. An example of immunopositive (“stable”) and immunonegative (“degraded”) carriers are marked by arrows and arrowheads, respectively. (A, B) Phase contrast shows all carriers associated with cells. Scale bars = 10 μm. Mean ± standard error of the mean. Comparison of anti-M6PR/NSM and anti-M6PR/IgG rendered p > 0.05.
Additionally, given that NSM functionalization of carriers improves endocytosis via ceramide generation and it is known that certain ceramides are involved in apoptosis65–67, we evaluated the potential effect of said functionalization on the number of cells and their viability. As shown in Fig. 8, neither parameter was affected in cells incubated for up to 24 h with anti-M6PR/NSM or control anti-M6PR/IgG carriers, compared to cells incubated in the absence of carriers. In contrast, incubation with H2O2, which is known to induce cell apoptosis through intracellular generation of ceramide,68 resulted in a significant reduction in both the number of cells and their viability (80.1% and 69.5% reduction at 24 h, respectively).
Figure 8. Safety of NSM-functionalized carriers.

Activated HUVECs were incubated in the absence or presence of anti-M6PR/NSM carriers or anti-M6PR/IgG carriers (50:50 surface-coverage) at 37°C for 3 h. At this time, as well as after a total time of 8 h or 24 h, cells were incubated with calcein AM (which fluoresces green in live cells) and ethidium homodimer-1 (which labels in red the nuclei of dead cells) to quantify: (A) the total number of cells and (B) the percentage of live cells (% viability) by microscopy. Incubation of cells with H2O2 served as a positive control for apoptosis. The gray horizontal bar in (A) represents the highest and lowest control values across time. A similar bar is shown for (B), yet the variability is too narrow and it appears as a line. Scale bars = 50 μm. Mean ± standard error of the mean. *Compares H2O2 to control (p< 0.05). No difference was found comparing anti-M6PR/IgG carriers or anti-M6PR/NSM carriers to control.
Effect of Sphingomyelinase-Functionalizion of Carriers on their In vivo Biodistribution
We finally aimed at exploring whether enhanced endocytosis of anti-M6PR carriers in cell culture by NSM functionalization of the carrier surface has the ability to impact carrier biodistribution in vivo. For this purpose, we used submicrometer (200-nm) carriers to avoid non-specific mechanical entrapment in capillaries with embolization. Anti-M6PR/NSM carriers or control anti-M6PR/IgG carriers were injected intravenously in mice, and their circulation and organ biodistribution were compared. As shown in Fig. 9A, both formulations showed a similar circulation pattern and disappeared considerably fast from the blood: e.g., 7.0 percent injected dose (%ID) for anti-M6PR/IgG and 7.3 %ID for anti-M6PR NSM, 30 min after injection.
Figure 9. Enhanced in vivo distribution of anti-M6PR carriers functionalized in mice.

(A) Circulation of anti-M6PR/NSM carriers (50:50 surface-coverage ratio) compared to control anti-M6PR/IgG carriers after intravenous injection in mice. (B) Enhancement (Δ) in the biodistribution of NSM-functionalized carriers was determined as the ratio of the specificity index of anti-M6PR/NSM carriers over that of anti-M6PR/IgG carriers, calculated as described in the Methods section. Mean ± standard error of the mean.
As per organ biodistribution, kidneys and lungs are known to express relative higher levels of M6PR compared to other tissues.44–46 Although the kidneys and lungs are often involved in non-specific clearance of circulating foreign material, they do not play such a key role regarding removal of particles as large as the ones used here (compared to the liver and spleen), as reviewed in the literature.7,25 In agreement with this (Suppl. Fig. 9), only 0.7 %ID and 1.0 %ID of control IgG carriers accumulated in the kidneys and lungs. Other organs that were also negative for IgG carriers were the heart (0.2 %ID) and brain (0.13 %ID). However, accumulation of IgG carriers was high in the liver and spleen (~55 %ID and 5.9 %ID, respectively), verifying that these are the main clearance organs for this type of particles. As compared to control IgG carriers, no changes were observed in the accumulation of anti-M6PR/NSM carriers in the heart and brain (0.2 %ID and 0.14 %ID), as expected, yet this was somewhat reduced in clearance organs (42.3 %ID in liver and 4.8 % ID in spleen), and increased in organs that express higher M6PR levels (1.3 %ID in lungs and 6.2 %ID in kidneys).
Then, to more accurately examine in vivo targeting and the potential effect of NSM, we used the specificity index (SI; see Methods section). This parameter normalizes accumulation of targeted carriers in an organ by accounting for the weight of the organ (to compare organs of different sizes) and the level of non-specific accumulation of control IgG carriers. Fig. 9B shows the comparison between the SI of anti-M6PR/NSM carriers to that of anti-M6PR/IgG carriers (ΔSI). When looking at this parameter, we found that surface functionalization of carriers with NSM resulted in slightly decreased accumulation in clearance organs (e.g., 0.8-fold difference in liver) with an increase in target organs (e.g., 1.5-fold in lungs and, mainly, 12-fold in kidneys). This is in accord with the relative expression level of M6PR in these organs44–46 and correlates well with cell culture observations showing enhanced endocytosis by influence of NSM on the carriers. Enhanced accumulation was also observed when targeting other receptors, i.e., TfR (Suppl. Fig. S10), where the presence of NSM on the carrier coat did not affect non-specific liver uptake but increased accumulation in lungs, which is expected due to expression of TfR in this organ.69
Although it is difficult to estimate if enhanced organ accumulation of anti-M6PR/NSM carriers or anti-TfR/NSM carriers in vivo is due to enhanced binding and/or endocytosis, based on cell culture data demonstrating that NSM did not contribute to binding, it is possible that endocytosis may be the primary mechanism for the observed outcome. Nevertheless, this result shows an advantage for NSM-functionalized carriers in vivo, which may improve targeted drug delivery strategies.
From a more focused perspective, M6PR is overexpressed in several pathological conditions, e.g., cancer, heart failure, amyotrophic lateral sclerosis, etc.43,44,46,47 and is often used for intracellular transport of therapeutics.41,48–51 Also, there is relatively high expression of TfR associated with various malignancies, it allows transport across the blood-brain barrier, and has been well documented in the context of targeted drug delivery.42,69,70 An example of an application of targeting these receptors is that of enzyme replacement therapies for treatment of genetic lysosomal storage disorders, where recombinant lysosomal enzymes are injected in circulation and need to be endocytosed by cells in the affected tissues.43,69,71 However, uptake via several clathrin-associated receptors is impaired in certain lysosomal disorders, leading to suboptimal therapeutic effects.72,73 Enhanced endocytosis by strategies such as the one explored here may help overcome this obstacle.
CONCLUSION
In summary (Fig. 10), surface functionalization of carriers with transducers of CAM-mediated endocytosis (i.e., sphingomyelinase) permitted us to highjack key signal transduction routes of said pathway (ceramide enrichment leading to engulfment and actin re-organization),31 independently from the receptors being targeted. This resulted in enhanced endocytosis of carriers targeted to receptors of more size-restrictive pathways (clathrin-associated M6PR or TfR), without affecting carrier binding or intracellular trafficking. This is, to the best of our knowledge, the first time that properties of an endocytic route are induced in cells independently from the receptors involved and even when the natural pathways they associate with are categorically different. Other strategies can also enhance intracellular delivery of carriers by surface functionalization, including manipulation of charged groups and their precise display, use of lipid motifs, fusogenic peptides, cell penetration counterparts, etc.7,74–76 In some cases, these strategies can provide endosomal escape and cytosolic delivery.7,74,75 These approaches may be useful in applications where drug carriers are first transported to the disease area by passive mechanisms (e.g., enhanced permeability and retention in tumors), where they can then come in contact with cells.3,6,7 Yet, these strategies are less amenable when recognition of selected cells or tissues is required, or when carriers need to be actively transported via a receptor-mediated mechanism across cell layers, for which specific ligand-mediated targeting is required.2,4,7 Hence, the implications of our finding may greatly impact drug delivery strategies by providing a means to enhance intracellular transport of receptor-targeted drug carriers and, in turn, their therapeutic potential, allowing wider design options with regard to carrier geometry. As an example, certain microcarriers (e.g., filomicelles, molded cylinders, carbon nanotubes, flat microdisks, etc.) have been shown to offer advantages of prolonged circulation, reduced non-specific clearance, and enhanced targeting specificity.14–17 Enhancing endocytosis of these vehicles may widen their translational application. In addition, we previously observed that for ICAM-1 targeting there is a negative relationship between carrier size and lysosomal trafficking, with larger carriers being retained in pre-lysosomal compartments for prolonged periods of time.30 This helped avoid rapid lysosomal degradation of therapeutics.30 Relatively low degradation of sphingomeylinase-functionalized microcarriers shown here suggests that such paradigm may be also achievable when targeting other receptors. Finally, although no cytotoxic effects were found in this study, full evaluation of potential side effects of this strategy is crucial since certain ceramides are involved in apoptosis or, when other signals are present, cell proliferation.65–67 The apoptotic activity of such ceramides is explored for tumor suppression 77–79 and this may be an ideal therapeutic focus of sphingomyelinase-functionalized carriers. Also, these apoptotic effects associate with mammalian enzymes (e.g., NSM-2) but not bacterial counterparts (used in this study),75,80,81 and cells can be induced to eliminate excess ceramide through the activity of glucosylceramide synthase, sphingomyelin synthases, and ceramidases.79,81 Future studies shall focus on optimizing this strategy and evaluating its safety.
Figure 10. Strategy of NSM-functionalization of drug carriers to bypass carrier size-restrictions and improve endocytosis.
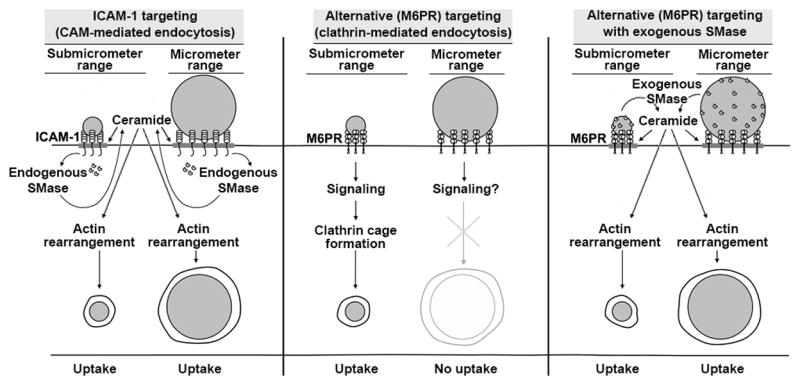
ICAM-1-targeted nano- and micro-carriers are both efficiently internalized by cells due to sphingomyelinase (SMase)-dependent generation of ceramide at carrier-binding sites on the plasmalemma, which is associated with CAM-mediated endocytosis (left panel). Ceramide improves carrier engulfment and membrane invagination, and acts as a second messenger toward actin re-organization, helping in endocytosis. In contrast, targeting drug carriers to receptors associated with more size-restrictive pathways, e.g., clathrin-associated M6PR, often enables intracellular transport of nano- but not micro-carriers (middle panel). Surface-functionalization of said carriers with elements mimicking the CAM-mediated pathway, namely exogenous SMases (such as NSM in this study), does not impact binding but supplies ceramide and actin re-organization, improving endocytosis of nano- and micro-carriers even when targeted to receptors different from ICAM-1 (right panel).
MATERIALS AND METHODS
Antibodies and Reagents
Polystyrene particles were from Polysciences (Warrington, PA). Mouse anti-human ICAM-1 (clone R6.5) and rat anti-mouse ICAM-1 (clone YN1) were from ATCC (Manassas, VA). Mouse anti-mannose-6-phosphate receptor (anti-M6PR; clone 2G11) was from Abcam (Cambridge, MA). Mouse anti-human transferrin receptor (anti-TfR; clone T56/14) and rat anti-mouse TfR (clone R17217) were from EMD Millipore Corporation (Billerica, MA) and Biolegend (San Diego, CA), respectively. Mouse anti-human EEA-1 was from Calbiochem (La Jolla, CA). Mouse IgM anti-ceramide and FITC-labeled goat anti-mouse IgM were from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse, rabbit and rat IgG, as well as Texas Red- and FITC-labeled goat anti-mouse IgG antibodies were from Jackson ImmunoResearch (Pike West Grove, PA). Neutral sphingomyelinase (NSM) from Bacillus cereus was from Sigma Aldrich (St. Louis, MO). Alexa Fluor 594 phalloidin was from Molecular Probes (Eugene, OR). Pierce iodination tubes were from Thermo Scientific (Rockford, IL). Unless otherwise stated, all other reagents were from Sigma-Aldrich (St. Louis, MO).
Preparation and Characterization of Polymer Carriers
Model polymer carriers were prepared by adsorbing onto the surface of polystyrene particles (100-nm, 1-μm, or 4.5-μm diameter) either control IgG, anti-ICAM, anti-M6PR, anti-TfR, combinations of anti-M6PR or anti-TfR and IgG (80:20 or 50:50 surface-coverage ratio), or combinations of anti-M6PR or anti-TfR and NSM (80:20 or 50:50 surface coverage ratio), as described previously.52,69 We have previously titrated the molar ratios to which antibodies and enzyme need to be combined in the coating mixture in order to achieve these values.69 Uncoated counterparts were removed by centrifugation. Carriers were resuspended in phosphate-buffered saline (PBS) containing 0.3% bovine serum albumin (BSA) and sonicated to avoid aggregation.69 The size and polydispersity of the preparations was assessed by dynamic light scattering (Malvern Zetasizer, Worcestershire, UK) or optical microscopy (see microscopy setting below). Particles 100-nm in diameter ranged from 180–200-nm after coating; hence, they are referred to as 200-nm in diameter. The coating density was assessed by coating carriers using protein counterparts labeled with 125I and measuring 125I content in a gamma counter (PerkinElmer Wizard2, Waltham, MA), as described previously.69 The characterization of these formulations is provided in Table 1.
Cell Culture
Human umbilical vein endothelial cells (HUVECs; Lonza, Walkersville, MD), were cultured in basal medium M199 (Invitrogen, Carlsbad, CA) supplemented with 15% fetal bovine serum, 15 μg/mL endothelial cell growth supplement, 2 mM L-glutamine, 100 μg/mL heparin, 100 U/mL penicillin, and 100 μg/mL streptomycin.31 Human epithelial colorectal adenocarcinoma Caco-2 cells (American Type Culture Collection, Manassas, VA) were cultured in DMEM (GibcoBRL, Grand Island, NY) supplemented with 15% fetal bovine serum, 4.50 g/L glucose, and the antibiotics described above.26 Cells were seeded on 12-mm2 gelatin-coated glass coverslips at a density of ~105cells/well and grown at 37°C, 5% CO2, and 95% relative humidity. In the case of HUVECs, cells were additionally activated with 10 ng/mL of tumor necrosis factor-α (TNF-α) for 16 h to mimic a disease status.
Binding and Internalization of Carriers
For binding experiments, activated cells were fixed with 2% paraformaldehyde to avoid confounding results of concomitant endocytosis. Fixed cells were then incubated at room temperature with ~2 x 107 carriers/mL (1-μm carriers), then non-bound carriers were washed. Carriers were counted by phase contrast microscopy.31
To determine internalization, live cells were incubated at 37°C with complete medium (see section above) in the absence or presence of pharmacological inhibitors of endocytic pathways, including 50 μM monodansylcadaverine (to inhibit clathrin-mediated uptake), 1 μg/mL filipin (to inhibit caveolae-mediated uptake), 3 mM amiloride (to inhibit CAM- and macropinocytic uptake), and 0.5 μM wortmannin (to inhibit macropinocytosis only). Cells were incubated in the presence of 200-nm carriers, 1-μm carriers, or 4.5-μm carriers. In some instances (indicated), incubation with carriers was conducted in the presence of either anti-M6PR or anti-ICAM as competitors to the cell surface receptor. Carriers 200-nm in diameter contained FITC in the polymer matrix, since this size is difficult to visualize by phase contrast microscopy. After washing off non-bound carriers, cells were fixed and incubated with Texas Red-labeled secondary antibody, which is accessible to surface-bound carriers but not internalized counterparts.31 Cell samples were then examined by optical microscopy. For 1-μm or 4.5-μm particles, total carriers were counted using phase contrast and surface-bound non-internalized carriers were counted using Texas Red fluorescence. For 200-nm particles, total carriers vs. non-internalized carriers were counted using FITC and Texas Red fluorescence, respectively, as previously described.20
F-actin and Ceramide Staining
Cells were first incubated for 30 min at 37°C with 1-μm or 4.5-μm carriers. Non-bound carriers were washed, and cells were fixed and blocked overnight with 2% BSA in PBS. Cells were permeabilized with 0.2% Triton X-100 and then F-actin was stained with red Alexa Fluor 594 phalloidin.31 Ceramide was immunostained using mouse anti-ceramide IgM, followed by green FITC-labeled goat anti-mouse IgM. Fluorescence intensity at carrier sites (identified by phase contrast) was compared to the fluorescence intensity of adjacent areas of the plasmalemma.31
Co-localization with Endosomes and Coat Degradation
Cells were incubated at 37°C with 200-nm or 1-μm carriers for either 3 h or 5 h. Non-bound carriers were washed, and cells were fixed and permeabilized. Early endosomes were immunostained using mouse anti-human EEA-1 and a secondary antibody labeled with Texas Red. Colocalization of Texas Red fluorescent signal with carriers (identified by phase contrast) indicated presence of carriers within this compartment.21 Independently, after permeabilization, the presence of the antibody coat on carriers was assessed by immunostaining using goat anti-mouse IgG conjugated to Texas Red. Absence of Texas Red fluorescent signal colocalizing with carriers (identified by phase contrast) indicated absence (degradation) of carrier coat, as validated previously.21
Cell viability
Cells were incubated in the absence or presence of anti-M6PR-IgG carriers or anti-M6PR/NSM carriers (50:50 ratio) for 3 h at 37°C. Carriers were then removed and the total number of cells and their viability were tested using the Live/Dead® kit (Invitrogen, Carlsbad, CA) at this time, as well as 7 h and 21 h thereafter (total time was 3 h, 8 h and 24 h after start of carrier incubation). Fluorescence microscopy was used to visualize and quantify calcein-positive (live, green) vs. ethidium homodimer-1-positive (dead, red) cells, while phase-contrast allowed visualization of the cell appearance. As a positive control for apoptosis, cells were incubated with medium containing 100 μM H2O2.
Microscopy analysis
Cells were analyzed using a 40X PlanApo objective and the Olympus IX81 inverted 3-axe automatic fluorescence microscope (Olympus Inc., Center Valley, PA). Samples were observed by phase contrast and fluorescence using filters from Semrock (Rochester, NY) in the Texas Red channel (excitation BP360-370 nm, dichroic DM570 nm, emission BA590-800+ nm) or FITC channel (excitation BP460-490 nm, dichroic DM505 nm, emission BA515-550 nm). Micrographs were taken using an Orca-ER camera from Hamamatsu (Bridgewater, NJ) and SlideBookTM 4.2 software from Intelligent Imaging Innovations (Denver, CO). Images were analyzed using Image-Pro 6.3 from Media Cybernetics Inc. (Bethesda, MD).
Biodistribution of Carriers in vivo
Male C57/B16 mice were anesthetized and injected intravenously either with control IgG carriers, anti-M6PR/IgG, anti-M6PR/NSM carriers, anti-TfR/IgG, or anti-TfR/NSM (~1.5 x 1013 particles/kg), all containing similar tracer amounts of 125I-IgG or 125I-NSM.69 Mice were sacrificed 30 minutes after injection, blood and organs samples (kidneys, liver, and lungs) were collected, and their weight and 125I content were determined to calculate the percent injected dose (%ID) and/or specificity (SI) of each targeted formulation. This parameter represents the ratio of percent injected dose per gram of tissue for anti-M6PR/IgG carriers or anti-M6PR/NSM carriers divided by the percent injected dose per gram of tissue for non-targeted IgG carriers. Experiments were performed in accordance with IACUC and University of Maryland regulations and the Guide for the Care and Use of Laboratory Animals of the U.S National Institutes of Health.
Statistics
Data are mean ± standard error of the mean. Each cell culture experiment contained duplicate samples (~105 cells/sample) and was repeated at least two independent times (total ≥4 samples). A minimum of 10 pictures were taken at locations selected randomly through each sample, where each picture encompasses ~10–15 cells. This renders ≥400–600 cells individually analyzed per condition. In vivo experiments were done using a minimum of 3 mice per condition. Significance was determined using the student’s t-test assuming a p level of 0.05.
Supplementary Material
SYNOPSIS.
Surface functionalization of drug carriers with biological elements (sphingomyelinases) associated with endocytosis mediated by ICAM-1 provides a means to bypass size-restrictions of drug carriers targeted to receptors associated with more size-restrictive pathways (e.g., clathrin-mediated). This strategy will enhance flexibility of design of drug carriers for intracellular delivery of therapeutics.
Acknowledgments
This study was funded by a grant from NIH (R01 HL098416) to SM and NIH Diversity Research Supplement (HL098416-S1) to DS.
ABBREVIATIONS
- Amil
amiloride
- BSA
bovine serum albumin
- CAM
cellular adhesion molecule
- Fil
filipin
- HUVECS
human umbilical vein endothelial cells
- ICAM-1
intercellular adhesion molecule-1
- M6P
mannose-6-phosphate
- M6PR
mannose-6-phosphate receptor
- MDC
monodansylcadaverine
- NHE1
sodium proton exchanger 1
- NSM
neutral sphingomyelinase
- TfR
transferrin receptor
- TNFα
tumor necrosis factor α
- Wort
wortmannin
Footnotes
Author Contributions.
MA and DS optimized methodology, performed and analyzed experiments, prepared figures, and wrote the manuscript; TB and IP contributed in vivo experiments; SM conceptualized and supervised the study, and contributed to writing and editing the manuscript. All authors have given approval to the final version of the manuscript.
Supporting Information Available: Supplementary Figures S1 to S10 complement the figures shown in this manuscript. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Hoffman AS. The Origins and Evolution of “Controlled” Drug Delivery Systems. J Control Release. 2008;132:153–163. doi: 10.1016/j.jconrel.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 2.Hans ML, Lowman AM. Biodegradable Nanoparticles for Drug Delivery and Targeting. Curr Opin Solid St M. 2002;6:319–327. [Google Scholar]
- 3.Torchilin VP. Multifunctional Nanocarriers. Adv Drug Delivery Rev. 2006;58:1532–1555. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Langer R. Drug Delivery-Drugs on Target. Science. 2001;293:58–59. doi: 10.1126/science.1063273. [DOI] [PubMed] [Google Scholar]
- 5.Debbage P. Targeted Drugs and Nanomedicine: Present and Future. Curr Pharm Des. 2009;15:153–172. doi: 10.2174/138161209787002870. [DOI] [PubMed] [Google Scholar]
- 6.Parveen S, Misra R, Sahoo SK. Nanoparticles: A Boon to Drug Delivery, Therapeutics, Diagnostics, and Imaging. Nanomed Nanotech Biol Med. 2012;8:147–166. doi: 10.1016/j.nano.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 7.Muro S. Challenges in Design and Characterization of Ligand-Targeted Drug Delivery Systems. J Control Release. 2012;164:125–137. doi: 10.1016/j.jconrel.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conner SD, Schmid SL. Regulated Portals of Entry into the Cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 9.Jones AT. Gateways and Tools for Drug Delivery: Endocytic Pathways and the Cellular Dynamics of Cell Penetrating Peptides. Int J Pharm. 2008;354:34–38. doi: 10.1016/j.ijpharm.2007.10.046. [DOI] [PubMed] [Google Scholar]
- 10.Viera OV, Botelho RJ, Grinstein S. Phagosome Maturation: Aging Gracefully. Biochem J. 2002;366:689–704. doi: 10.1042/BJ20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Swanson JA, Watts C. Macropinocytosis. Trends Cell Biol. 1995;5:424–428. doi: 10.1016/s0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- 12.Bareford LM, Swaan PW. Endocytic Mechanism for Targeted Drug Delivery. Adv Drug Delivery Rev. 2007;59:748–758. doi: 10.1016/j.addr.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rejman J, Oberle V, Zuhorn IS, Hoekstra D. Size-Dependent Internalization of Particles via the Pathways Of Clathrin- And Caveolae-Mediated Endocytosis. Biochem J. 2004;377:159–169. doi: 10.1042/BJ20031253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gratton SEA, Roff PA, Pohlhaus PD, Luft C, Madden VJ, Napier ME, DeSimone JM. The Effect of Particle Design on Cellular Internalization Pathways. PNAS. 2008;105:11613–11618. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doshi N, Prabhakarpandian B, Rea-Ramsey A, Pant K, Sundaram S, Mitragotri S. Flow and Adhesion of Drug Carriers in Blood Vessels Depend on their Shape: A Study Using Model Synthetic Microvascular Networks. J Control Release. 2010;146:196–200. doi: 10.1016/j.jconrel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Shape Effects of Filaments versus Spherical Particles in Flow and Drug Delivery. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Charoenphol P, Huang RB, Eniola-Adefeso O. Potential Role of Size and Hemodynamics in the Efficacy of Vascular-Targeted Spherical Drug Carriers. Biomaterials. 2010;31:1392–1402. doi: 10.1016/j.biomaterials.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Stan RV. Endocytosis Pathways in Endothelium: How Many? Am J Physiol Lung Cell Mol Physiol. 2006;290:806–808. doi: 10.1152/ajplung.00533.2005. [DOI] [PubMed] [Google Scholar]
- 19.Atochina EN, Balyasnikova IV, Danilov SM, Granger DN, Fisher AB, Muzykantov VR. Immunotargeting of Catalase to ACE or ICAM-1 Protects Perfused Rat Lungs Against Oxidative Stress. Am J Phsyiol. 1998;275:806–817. doi: 10.1152/ajplung.1998.275.4.L806. [DOI] [PubMed] [Google Scholar]
- 20.Muro S, Wiewrodt R, Thomas A, Koniaris L, Albelda SM, Muzykantov VR, Koval M. A Novel Endocytic Pathway Induced by Clustering Endothelial ICAM-1 or PECAM-1. J Cell Sci. 2003;116:1599–1609. doi: 10.1242/jcs.00367. [DOI] [PubMed] [Google Scholar]
- 21.Muro S, Gajewski C, Koval M, Muzykantov VR. ICAM-1 Recycling in Endothelial Cells: A Novel Pathway for Sustained Intracellular Delivery and Prolonged Effects of Drugs. Blood. 2005;105:650–658. doi: 10.1182/blood-2004-05-1714. [DOI] [PubMed] [Google Scholar]
- 22.Bloemen PG, Henricks PA, vanBloois L, van den Tweel MC, Bloem AC, Nijkamp FP, Crommelin DJ, Storm G. Adhesion Molecules: A New Target for Immunoliposome-Mediated Drug Delivery. FEBS Lett. 1995;357:140–144. doi: 10.1016/0014-5793(94)01350-a. [DOI] [PubMed] [Google Scholar]
- 23.Villanueva FS, Jankowski RJ, Klibanov S, Pina ML, Alber SM, Watkins SC, Brandenburger GH, Wagner WR. Microbubbles Targeted to Intercellular Adhesion Molecule-1 Bind to Activated Coronary Artery Endothelial Cells. Circulation. 1998;98:1–5. doi: 10.1161/01.cir.98.1.1. [DOI] [PubMed] [Google Scholar]
- 24.Chittasupho C, Xie SX, Baoum A, Yakovleva T, Siahaan TJ, Berkland CJ. ICAM-1 Targeting of Doxorubicin-Loaded PLGA Nanoparticles to Lung Epithelial Cells. Eur J Pharm Sci. 2009;37:141–150. doi: 10.1016/j.ejps.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muzykantov V. Targeting Drugs to Pulmonary Endothelium. Expert Opin Drug Deliv. 2005;2:909–926. doi: 10.1517/17425247.2.5.909. [DOI] [PubMed] [Google Scholar]
- 26.Ghafarrian R, Bhowmick T, Muro S. Transport of Nanocarriers across Gastrointestinal Epithelial Cells by a New Transcellular Route Induced by Targeting ICAM-1. J Control Release. 2012;163:25–33. doi: 10.1016/j.jconrel.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhowmick T, Berk E, Cui X, Muzykantov VR, Muro S. Effect of Flow on Endothelial Endocytosis of Nanocarriers Targeted to ICAM-1. J Control Release. 2012;157:485–492. doi: 10.1016/j.jconrel.2011.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rothlein R, Dustin ML, Marlin SD, Springer TA. A Human Intercellular Adhesion Molecule (ICAM-1) Distinct from LFA-1. J Immunol. 1986;137:1270–1274. [PubMed] [Google Scholar]
- 29.Muro S. Endothelial Biomedicine. 1. Cambridge Univ. Press; New York: 2007. Intercellular Adhesion Molecule-1 and Vascular Cell Adhesion Molecule-1; pp. 1058–1070. [Google Scholar]
- 30.Muro S, Garnacho C, Champion JA, Leferovich J, Gajewski C, Schuchman EH, Mitragotri S, Muzykantov VR. Control of Endothelial Targeting and Intracellular Delivery of Therapeutic Enzymes by Modulating the Size and Shape of ICAM-1-Targeted Carriers. Mol Ther. 2008;16:1450–1458. doi: 10.1038/mt.2008.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Serrano D, Bhowmick T, Chadha R, Garnacho C, Muro S. Intercellular Adhesion Molecule 1 Engagement Modulates Sphingomyelinase and Ceramide, Supporting Uptake of Drug Carriers by the Vascular Endothelium. Arterioscler Thromb Vasc Biol. 2012;32:1178–1185. doi: 10.1161/ATVBAHA.111.244186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muro S, Mateescu M, Gajewski C, Robinson M, Muzykantov VR, Koval M. Control of Intracellular Trafficking of ICAM-1 Targeted Nanocarriers by Endothelial Na+/H+ Exchanger Proteins. Am J Physiol Lung Cell Mol Physiol. 2006;290:809–817. doi: 10.1152/ajplung.00311.2005. [DOI] [PubMed] [Google Scholar]
- 33.Zeidan YH, Jenkins RW, Hannun YA. Remodeling Of Cellular Cytoskeleton by the Acid Sphingomyelinase/Ceramide Pathway. J Cell Biol. 2008;181:335–350. doi: 10.1083/jcb.200705060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holopainen JM, Subramanian M, Kinnunen PKJ. Sphingomyelinase Induces Lipid Microdomain Formation in a Fluid Phosphotidylcholine/sphingomyelin Membrane. Biochemistry. 1998;37:17562–17570. doi: 10.1021/bi980915e. [DOI] [PubMed] [Google Scholar]
- 35.Holopainen JM, Angelova MI, Kinnuen PKJ. Vectorial Budding of Vesicles by Asymmetrical Enzymatic Formation of Ceramide in Giant Liposomes. Biophysical J. 2000;78:830–838. doi: 10.1016/S0006-3495(00)76640-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grassme H, Jendrossek V, Bock J, Riehle A, Gulbins E. Ceramide-Rich Membrane Rafts Mediate CD40 Clustering. J Immunol. 2002;168:298–307. doi: 10.4049/jimmunol.168.1.298. [DOI] [PubMed] [Google Scholar]
- 37.Zha X, Pierini LM, Leopold PL, Skiba PJ, Tabas I, Maxfield FR. Sphingomyelinase Treatment Induces ATP-Independent Endocytosis. J Cell Biol. 1998;140:39–47. doi: 10.1083/jcb.140.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruiz-Arguiello MB, Goni FM, Alonso A. Vesicle Membrane Fusion Induced by the Concerted Activities of Sphingomyelinase and Phospholipase C. J Biol Chem. 1998;273:22977–22982. doi: 10.1074/jbc.273.36.22977. [DOI] [PubMed] [Google Scholar]
- 39.Grassme H, Riethmuller J, Gulbins E. Biological Aspects of Ceramide-Enriched Membrane Domains. Prog Lipid Res. 2007;46:161–170. doi: 10.1016/j.plipres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Grassme H, Becker KA, Zhang Y, Gulbins E. Ceramide in Bacterial Infections and Cystic Fibrosis. J Biol Chem. 2008;389:1371–1379. doi: 10.1515/BC.2008.162. [DOI] [PubMed] [Google Scholar]
- 41.Dahms NM, Olson LJ, Kim JP. Strategies for Carbohydrate Recognition by the Mannose 6-Phosphate Receptor. Glycobiology. 2008;18:664–678. doi: 10.1093/glycob/cwn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dautry-Varsat A. Receptor-Mediated Endocytosis: The Intracellular Journey of Transferrin and its Receptor. Biochimie. 1986;68:375–381. doi: 10.1016/s0300-9084(86)80004-9. [DOI] [PubMed] [Google Scholar]
- 43.Korenfeld S. Structure and Function of the Mannose 6-Phosphate/Insulinlike Growth Factor II Receptors. Annu Rev Biochem. 1992;61:307–330. doi: 10.1146/annurev.bi.61.070192.001515. [DOI] [PubMed] [Google Scholar]
- 44.Valentino K, Phan H, Ocrant I, Rosenfeld RG. Distribution of Insulin-Like Growth Factor II Receptor Immunoreactivity in Rat Tissues. Endocrinology. 1987;122:2753–2763. doi: 10.1210/endo-122-6-2753. [DOI] [PubMed] [Google Scholar]
- 45.Taylor JE, Scott CD, Baxter RC. Comparison of Receptors for Insulin-Like Growth Factor II from Various Rat Tissues. J Endocrinol. 1987;115:35–41. doi: 10.1677/joe.0.1150035. [DOI] [PubMed] [Google Scholar]
- 46.Funk B, Kessler U, Eisenmenger W, Hansmann A, Kolb HJ, Kiess W. Expression of the Insulin-Like Growth Factor-II/Mannose-6-Phosphate Receptor in Multiple Human Tissues During Fetal Life and Early Infancy. J Clin Endocrinol Metab. 1992;75:424–431. doi: 10.1210/jcem.75.2.1379254. [DOI] [PubMed] [Google Scholar]
- 47.Lee JS, Weiss J, Martin JL, Scott CD. Increased Expression of the Mannose 6-phosphate/Insulin-Like Growth Factor-II Receptor in Breast Cancer Cells Alters Tumorigenic Properties in Vitro and in Vivo. Int J Cancer. 2003;107:564–570. doi: 10.1002/ijc.11453. [DOI] [PubMed] [Google Scholar]
- 48.Wei Y, Li J, Huang J, Zhang X, Zhao H, Cui C, Li Y, Hu S. Elevation of IGF-2 Receptor and the Possible Underlying Implications in End-Stage Heart Failure Patients Before and After Heart Transplantation. J Cell Mol Med. 2012;16:1038–1046. doi: 10.1111/j.1582-4934.2011.01414.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ajantsan AR, Aoki M, Warita H, Ki NSZ, Itoyama YT. Up-Regulation of Insulin-Like Growth Factor-II Receptor in Reactive Astrocytes in the Spinal Cord of Amyotrophic Lateral Sclerosis Transgenic Rats. Tohoku J Exp Med. 2008;214:303–310. doi: 10.1620/tjem.214.303. [DOI] [PubMed] [Google Scholar]
- 50.Gary-Bobo M, Nirde P, Jeanjean A, Morere A, Garcia M. Mannose 6-Phosphate Receptor Targeting and its Applications in Human Diseases. Curr Med Chem. 2007;14:2945–2953. doi: 10.2174/092986707782794005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prakash J, Beljaars L, Harapanahalli AK, Zeinstra-Smith M, Jager-Krikken AD, Hessing M, Steen H, Poelstra K. Tumor-Targeted Intracellular Delivery of Anticancer Drugs Through the Mannose-6-Phosphate/Insulin-Like Growth Factor II Receptor. Int J Cancer. 2010;126:1966–1981. doi: 10.1002/ijc.24914. [DOI] [PubMed] [Google Scholar]
- 52.Muro S, Dziubla T, Qiu W, Leferovich J, Cui X, Berk E, Muzykantov VR. Endothelial Targeting of High-Affinity Multivalent Polymer Nanocarriers Directed to Intercellular Adhesion Molecule 1. J Pharmacol Exp Ther. 2006;317:1161–1169. doi: 10.1124/jpet.105.098970. [DOI] [PubMed] [Google Scholar]
- 53.Garnacho C, Shuvaev V, Thomas A, McKenna L, Sun J, Koval M, Albelda S, Muzykantov V, Muro S. RhoA Activation and Actin Reorganization Involved in Endothelial CAM-mediated Endocytosis of Anti-PECAM Carriers: Critical Role for Tyrosine 686 in the Cytoplasmic Tail of PECAM-1. Blood. 2008;111:3024–3033. doi: 10.1182/blood-2007-06-098657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grau GE, Lambert PH, Vassalli P, Piguet PF. Tumor Necrosis Factor (TNF) and Pathology; its Relationships with other Cytokines. Schweiz Med Wochenschr. 1989;119:1756–1761. [PubMed] [Google Scholar]
- 55.Garnacho C, Albelda SM, Muzykantov VR, Muro S. Differential Intra-Endothelial Delivery of Polymer Nanocarriers Targeted to Distinct PECAM-1 Epitopes. J Control Release. 2008;130:226–233. doi: 10.1016/j.jconrel.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shakor ABA, Atia MM, Kwiatkowska K, Sobota A. Cell Surface Ceramide Controls Translocation of Transferrin Receptor to Clathrin-Coated Pits. Cell Signalling. 2012;24:677–684. doi: 10.1016/j.cellsig.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 57.Byrd JC, MacDonald RG. Mechanisms for High Affinity Mannose 6-Phosphate Ligand Binding to the Insulin-Like Growth Factor II/Mannose 6-Phosphate Receptor. J Biol Chem. 2000;275:18638–18646. doi: 10.1074/jbc.M000010200. [DOI] [PubMed] [Google Scholar]
- 58.Vyas SP, Sihorkar V. Endogenous Carriers and Ligands in Non-Immunogenic Site-Specific Drug Delivery. Adv Drug Deliv Rev. 2000;43:101–164. doi: 10.1016/s0169-409x(00)00067-3. [DOI] [PubMed] [Google Scholar]
- 59.Shuvaev VV, Illies MA, Simone E, Zaitsev S, Kim Y, Cai S, Mahmud A, Dziubla T, Muro S, Discher DE, et al. Endothelial Targeting of Antibody-Decorated Polymeric Filomicelles. ACS Nano. 2011;5:6991–6999. doi: 10.1021/nn2015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Collins A, Warrington A, Taylor KA, Svitkina T. Structural Organization of the Actin Cytoskeleton at the Sites of Clathrin Mediated Endocytosis. Curr Biol. 2011;21:1167–1175. doi: 10.1016/j.cub.2011.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanzaki M, Pessin JE. Caveolin-Associated Filamentous Actin (Cav-actin) Defines a Novel F-actin Structure in Adipocytes. J Biol Chem. 2002;277:25867–25869. doi: 10.1074/jbc.C200292200. [DOI] [PubMed] [Google Scholar]
- 62.Coppolino MG, Krause M, Hagendorff P, Monner DA, Trimble W, Grinstein S, Wehland J, Sechi AS. Evidence for a Molecular Complex Consisting of Fyb/SLAP, SLP-76, Nck, VASP and WASP that Links the Actin Cytoskeleton to Fc Gamma Receptor Signalling During Phagocytosis. J Cell Sci. 2001;114:4307–4318. doi: 10.1242/jcs.114.23.4307. [DOI] [PubMed] [Google Scholar]
- 63.Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid E. Dynamic Interaction of VCAM-1 and ICAM-1 with Moesin and Ezrin in a Novel Endothelial Docking Structure for Adherent Leukocytes. J Cell Biol. 2002;157:1233–1245. doi: 10.1083/jcb.200112126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Serrano D, Muro S. Mechanobiology of the Endothelium. Science Publishers; Maryland: 2013. Endothelial Cell Adhesion Molecules and Drug Delivery Applications. In Press. [Google Scholar]
- 65.Kolesnick R, Fuks Z. Ceramide: A Signal for Apoptosis or Mitogenesis. J Exp Med. 1995;181:1949–1952. doi: 10.1084/jem.181.6.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Levade T, Jaffrezou J. Signaling Sphingomyelinases: Which, Where, How and Why. Biochim Biophys Acta. 1999;1438:1–17. doi: 10.1016/s1388-1981(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 67.Morad SA, Cabot MC. Ceramide-Orchestrated Signaling in Cancer Cells. Nat Rev. 2013;13:51–65. doi: 10.1038/nrc3398. [DOI] [PubMed] [Google Scholar]
- 68.Goldkorn T, Balaban N, Shannon M, Chea V, Matsukuma K, Gilchrist D, Wang H, Chan C. H2O2 Acts on Cellular Membranes to Generate Ceramide Signaling Initiate Apoptosis in Tracheobronchial Epithelial Cells. J Cell Sci. 1998;111:3209–3220. doi: 10.1242/jcs.111.21.3209. [DOI] [PubMed] [Google Scholar]
- 69.Papademetriou IT, Garnacho C, Schuchman EH, Muro S. In Vivo Performance of Polymer Nanocarriers Dually-Targeted to Epitopes of the Same or Different Receptors. Biomaterials. 2013;34:3459–3466. doi: 10.1016/j.biomaterials.2013.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pardridge WM. Biopharmaceutical Drug Targeting to the Brain. J Drug Target. 2010;18:157–167. doi: 10.3109/10611860903548354. [DOI] [PubMed] [Google Scholar]
- 71.Muro S. New Biotechnological and Nanomedicine Strategies for Treatment of Lysosomal Storage Disorders. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2010;2:189–204. doi: 10.1002/wnan.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cardone M, Porto C, Tarallo A, Vicinanze M, Rossi B, Polishchuk E, Donaudy F, Andria G, Matteis M, Parenti G. Abnormal Mannose-6-Phosphate Receptor Trafficking Impairs Recombinant Alpha-Glucosidase Uptake in Pompe Disease Fibroblasts. Pathogenetics. 2008;1:6. doi: 10.1186/1755-8417-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dhami R, Schuchman EH. Mannose 6-Phosphate Receptor Mediated Uptake is Defective in Acid Sphingomyelinase-Deficient Macrophages: Implications for Niemann-Pick Disease Enzyme Replacement Therapy. J Biol Chem. 2004;279:1526–1532. doi: 10.1074/jbc.M309465200. [DOI] [PubMed] [Google Scholar]
- 74.Ravindran S, Snee PT, Ramachandran A, George A. Acidic Domain in Dentin Phosphophoryn Facilitates Cellular Uptake: Implications in Targeted Protein Delivery. J Biol Chem. 2013;288:16098–16109. doi: 10.1074/jbc.M113.450585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Boeneman K, Delehanty JB, Blanco-Canosa JB, Susumu K, Stewart MH, Oh E, Huston AL, Dawson G, Ingale S, Walters R, et al. Selecting Improved Peptidyl Motifs for Cytosolic Delivery of Disparate Protein and Nanoparticle Materials. ACS Nano. 2013;7:3778–3796. doi: 10.1021/nn400702r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Han H-S, Martin JD, Lee J, Harris DH, Fukumura D, Jain RK, Bawendi M. Spatial Charge Configuration Regulates Nanoparticle Transport Binding. In Vivo Angew Chem Int Ed. 2013;52:1414–1419. doi: 10.1002/anie.201208331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Henry B, Moller C, Dimanche-Boitrel MT, Gulbins E, Becker KA. Targeting the Ceramide System in Cancer. Cancer Lett. 2013;332:286–294. doi: 10.1016/j.canlet.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 78.Kartal-Yandim M, Apohan E, Baran Y. Therapeutic Potential of Targeting Ceramide/Glucosylceramide Pathway in Cancer. Cancer Chemother Pharmacol. 2013;71:13–20. doi: 10.1007/s00280-012-1984-x. [DOI] [PubMed] [Google Scholar]
- 79.Barth BM, Cabot MC, Kester M. Ceramide-Based Therapeutics for the Treatment of Cancer. Anticancer Agents Med Chem. 2011;11:911–919. doi: 10.2174/187152011797655177. [DOI] [PubMed] [Google Scholar]
- 80.Zhang P, Liu B, Jenkins GM, Hannun YA, Obeid LM. Expression of Neutral Sphingomyelinase Identifies a Distinct Pool of Sphingomyelin Involved in Apoptosis. J Biol Chem. 1997;272:9609–9612. doi: 10.1074/jbc.272.15.9609. [DOI] [PubMed] [Google Scholar]
- 81.Wu BX, Clarke CJ, Hannun YA. Mammalian Neutral Sphingomyelinases: Regulation and Roles in Cell Signaling Responses. Neuro Mol Med. 2010;12:320–330. doi: 10.1007/s12017-010-8120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.