

Abstract
Male-mediated developmental toxicity has been of concern for many years. The public became aware of male-mediated developmental toxicity in the early 1990s when it was reported that men working at Sellafield might be causing leukemia in their children. Human and animal studies have contributed to our current understanding of male-mediated effects. Animal studies in the 1980s and 1990s suggested that genetic damage after radiation and chemical exposure might be transmitted to offspring. With the increasing understanding that there is histone retention and modification, protamine incorporation into the chromatin and DNA methylation in mature sperm and that spermatozoal RNA transcripts can play important roles in the epigenetic state of sperm, heritable studies began to be viewed differently. Recent reports using molecular approaches have demonstrated that DNA damage can be transmitted to babies from smoking fathers, and expanded simple tandem repeats minisatellite mutations were found in the germline of fathers who were exposed to radiation from the Chernobyl nuclear power plant disaster. In epidemiological studies, it is possible to clarify whether damage is transmitted to the sons after exposure of the fathers. Paternally transmitted damage to the offspring is now recognized as a complex issue with genetic as well as epigenetic components.
Keywords: genetic and epigenetic effects, human exposure to chemicals and radiation, male-mediated animal studies, paternal exposure to chemicals and radiation
INTRODUCTION
Miller and coworkers asked whether a human sperm is more than the sum of its parts: DNA, protamines, histones and epigenetics.1 Haploid male germ cells package their DNA into a volume that is typically 10% or less than that of a somatic cell nucleus. To achieve this compaction, spermatozoa replace most of their histones with protamines. Such compaction helps to optimize the nuclear shape and hence supports the gametes’ swimming ability for the journey across the female reproductive tract to the oocyte. Thus, the human haploid sperm and ovum differ in configuration but not in DNA content.
The scope of paternal contributions during early embryonic development has long been considered limited. Dramatic changes in chromatin structure throughout spermatogenesis have been thought to leave the sperm void of complex layers of epigenetic regulation over the DNA blueprint, thus leaving the balance of that regulation to the oocyte. However, recent work in the fields of epigenetics and male-factor infertility has provided additional information that can be used to reinterpret this long-held and now controversial dogma. Histone retention and modification, protamine incorporation into the chromatin, DNA methylation and spermatozoal RNA transcripts appear to play important roles in the epigenetic state of mature sperm.2
GENOMIC PATERNAL CONTRIBUTIONS
The human diploid genome is not constant but continuously changes, giving rise to an average of 70 de novo mutations per generation, which are mostly paternal in origin.3 When compared with the mother, an increased number of mutations are conveyed by the father to their common children. The father's age is the main factor in the number of new mutations in the offspring. Kong et al.4 determined that this age effect contributes two additional mutations per year or a doubling of the paternal mutation rate every 16.5 years. Hence, it is essential to consider the father's age and to treat the mutation rate not as a constant factor but as a time-dependent variable. Nonetheless, detrimental environmental impacts by genotoxins might increase the rate of new paternal mutations, the expression patterns of regulatory small non-coding RNAs (sncRNAs) and epigenetic modifications.
Recently, Laubenthal et al.5 demonstrated in a human triad cohort of mothers, fathers and babies that paternal preconception cigarette smoking induced male-mediated transgenerational alterations in fetal lymphocytes in the offspring. This result demonstrates that toxicant exposure before and during conception is relevant to children's genomic stability. Paternal exposure to cigarette smoke resulting in the induction of germline mutations has also been recently assessed by focusing on minisatellite loci such as CEB1 and B6.7. Mutation frequencies for CEP1 but not B6.7 significantly increased in the offspring when fathers smoked 6 months before pregnancy, indicating that a healthy lifestyle without smoking coincides with lower CEP1 mutation frequencies. Thus, heritable mutations in repetitive DNA sequences can be a consequence of a harmful paternal lifestyle.6
A major component of cigarette smoke is the polycyclic aromatic hydrocarbon benzo[a]pyrene (BaP). Hence, smokers exhibit a significant increase in BaP-DNA adducts in sperm7 as well as increased DNA damage in mature spermatozoa due to exposure to oxidative stress.8 Male germ cell exposure to genotoxins thus leads to sperm DNA damage. Despite the fact that spermatozoa are largely transcriptionally inactive due to chromatin remodelling, mRNA profiles from the ejaculated sperm of cigarette smokers revealed increased expression of the germ cell-specific gene for protamine 2 (PRM2) as well as a 5.4-fold upregulation of the germ cell-specific transcription factor SALF and a 7.4-fold downregulation of the zinc finger encoding gene TRIM26.6 Subsequent analysis of transcription factor networks suggested that apoptosis is inhibited in smokers.9 An unbalanced PRM2/PRM1 ratio and altered levels of protamine may lead to infertility and increased DNA damage.10 Growing evidence in animal models suggests that fertilization of DNA-fragmented spermatozoa using intracytoplasmic sperm injection leads to immediate adverse effects regarding gene transcription and methylation as well as long-term pathologies in the embryo, fetus and the offspring.11 These findings demonstrate that sperm that might potentially fertilize an oocyte can carry damaged DNA as well as altered mRNA profiles into the next generation.
In recent years the importance of sncRNA has become very evident. sncRNAs, like micro-RNAs (miRNAs), play a crucial role in regulating gene expression and epigenetic patterns. Sperm not only function as the means to transport the male DNA complement to the oocyte but also to deliver a plethora of other factors crucial for early embryogenesis. In addition to the oocyte activation factor, centrosomes and mRNA, mature spermatozoa also contain sncRNA of 18–30 nucleotides in length.12,13 The main fraction of sncRNAs is miRNA mediating posttranscriptional degradation as another level of gene expression control. Furthermore, piwi-interacting RNAs specific to sperm play an essential role in masking repetitive and transposon elements in the paternal pronucleus after fertilization.14 Although >95% of the sperm DNA is tightly packed with protamines, the rest remains associated with nucleosomes at loci important for development and signaling, including clusters of HOX genes, imprinted genes and miRNAs.13,15 Deep sequencing analysis of sncRNAs recently revealed the diversity of miRNAs and piwi-interacting RNAs in the human epididymis and the maturing sperm.16 Although a sperm contributes only 5–10 fg of total RNA to the 1 ng of maternal RNA within a fertilized oocyte,17 sncRNAs (approximately 0.3 fg per sperm12) may be crucial developmental and epigenetic modifiers controlling chromatin remodelling and gene expression while protecting the genome against intrusion.12,18
For the genotoxin and carcinogen BaP, transcriptomic analysis revealed a significant effect on altered gene expression patterns in human sperm.9 Although performed in HepG2 cell lines but not in germ cells, the impact of this toxic compound on miRNA networks revealed that various miRNAs were expressed in response to BaP exposure, together with several alterations in mRNA levels. These mRNAs themselves were targets of altered miRNAs. Eight miRNAs, e.g. miR-29b, miR-26a-1 and miR-122, were identified as being involved in BaP-responsive pathways associated with the genotoxicity, specifically participating in DNA damage response/repair, cell cycle arrest and apoptotic signaling.19 Increases in miR-29b after neonatal hormonal disruption are commonly associated with a decrease in DNA methyltransferase (DNMT1) expression and a decrease in the levels of antiapoptotic Mcl-1, resulting in increased germ cell apoptosis in adult rats and thus perinatally programming these cells for cell death.20 Such programming might well happen when altered miRNA expression patterns arise in male germ cells due to paternal exposure to genotoxins that subject filial germ cells to increased apoptosis and therefore to a higher risk of infertility.
Environmental chemical toxins such as heavy metals, heavy air pollution, cigarette smoking and compounds such as bisphenol A, diethylstilbestrol, dioxin and many others can affect the function of miRNAs as negative regulators by posttranscriptional silencing/suppression of their target gene expression.21,22 Hou et al.21 also hypothesized that oxidation as well as inflammation reactions play a crucial role in miRNA alterations. Furthermore, in addition to the effect of toxins on offspring development, paternal food deprivation can cause altered physiological parameters. The influence of food deprivation was confirmed in a study on male mice,23 which demonstrated that food deprivation altered the time of maximum response to ethyl methane sulfonate by 1 week in a dominant lethal assay. Another study24 demonstrated that food deprivation decreased the average serum glucose levels in male and female offspring. Food deprivation also changed the levels of corticosterone and insulin-like growth factor 1, demonstrating a transgenerational male-mediated effect regarding parameters of metabolism and growth in the filial generation.24
EPIGENETIC PATERNAL CONTRIBUTIONS
Small interfering RNAs as well as miRNAs, both key players in the epigenetic machinery, are often considered to be involved in epigenetic modifications, e.g., in gene silencing at both the transcriptional and posttranscriptional levels, and to mediate epigenetic DNA and histone modifications.25 Epigenetics is the study of heritable changes in gene expression that are predominantly caused by modifications of the nuclear chromatin rather than by changes in the underlying DNA sequence.26 In humans, the most widely studied epigenetic modification is the methylation of cytosine residues at the carbon 5 position (5-mC) within the CpG dinucleotides mediated by a family of DNMTs. Dnmt1 is the most abundant DNMT and is considered the key maintenance methyltransferase in mammals. Histone modifications are the other major epigenetic modification, which consist of reversible posttranslational modifications of the residues at the N-terminal tails of histones.26 Histone modifications include acetylation, methylation, ubiquitylation and phosphorylation. For example, acetylation of the K9 and K14 lysines of the tail of histone H3 by histone acetyltransferases is highly correlated with transcriptional competence. These two main epigenetic mechanisms, DNA methylation and histone modifications, are known to have direct effects on controlling gene expression.
To establish the role of epigenetic factors in male-mediated developmental effects, the environmental factors that induce epigenetic modifications in the germ cells must be shown to be transmitted across generations. Most of the histones in sperm are replaced by protamines, which allow negatively charged DNA to condense and fit into the sperm head owing to their large number of positively charged arginine residues.27 The remaining histones in the late spermatid chromatin exhibit modifications. Acetylated histone H4 seems to be involved in the histone-to-protamine transition. These modified histones are not randomly distributed in the sperm genome, but instead are found to be enriched at the specific loci of developmental importance.27 In human sperm, approximately 85% of DNA-binding histones are replaced by protamines, with the remaining histones exhibiting epigenetic modifications.26
DNA epigenetic modifications are known to be essential for spermatogenesis. During sperm development, mammalian cells undergo an almost complete reprogramming of DNA methylation patterns. Therefore, environmentally induced epigenetic modifications, which are transmitted to offspring, must escape the DNA epigenetic reprogramming when genome-wide methylation patterns are subjected to extensive demethylation and remethylation.28 Later on, germ cell methylation patterns are erased again during a second wave of epigenetic reprogramming that occurs during preimplantation development.29 Testicular tissue has been shown to exhibit eight times more hypomethylated loci than any other somatic tissue.26 In male germ cells, DNA methylation patterns are largely established and reprogrammed by both demethylation and de novo methylation events. Demethylation of DNA is almost absent in non-mitotic germ cells, whereas de novo methylation events take place within non-repetitive sequences.29 The DNA methylation pattern of male germ cells does not always reflect the gene expression pattern, but may be involved in germ cell-specific chromatin organization.30 In contrast, DNA hypermethylation may be associated with poor sperm motility due to insufficient removal of methyl groups rather than the result of de novo methylation events.31
Environmental exposures have been found to result in altered DNA methylation and in the activity of enzymes involved in regulating epigenetic modifications. These effects may arise as a result of differential methylation at imprinted genes in the male germ line.28
Vinclozolin and its metabolites are known to be endocrine disruptors and act as androgen receptor antagonists. Endocrine disruptors are hormonally active environmental toxins that can influence sperm viability. Following vinclozolin exposure, the gene expression of nearly 400 genes in male offspring was altered in the hippocampus when measured in three generations, demonstrating that many genes can retain an imprint or memory of the initial environmental exposure.32 In the last decade, growing evidence has suggested that epigenetic modifications, including DNA methylation, may have functional consequences for the offspring and contribute to the regulation of gene transcription in the embryo post-fertilization.33 DNA methylation patterns are first established in a precise manner during gametogenesis. The gene DNMT1 for the enzyme Dnmt1 is expressed at high levels in spermatogonia and in leptotene spermatocytes, but is not detectable in the nucleus of pachytene spermatocytes. This is consistent with a role for DNMTs in setting or maintaining paternal methylation patterns during the early stages of spermatogenesis. Recently, Marques et al.34 demonstrated that de novo and maintenance DNMTs, including Dnmt1, are expressed in human spermatogenesis during proliferation, meiosis, cell divisions and spermatid differentiation as well as in mature sperm. The dynamics of DNA methylation and the expression of the different DNMT enzymes are tightly regulated during spermatogenesis.33
Current research has tended to focus on more molecular techniques. However, most of the early pioneering work on male-mediated developmental toxicity was conducted in animal studies. With the active animal liberation front, there is a recent trend to reduce, refine and replace animal studies.35,36 In addition, the expense involved in large animal studies has become almost prohibitive, and many of the large research animal houses in different parts of the world have closed. However, it is in such animal studies that many of the basic principles of heritable transmission of damage to offspring were determined after both chemical and radiation exposure of males.
PATERNAL EXPOSURE TO CHEMICALS
Regulatory testing requires that the female is tested for teratogenic effects, but because the male contributes half of the genetic information to developing offspring, males could also be examined for induced ‘teratogenic’ effects (congenital malformations). Transplacental carcinogenesis is recognized in the female, but carcinogenesis mediated through the male germ cells is not so well-appreciated or understood. The public have become more aware that the exposure of males to certain agents can adversely affect their offspring. For example, smoking parents could give rise to tumors in the F1 generation37,38,39 and produce minisatellite mutations in the offspring; thus, heritable mutations in repetitive DNA sequences can be a consequence of a harmful paternal lifestyle.6
Lefebvre et al.40 demonstrated that the paternally-transmitted and paternally-imprinted gene, MEST, is involved in normal maternal behavior. MEST-deficient females exhibit abnormal behavior and intrauterine and postnatal growth retardation of progeny. This phenotype lends additional evidence to the importance of the male in the successful development of the future generation. Congenital malformations and tumors can be studied after the exposure of males in an extended dominant-lethal assay, wherein untreated females are mated to treated males and then examined the day before term, as opposed to mid-term in the conventional study.41 At this stage, congenital malformations, such as hydrocephaly, exencephaly, cleft palate, open eye, runts (dwarfs), edema, anasarca and gastrochisis, can be observed. Some of these abnormalities have similar manifestations in humans. The fetuses can also be examined for skeletal malformations using alizarin staining. If the F0 treated and control males are mated with more than one female, then in the F1 generation, litters of the extra female(s) can be examined for the same effects in live-born offspring, confirming the original observation. Litters can also be allowed to develop to adulthood, when tumors can be observed and karyotype analysis can be performed on fetuses and adult offspring. By using this type of study design, we have examined the following compounds using chronic and acute exposure: cyclophosphamide, 1,3-butadiene and urethane (ethyl carbamate) (Table 1).42,43,44,45,46,47,48,49
Table 1.
Responses in different studies in rats and mice
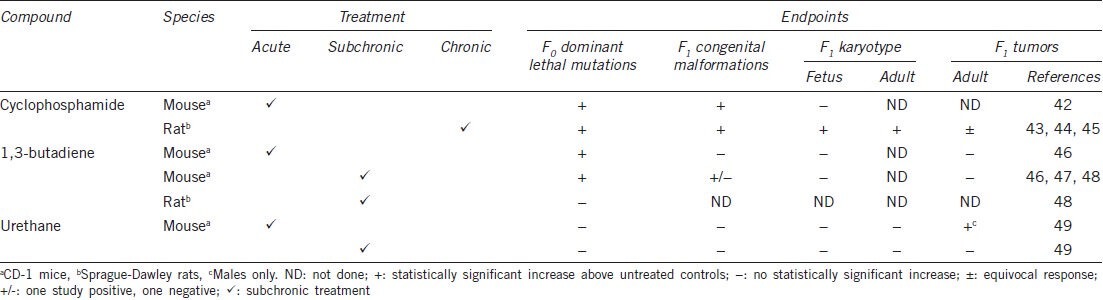
Cyclophosphamide was positive for all endpoints in the rat after chronic gavage exposure.43,44 This effect has also been shown by others45 and led to the belief that chronic exposure might be a more realistic model than acute exposure, as man is chronically exposed in the workplace and environmentally. The colorless gas 1,3-butadiene also induced congenital malformations and was significant in mice in one study46 but not in another,47 with no increase in tumors after subchronic inhalation exposure.46 In the rat, no dominant lethality was observed.48 However, for urethane, there were negative results for dominant lethality and congenital malformations after subchronic exposure in the drinking water, although an increase in tumors in males was observed after acute intraperitoneal treatment.49 A Japanese study in ICR mice after acute intraperitoneal treatment also reported negative results for dominant lethal mutations, confirming results by other workers, but demonstrated an increase in congenital malformations, tumors in the F1 generation and transmitted tumors in the F2 and F3 generations.50 It is assumed that such effects outlined above have been produced as a result of alterations induced in the male germ cells because of their heritability through the generations. However, an epigenetic component cannot be ruled out.
Environmental toxic substances such as urethane, which had been injected subcutaneously to 50 million people as a co-solvent of analgesics and dioxin (an endocrine disruptor), have been associated with adverse effects in the progeny of mice after potential exposures. There are some reports of congenital malformations in the progeny of fathers who had been exposed. Dioxin induces mutations at expanded simple tandem repeat loci in mice at low frequencies as well as congenital malformations (reviewed by Nomura).51
Recently, genomic damage has been measured in a human model in mothers, fathers and baby triads. The study demonstrated that fathers can transmit DNA damage (single-strand breaks as measured in the Comet assay and double-strand breaks as measured by gH2AX) to their babies.5
As tumors can manifest without dominant lethal mutations,49 the different endpoints may be independent genetic (germ cell transmissible) events and might be species- and/or strain-dependent. The question of acute vs chronic exposure might also be agent/compound-dependent. The exact time of mating, within the week after treatment and local husbandry conditions can have an effect on the observed responses. To obtain sufficient numbers of offspring for analysis, there is a delicate balance between death through dominant lethality and the survival of normal and malformed offspring, creating a ‘window’ for detection of effects. As with any toxicological model, careful control of parameters is required. However, it is a useful model for examining inherited congenital malformations and tumors that can be attributed to exposure of the male.
There can also be difficulties in detecting reproductive effects directly in humans, e.g., when interviewing for reproductive outcomes. This fact can be illustrated by reproductive studies with vinyl chloride. Personal interviews and/or questionnaires are a primary source of data for monitoring programs. In gathering information covering reproductive events, studies based on husbands’ indirect reports yielded considerably lower figures for pregnancy loss52 than those based on interviews with wives.53 Individuals clearly have a much better recall for events in their own lives, and the circumstances of pregnancy are far more significant for a woman than a man. Hence, gathering information directly from the wives of employees would be a valuable technique in industrial male reproductive monitoring programs. Therefore, even human models are not perfect, and animal models can provide useful information for man.
As with radiation exposure, to be discussed later, men taking anticancer drugs (many of which are radiomimetic) are too unwell to mate. If they wait until the damaged cells are removed through spermatogenic cycling, limited or no abnormalities can occur. This is not the case for treated male animals, which mate regardless of treatment, and late congenital ‘teratogenic-like’ anomalies can occur.42,43,44,46,47,48,49 A report is available on congenital anomalies in the children of cancer survivors.54 Their findings offer strong evidence that the children of cancer survivors are not at a significantly increased risk for congenital anomalies stemming from their parent's exposure to mutagenic cancer treatments. This information is important for counseling cancer survivors planning to have children. Mulvihill55 discusses preconception exposure to mutagens covering medical and other exposures to radiation and chemicals. Winther et al.56 examined genetic disease in the children of 472 Danish survivors of childhood and adolescent cancer and their 1037 pregnancies. The study reported that mutagenic chemotherapy and radiotherapy doses to the gonads were not associated with genetic defects in the children of cancer survivors. However, larger studies may be needed to further explore the potential associations between high-dose pelvic irradiation and specific adverse pregnancy outcomes. Thus, the exposure of rodent male germ cells to chemicals (and radiation, discussed later) can result in embryonic death, mutation, malformations and cancer, as we have confirmed above for chemicals and other adverse effects (e.g. functional disorders) in the offspring.51 Another area of concern is chromosomal mosaicism, which has been studied in mice.
CHROMOSOMAL MOSAICISM
Chromosomal mosaicism could occur in human preimplantation embryos and could result in spontaneous abortions.57 Authors used multicolor fluorescence in situ hybridization painting to investigate whether paternally transmitted chromosomal aberrations result in mosaicism in mouse two-cell embryos. Paternal exposure to acrylamide, an important industrial chemical also found in tobacco smoke and generated during the cooking process of starchy foods, produced significant increases in chromosomally defective two-cell embryos. The effects were transient, primarily affecting the postmeiotic stages of spermatogenesis. Comparisons with their previous study of zygotes demonstrated similar frequencies of chromosomally abnormal zygotes and two-cell embryos, suggesting that there was no apparent selection against numerical or structural chromosomal aberrations. However, the majority of affected two-cell embryos were mosaics, exhibiting different chromosomal abnormalities in the two blastomeric metaphases. Chromosomal aberrations analyzed in zygotes and two-cell embryos exhibited a tendency for loss of acentric fragments during the first mitotic division of embryogenesis, whereas both dicentrics and translocations apparently underwent proper segregation. These findings suggested that embryonic development can proceed up to the end of the second cell cycle of development in the presence of abnormal paternal chromosomes and that even dicentrics can persist through cell division. The high incidence of chromosomally mosaic two-cell embryos also suggested that the first mitotic division of embryogenesis is prone to missegregation errors and that paternally transmitted chromosomal abnormalities increase the risk of missegregation, leading to embryonic mosaicism.
PATERNAL EXPOSURE TO IONIZING RADIATION
The hazards associated with the dangers of exposure to ionizing radiation have been recognized for nearly a century, but much interest was aroused when a cluster of leukemia cases was identified in the 1980s in young children living in Seascale close to the nuclear processing plant at Sellafield in West Cumbria, UK.58 Clusters were sought and identified in the vicinity of other nuclear establishments in the UK and other countries,59,60,61 although leukemia clusters were not found exclusively in the vicinity of nuclear reactors.62 The reports of Gardner et al.63,64 however, suggested that occupational exposure of men at Sellafield might be linked to increased susceptibility to leukemia or non-Hodgkin's lymphoma in children. COMARE,65 the Committee on Medical Aspects of Radiation in the Environment appointed by the UK Department of Health, could find no epidemiological evidence from studies in other locations, including studies elsewhere in Cumbria,66 near a similar plant at Dounreay in Scotland,67 around the Aldermaston and Burghfield nuclear weapons establishments in England59 or from Ontario.68,69,70 Despite these negative surveys, the Gardner report was used in a civil court case on behalf of two of the alleged victims of paternal irradiation at Seascale against British Nuclear Fuels.71 The case foundered on ‘the balance of probabilities’.72 A survey by Parker et al. and parallel studies performed by the Great Britain Health Safety Executive73,74,75 corroborated this report; see also Draper et al.76 Gardner et al.63,64 acknowledged that possible exposure to internally incorporated radionuclides had not been taken into account and that such sources could possibly explain their findings. The relative risk factor for leukemia and non-Hodgkin's Lymphoma for all surveys other than Seascale were not significantly different from 1.0;77,78 however, for children born to exposed residents of Seascale, the risk was 36 times higher than the control level.
In 1993, Roman and his colleagues,59 in a small case-control study among children living in West Berkshire, North Hampshire, UK, also came to the conclusion that preconception, paternal irradiation might lead to an increased risk of cancer. As early as 1966, Graham et al.79 came to the same conclusion from a study following diagnostic X-irradiations, but after maternal exposure. However, the Oxford Survey80 found no association in 4542 children who died of cancer with parental exposure. Additionally, studies of the population after the atomic bombs in Hiroshima and Nagasaki revealed no increase in malignancy with increasing parental gonadal dose.81,82,83 Furthermore, in 1800 offspring from male cancer patients who received radiotherapy in the UK84 and the USA,39,85 only heritable retinoblastomas were found. In the human situation, e.g. after radiation therapy, males are advised not to indulge in sexual practices for a few months to allow progression of newly formed germ cells through the next spermatogenic cycle, and often men do not feel well enough for sexual activity.54,56 Thus, the animal models do not exactly reflect the human situation because animals are constantly mating. This might help explain the generally negative results in cancer therapy patients. Dubrova et al.86 reported that radiation exposure post-Chernobyl was found to have induced heritable mutations in the male germ line (cf. with recent results of Laubenthal et al.5 and Linschooten et al.6 reviewed above after smoking and BaP exposure).
There is evidence for the transmission of abnormalities and susceptibility after paternal exposure from experimental X-ray studies in mice.50,87 In the study by Nomura, the tumors were clearly heritable, as shown by F2 transmission. There were increases in leukemia up to 18-fold for one strain, when radiation was given at the spermatogonial stage.88 These findings have been criticized89,90 because of a lack of simultaneous controls, the seasonal variation in tumourigenicity, the small level of pneumonia in the experimental groups but not the control group, and a possible stimulation of preexisting tumors predisposing mutations. Furthermore, Cattanach et al.91 in experiments that mimicked Nomura's, but did not use the same strain, could not confirm his findings; and Cosgrove et al.92 found that offspring of males irradiated as spermatogonia lived normal lifespans. Takahashi et al.93 reported inheritance by first generation male offspring but not female offspring of a cancer-prone genetic trait after irradiation with californium-252 neutrons of the fathers. Nomura94 found that a subsequent challenge to the offspring with urethane, known to induce lung tumors, stimulated large clusters of tumor nodules. Cattanach et al.95 again mimicking Nomura's work, failed to corroborate these findings. However, similar studies demonstrated96,97 that a second insult could promote skin cancers.
Lord et al.98,99 treated male mice with plutonium-239 and generated offspring in which hematopoiesis was assayed. For the secondary insult, female mice were treated with either methyl nitrosourea (a leukemia-inducing drug) or a leukemia-inducing sublethal dose of 3.3 Gy g-rays. To overcome the criticisms of earlier work, parallel control groups and two different mouse strains with zero spontaneous leukemia incidence rates were used at two different centers. Each mouse could be considered as individual. The inter-animal variation was larger than usual, and the balance of hematopoiesis in the offspring was disturbed in a significant number of mice. These mice also exhibited a significant trend to higher levels of chromosomal aberrations in bone marrow cells. In mice secondarily insulted with methyl nitrosourea or sub-lethal radiation, there was a significant increase in the rates of lymphohematopoietic malignancy and a change in the disease patterns, e.g., myeloid leukemia now predominated, whereas in methyl nitrosourea-treated mice, myeloid leukemia normally only developed secondarily to thymic tumors. Thus, overall, experimental evidence from radiation studies does suggest the possibility of transmission of paternally mediated congenital effects.
EPIDEMIOLOGICAL APPROACHES
Epidemiological studies on male-mediated developmental toxicity may focus on links between the environmental exposure of the male and pregnancy failures or disorders in the offspring. In contrast, the focus may be on biological markers of male fecundity and developmental disorders. Only rarely is there a combination of these two approaches.100 The demonstration of the effects of male exposure on the male reproductive system and subsequent demonstration of links between the observed abnormalities of the male system would provide strong evidence for causal associations between exposure and outcomes.100
A study by Ramlau-Hansen et al.101 examined male-mediated infertility in the sons of building painters and gardeners compared with the sons of bricklayers, carpenters and electricians in a Danish nationwide register-based follow-up study of men born between 1965 and 1984 (n = 22 978). Their fathers had worked as gardeners, painters, bricklayers, carpenters or electricians in the year before their sons’ births. Cases of infertility were identified by Danish registers, and participants were followed for 24 years after their 20th birthday. The sons of gardeners did not exhibit an increased risk, and the hazard ratios for the sons of painters fluctuated around the null value. When compared with the others, neither group exhibited an increased risk of infertility among the next generation of males.
Epidemiological studies of paternal exposure to organic solvents and pesticides have been associated with birth defects, spontaneous abortion and childhood cancer, and these associations may be due to a direct effect on sperm DNA. Alternatively, however, an indirect effect from toxicants transmitted to the mother from contaminated clothing or via seminal fluid cannot be excluded.102,103,104
A report from the ‘Born in Bradford’ study (UK) by Sheridan et al.105 investigated a prospective multi-ethnic birth cohort of 13 776 babies and their families, in which recruitment was undertaken between 2007 and 2011. Of 11 396 babies for whom questionnaire data were available, 386 (3%) exhibited a congenital anomaly (causing infant death or disability). The rates for congenital anomalies were 305.7 per 10 000 live births compared with a national rate of 165.9 per 10 000. The risk was greater for families of Pakistani origin than for those of white British origin. Approximately 31% of anomalies in children of Pakistani origin could be attributed to consanguinity. In these cousin marriages, the risk remained even after adjustment for deprivation. The extent of the contribution of the father is unclear, and the study was not designed as a male-mediated study, but high levels of parental educational attainment reduced the risk in all ethnic groups.105
QUESTIONS FROM THE PANEL
Q1: What is known about the late effects in the offspring?
A1a: In rodent models (rats and mice) where the male has been treated and mated with untreated females as in a conventional dominant lethal assay, congenital male ‘teratogenic’ defects can be detected the day before term.41 They cannot be detected at mid-term as is the usual time for sacrifice. This late killing was suggested as a way of detecting ‘teratogenic’ defects in the male. They can be measured and scored in the same way as teratogenic defects after in utero exposure of females.42,43,44,46,47,48,49
A1b: If delayed effects in the offspring are considered as childhood cancers, cognitive developmental defects, growth disorders and the like, then in humans after radiation it was thought that exposed fathers had given rise to leukemia in their children.63,64 However, the case was thrown out of court because it foundered on ‘the balance of probabilities’.72 On examining for late effects in offspring of fathers treated for cancer, these late effects were not found.54,56,80,81,82,83 Only heritable blastomas were found.84,85 Generally, treated fathers are advised to wait for several months after treatment or until they feel well enough to mate. This allows damaged sperm to be removed during the spermatogenic cycle. In contrast, in the case of chronic low dose exposure in the workplace or environmentally, then mating is continuous and there may be more of a chance of delayed effects occurring. However, the sons of Danish gardeners did not have an increased risk, and hazard ratios for sons of painters fluctuated around the null value. Both groups when compared to the sons of bricklayers, carpenters and electricians were not related to an increased risk of infertility among the next generation of males.101
Q2: Is there only a suggestion or is there any proof of male-mediated developmental toxicity in humans?
A2: Laubenthal et al.5 showed that paternal preconceptional cigarette smoking induced male-mediated transgenerational alterations in fetal lymphocytes in the offspring and Dubrova et al.86 showed minisatellite mutations in the germ line of exposed fathers after the Chernobyl accident, but strong evidence that male environmental exposure may cause disease in humans is still limited.
CONCLUSIONS
Paternally transmitted damage to the offspring is recognized as a complex issue with genetic as well as epigenetic components.32,106 From the human data, we know that fathers should not smoke during conception because DNA damage can be transmitted to the cells of the offspring.5 Cigarette smoke behaves as a human germ cell mutagen, as was also confirmed by Linschooten et al.6 who demonstrated that paternal smoking caused increased heritable germ line minisatellite mutations. Dubrova et al.86 demonstrated that minisatellite mutations were found in survivors exposed to post-Chernobyl radiation and that radiation also behaves as a human germ cell mutagen. To detect male-mediated effects, it is necessary to consider the exposure dose and the phase at which the sensitive germ cell responds. In human cancer studies, treated individuals are usually too ill to participate in sexual activity after treatment and wait for a recovery period, during which exposed damaged cells may have been removed through spermatogenic cycling. This is not the case for animal studies, in which mating is continuous regardless of treatment. This difference may help explain the negative results in the human cancer studies.55,56
Some occupational or environmental exposures that exist as low-dose chronic exposures could, however, have a different impact. Epidemiological studies have suggested that the sons of gardeners and painters do not have an increased risk of infertility even though paternal exposure to organic solvents and pesticides has been shown to be associated with birth defects, spontaneous abortion and childhood cancer.102,103,104 Ethnicities and the associated different dietary and cultural habits can affect congenital anomaly rates.105 The large animal studies of the 1980s and 1990s demonstrated that damage could be transmitted through the father to both the first42,43,44,46,47,48,49 and second generation.50 It was originally assumed that this transmission was limited to genetically-based inheritance. However, it is possible that there may be epigenetic components of inheritance that scientists were not wholly aware of at the time. Future studies should help delineate the differences between the genetic and epigenetic components.
AUTHOR CONTRIBUTIONS
DA was responsible for the concept and framework of the paper. DA, TES and AB participated in collecting and evaluating the data and writing the first draft. DA was responsible for the final editing. All authors read and approved the final manuscript.
COMPETING INTERESTS
The authors declare that they have no competing interests.
ACKNOWLEDGMENTS
DA is grateful to ICI Plc and MAFF (Ministry of Agriculture Fisheries and Food) for the animal studies she conducted in the 1980s and 1990s and more recently to the European Union Integrated Project New Generis, 6th Framework Programme, Priority 5: food Quality and Safety, contract FOOD CT 2005 016320 (http://www.newgeneris.org), for the mother, father and baby triad study.
REFERENCES
- 1.Miller D, Brinkworth M, Iles D. Paternal DNA packaging in spermatozoa: more than the sum of its parts? DNA, histones, protamines and epigenetics. Reproduction. 2010;139:287–301. doi: 10.1530/REP-09-0281. [DOI] [PubMed] [Google Scholar]
- 2.Jenkins TG, Carrell DT. The sperm epigenome and potential implications for the developing embryo. Reproduction. 2012;143:727–34. doi: 10.1530/REP-11-0450. [DOI] [PubMed] [Google Scholar]
- 3.Keightley PD. Rates and fitness consequences of new mutations in humans. Genetics. 2012;190:295–304. doi: 10.1534/genetics.111.134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012;488:471–5. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laubenthal J, Zlobinskaya O, Poterlowicz K, Baumgartner A, Gdula MR, et al. Cigarette smoke-induced transgenerational alterations in genome stability in cord blood of human F1 offspring. FASEB J. 2012;26:3946–56. doi: 10.1096/fj.11-201194. [DOI] [PubMed] [Google Scholar]
- 6.Linschooten JO, Verhofstad N, Gutzkow K, Olsen AK, Yauk C, et al. Paternal lifestyle as a potential source of germline mutations transmitted to offspring. FASEB J. 2013;27:2873–9. doi: 10.1096/fj.13-227694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sipinen V, Laubenthal J, Baumgartner A, Cemeli E, Linschooten JO, et al. In vitro evaluation of baseline and induced DNA damage in human sperm exposed to benzo[a] pyrene or its metabolite benzo[a] pyrene-7,8-diol-9,10-epoxide, using the comet assay. Mutagenesis. 2010;25:417–25. doi: 10.1093/mutage/geq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linschooten JO, Laubenthal J, Cemeli E, Baumgartner A, Anderson D, et al. Incomplete protection of genetic integrity of mature spermatozoa against oxidative stress. Reprod Toxicol. 2011;32:106–11. doi: 10.1016/j.reprotox.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Linschooten JO, Van Schooten FJ, Baumgartner A, Cemeli E, Van Delft J, et al. Use of spermatozoal mRNA profiles to study gene-environment interactions in human germ cells. Mutat Res. 2009;667:70–6. doi: 10.1016/j.mrfmmm.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 10.Oliva R. Protamines and male infertility. Hum Reprod Update. 2006;12:417–35. doi: 10.1093/humupd/dml009. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Gonzalez R, Moreira PN, Perez-Crespo M, Sanchez-Martin M, Ramirez MA, et al. Long-term effects of mouse intracytoplasmic sperm injection with DNA-fragmented sperm on health and behavior of adult offspring. Biol Reprod. 2008;78:761–72. doi: 10.1095/biolreprod.107.065623. [DOI] [PubMed] [Google Scholar]
- 12.Krawetz SA, Kruger A, Lalancette C, Tagett R, Anton E, et al. A survey of small RNAs in human sperm. Hum Reprod. 2011;26:3401–12. doi: 10.1093/humrep/der329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar M, Kumar K, Jain S, Hassan T, Dada R. Novel insights into the genetic and epigenetic paternal contribution to the human embryo. Clinics (Sao Paulo) 2013;68(Suppl 1):5–14. doi: 10.6061/clinics/2013(Sup01)02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Donnell KA, Boeke JD. Mighty Piwis defend the germline against genome intruders. Cell. 2007;129:37–44. doi: 10.1016/j.cell.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammoud SS, Nix DA, Zhang H, Purwar J, Carrell DT, et al. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–8. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y, Wang HY, Wan FC, Liu FJ, Liu J, et al. Deep sequencing analysis of small non-coding RNAs reveals the diversity of microRNAs and piRNAs in the human epididymis. Gene. 2012;497:330–5. doi: 10.1016/j.gene.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 17.Boerke A, Dieleman SJ, Gadella BM. A possible role for sperm RNA in early embryo development. Theriogenology. 2007;68(Suppl 1):S147–55. doi: 10.1016/j.theriogenology.2007.05.058. [DOI] [PubMed] [Google Scholar]
- 18.Khraiwesh B, Arif MA, Seumel GI, Ossowski S, Weigel D, et al. Transcriptional control of gene expression by microRNAs. Cell. 2010;140:111–22. doi: 10.1016/j.cell.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 19.Lizarraga D, Gaj S, Brauers KJ, Timmermans L, Kleinjans JC, et al. Benzo[a] pyrene-induced changes in microRNA-mRNA networks. Chem Res Toxicol. 2012;25:838–49. doi: 10.1021/tx2003799. [DOI] [PubMed] [Google Scholar]
- 20.Meunier L, Siddeek B, Vega A, Lakhdari N, Inoubli L, et al. Perinatal programming of adult rat germ cell death after exposure to xenoestrogens: role of microRNA miR-29 family in the down-regulation of DNA methyltransferases and Mcl-1. Endocrinology. 2012;153:1936–47. doi: 10.1210/en.2011-1109. [DOI] [PubMed] [Google Scholar]
- 21.Hou L, Wang D, Baccarelli A. Environmental chemicals and microRNAs. Mutat Res. 2011;714:105–12. doi: 10.1016/j.mrfmmm.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonkoly E, Pivarcsi A. MicroRNAs in inflammation and response to injuries induced by environmental pollution. Mutat Res. 2011;717:46–53. doi: 10.1016/j.mrfmmm.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Brinkworth MH, Anderson D, McLean AE. Effects of dietary imbalances on spermatogenesis in CD-1 mice and CD rats. Food Chem Toxicol. 1992;30:29–35. doi: 10.1016/0278-6915(92)90133-6. [DOI] [PubMed] [Google Scholar]
- 24.Anderson LM, Riffle L, Wilson R, Travlos GS, Lubomirski MS, et al. Preconceptional fasting of fathers alters serum glucose in offspring of mice. Nutrition. 2006;22:327–31. doi: 10.1016/j.nut.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24–9R. doi: 10.1203/pdr.0b013e3180457684. [DOI] [PubMed] [Google Scholar]
- 26.Schagdarsurengin U, Paradowska A, Steger K. Analysing the sperm epigenome: roles in early embryogenesis and assisted reproduction. Nat Rev Urol. 2012;9:609–19. doi: 10.1038/nrurol.2012.183. [DOI] [PubMed] [Google Scholar]
- 27.Arpanahi A, Brinkworth M, Iles D, Krawetz SA, Paradowska A, et al. Endonuclease-sensitive regions of human spermatozoal chromatin are highly enriched in promoter and CTCF binding sequences. Genome Res. 2009;19:1338–49. doi: 10.1101/gr.094953.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Curley JP, Mashoodh R, Champagne FA. Epigenetics and the origins of paternal effects. Horm Behav. 2011;59:306–14. doi: 10.1016/j.yhbeh.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molaro A, Hodges E, Fang F, Song Q, McCombie WR, et al. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell. 2011;146:1029–41. doi: 10.1016/j.cell.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM. A unique configuration of genome-wide DNA methylation patterns in the testis. Proc Natl Acad Sci U S A. 2007;104:228–33. doi: 10.1073/pnas.0607521104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Houshdaran S, Cortessis VK, Siegmund K, Yang A, Laird PW, et al. Widespread epigenetic abnormalities suggest a broad DNA methylation erasure defect in abnormal human sperm. PloS One. 2007;2:e1289. doi: 10.1371/journal.pone.0001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skinner MK. What is an epigenetic transgenerational phenotype. F3 or F2? Reprod Toxicol. 2008;25:2–6. doi: 10.1016/j.reprotox.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakai Y, Suetake I, Itoh K, Mizugaki M, Tajima S, et al. Expression of DNA methyltransferase (Dnmt1) in testicular germ cells during development of mouse embryo. Cell Struct Funct. 2001;26:685–91. doi: 10.1247/csf.26.685. [DOI] [PubMed] [Google Scholar]
- 34.Marques CJ, Joao Pinho M, Carvalho F, Bieche I, Barros A, et al. DNA methylation imprinting marks and DNA methyltransferase expression in human spermatogenic cell stages. Epigenetics. 2011;6:1354–61. doi: 10.4161/epi.6.11.17993. [DOI] [PubMed] [Google Scholar]
- 35.Balls M, Fentem JH. The Fund for the Replacement of Animals in Medical Experiments (FRAME): 23 years of campaigning for the Three Rs. In: Reinhardt CA, editor. Alternatives to Animal Testing: New Ways in the Biomedical Sciences, Trends and Progress. Germany: VCH Verlagsgesellschaft; 1994. pp. 45–55. [Google Scholar]
- 36.Russell WM, Burch RL, Hume CW. Potters Bar: Universities Federation for Animal Welfare; 1992. The principles of humane experimental technique. [Google Scholar]
- 37.Sorahan T, Prior P, Lancashire RJ, Faux SP, Hulten MA, et al. Childhood cancer and parental use of tobacco: deaths from 1971 to 1976. Br J Cancer. 1997;76:1525–31. doi: 10.1038/bjc.1997.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sorahan T, Lancashire RJ, Hulten MA, Peck I, Stewart AM. Childhood cancer and parental use of tobacco: deaths from 1953 to 1955. Br J Cancer. 1997;75:134–8. doi: 10.1038/bjc.1997.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ji BT, Shu XO, Linet MS, Zheng W, Wacholder S, et al. Paternal cigarette smoking and the risk of childhood cancer among offspring of nonsmoking mothers. J Natl Cancer Inst. 1997;89:238–44. doi: 10.1093/jnci/89.3.238. [DOI] [PubMed] [Google Scholar]
- 40.Lefebvre L, Viville S, Barton SC, Ishino F, Keverne EB, et al. Abnormal maternal behaviour and growth retardation associated with loss of the imprinted gene Mest. Nat Genet. 1998;20:163–9. doi: 10.1038/2464. [DOI] [PubMed] [Google Scholar]
- 41.Knudsen I, Hansen EV, Meyer OA, Poulsen E. A proposed method for the simultaneous detection of germ-cell mutations leading to fetal death (dominant lethality) and of malformations (male teratogenicity) in mammals. Mutat Res. 1977;48:267–70. doi: 10.1016/0027-5107(77)90168-3. [DOI] [PubMed] [Google Scholar]
- 42.Jenkinson PC, Anderson D, Gangolli SD. Increased incidence of abnormal foetuses in the offspring of cyclophosphamide-treated male mice. Mutat Res. 1987;188:57–62. doi: 10.1016/0165-1218(87)90115-7. [DOI] [PubMed] [Google Scholar]
- 43.Jenkinson PC, Anderson D. Malformed foetuses and karyotype abnormalities in the offspring of cyclophosphamide and allyl alcohol-treated male rats. Mutat Res. 1990;229:173–84. doi: 10.1016/0027-5107(90)90091-h. [DOI] [PubMed] [Google Scholar]
- 44.Francis AJ, Anderson D, Evans JG, Jenkinson PC, Godbert P. Tumours and malformations in the adult offspring of cyclophosphamide-treated and control male rats-preliminary communication. Mutat Res. 1990;229:239–46. doi: 10.1016/0027-5107(90)90097-n. [DOI] [PubMed] [Google Scholar]
- 45.Trasler JM, Hales BF, Robaire B. Paternal cyclophosphamide treatment of rats causes fetal loss and malformations without affecting male fertility. Nature. 1985;316:144–6. doi: 10.1038/316144a0. [DOI] [PubMed] [Google Scholar]
- 46.Anderson D, Edwards AJ, Brinkworth MH, Hughes JA. Male-mediated F1 effects in mice exposed to 1,3-butadiene. Toxicology. 1996;113:120–7. doi: 10.1016/0300-483x(96)03436-1. [DOI] [PubMed] [Google Scholar]
- 47.Brinkworth MH, Anderson D, Hughes JA, Jackson LI, Yu TW, et al. Genetic effects of 1,3-butadiene on the mouse testis. Mutat Res. 1998;397:67–75. doi: 10.1016/s0027-5107(97)00196-6. [DOI] [PubMed] [Google Scholar]
- 48.Anderson D, Hughes JA, Edwards AJ, Brinkworth MH. A comparison of male-mediated effects in rats and mice exposed to 1,3-butadiene. Mutat Res. 1998;397:77–84. doi: 10.1016/s0027-5107(97)00197-8. [DOI] [PubMed] [Google Scholar]
- 49.Edwards AJ, Anderson D, Brinkworth MH, Myers B, Parry JM. An investigation of male-mediated F1 effects in mice treated acutely and sub-chronically with urethane. Teratog Carcinog Mutagen. 1999;19:87–103. [PubMed] [Google Scholar]
- 50.Nomura T. Parental exposure toxrays and chemicals induces heritable tumours and anomalies in mice. Nature. 1982;296:575–7. doi: 10.1038/296575a0. [DOI] [PubMed] [Google Scholar]
- 51.Nomura T. Transgenerational effects from exposure to environmental toxic substances. Mutat Res. 2008;659:185–93. doi: 10.1016/j.mrrev.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 52.Infante PF, Wagoner JK, McMichael AJ, Waxweiler RJ, Falk H. Genetic risks of vinyl chloride. Lancet. 1976;1:734–5. doi: 10.1016/s0140-6736(76)93103-2. [DOI] [PubMed] [Google Scholar]
- 53.Buffler PA, Aase JM. Genetic risks and environmental surveillance: epidemiological aspects of monitoring industrial populations for environmental mutagens. J Occup Med. 1982;24:305–14. [PubMed] [Google Scholar]
- 54.Signorello LB, Mulvihill JJ, Green DM, Munro HM, Stovall M, et al. Congenital anomalies in the children of cancer survivors: a report from the childhood cancer survivor study. J Clin Oncol. 2012;30:239–45. doi: 10.1200/JCO.2011.37.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mulvihill JJ. Preconception exposure to mutagens: medical and other exposures to radiation and chemicals. J Community Genet. 2012;3:205–11. doi: 10.1007/s12687-012-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winther JF, Olsen JH, Wu H, Shyr Y, Mulvihill JJ, et al. Genetic disease in the children of Danish survivors of childhood and adolescent cancer. J Clin Oncol. 2012;30:27–33. doi: 10.1200/JCO.2011.35.0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marchetti F, Bishop J, Lowe X, Wyrobek AJ. Chromosomal mosaicism in mouse two-cell embryos after paternal exposure to acrylamide. Toxicol Sci. 2009;107:194–205. doi: 10.1093/toxsci/kfn209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Black D. London: HMSO; 1984. Investigation of the possible increased incidence of cancer in West Cumbria: report of the Independent Advisory Group. [Google Scholar]
- 59.Roman E, Watson A, Beral V, Buckle S, Bull D, et al. Case-control study of leukaemia and non-Hodgkin's lymphoma among children aged 0-4 years living in west Berkshire and north Hampshire health districts. BMJ. 1993;306:615–21. doi: 10.1136/bmj.306.6878.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cook-Mozaffari PJ, Darby SC, Doll R, Forman D, Hermon C, et al. Geographical variation in mortality from leukaemia and other cancers in England and Wales in relation to proximity to nuclear installations, 1969-78. Br J Cancer. 1989;59:476–85. doi: 10.1038/bjc.1989.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bithell JF, Dutton SJ, Draper GJ, Neary NM. Distribution of childhood leukaemias and non-Hodgkin's lymphomas near nuclear installations in England and Wales. BMJ. 1994;309:501–5. doi: 10.1136/bmj.309.6953.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buckley JD, Robison LL, Swotinsky R, Garabrant DH, LeBeau M, et al. Occupational exposures of parents of children with acute nonlymphocytic leukemia: a report from the Childrens Cancer Study Group. Cancer Res. 1989;49:4030–7. [PubMed] [Google Scholar]
- 63.Gardner MJ, Snee MP, Hall AJ, Powell CA, Downes S, et al. Results of case-control study of leukaemia and lymphoma among young people near Sellafield nuclear plant in West Cumbria. BMJ. 1990;300:423–9. doi: 10.1136/bmj.300.6722.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gardner MJ, Hall AJ, Snee MP, Downes S, Powell CA, et al. Methods and basic data of case-control study of leukaemia and lymphoma among young people near Sellafield nuclear plant in West Cumbria. BMJ. 1990;300:429–34. doi: 10.1136/bmj.300.6722.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.COMPARE. Possible effects of paternal preconception irradiation in cancer, Committee on Medical Aspects of Radiation in the Environment: committee on Medical Aspects of Radiation in the Environment. 1996 [Google Scholar]
- 66.Wakeford R, Parker L. Leukaemia and non-Hodgkin's lymphoma in young persons resident in small areas of West Cumbria in relation to paternal preconceptional irradiation. Br J Cancer. 1996;73:672–9. doi: 10.1038/bjc.1996.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Urquhart JD, Black RJ, Muirhead MJ, Sharp L, Maxwell M, et al. Case-control study of leukaemia and non-Hodgkin's lymphoma in children in Caithness near the Dounreay nuclear installation. BMJ. 1991;302:687–92. doi: 10.1136/bmj.302.6778.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kinlen LJ. Childhood leukaemia and non-Hodgkins lymphoma in young people living close to nuclear reprocessing sites. Biomed Pharmacother. 1993;47:429–34. doi: 10.1016/0753-3322(93)90338-l. [DOI] [PubMed] [Google Scholar]
- 69.Kinlen LJ. Can paternal preconceptional radiation account for the increase of leukaemia and non-Hodgkin's lymphoma in Seascale? BMJ. 1993;306:1718–21. doi: 10.1136/bmj.306.6894.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kinlen LJ, Clarke K, Balkwill A. Paternal preconceptional radiation exposure in the nuclear industry and leukaemia and non-Hodgkin's lymphoma in young people in Scotland. BMJ. 1993;306:1153–8. doi: 10.1136/bmj.306.6886.1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wakeford R, Tawn EJ. Childhood leukaemia and Sellafield: the legal cases. J Radiol Prot. 1994;14:293–316. [Google Scholar]
- 72.Doll R, Evans HJ, Darby SC. Paternal exposure not to blame. Nature. 1994;367:678–80. doi: 10.1038/367678a0. [DOI] [PubMed] [Google Scholar]
- 73.HSE. HSE investigation of leukaemia and other cancers in the children of male workers at Sellafield: review of results published in October 1993: Great Britain, Health and Safety Executive. 1994 [Google Scholar]
- 74.HSE. HSE investigation of leukaemia and other cancers in the children of male workers at Sellafield: HSE Books. 1993 [Google Scholar]
- 75.Parker L, Craft AW, Smith J, Dickinson H, Wakeford R, et al. Geographical distribution of preconceptional radiation doses to fathers employed at the Sellafield nuclear installation, West Cumbria. BMJ. 1993;307:966–71. doi: 10.1136/bmj.307.6910.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Draper GJ, Little MP, Sorahan T, Kinlen LJ, Bunch KJ, et al. Cancer in the offspring of radiation workers: a record linkage study. BMJ. 1997;315:1181–8. [PMC free article] [PubMed] [Google Scholar]
- 77.Little MP, Wakeford R, Charles MW. A comparison of the risks of leukaemia in the offspring of the Sellafield workforce born in Seascale and those born elsewhere in West Cumbria with the risks in the offspring of the Ontario and Scottish workforces and the Japanese bomb survivors. J Radiol Prot. 1994;14:187–201. [Google Scholar]
- 78.Little MP, Wakeford R, Charles MW, Andersson M. A comparison of the risks of leukaemia and non-Hodgkin›s lymphoma in the first generation offspring (F1) of the Danish Thorotrast patients with those observed in other studies of parental pre-conception irradiation. J Radiol Prot. 1996;16:25–36. [Google Scholar]
- 79.Graham S, Levin ML, Lilienfeld AM, Schuman LM, Gibson R, et al. Preconception, intrauterine, and postnatal irradiation as related to leukemia. Natl Cancer Inst Monogr. 1966;19:347–71. [PubMed] [Google Scholar]
- 80.Kneale GW, Stewart AM. Pre-conception X-rays and childhood cancers. Br J Cancer. 1980;41:222–6. doi: 10.1038/bjc.1980.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshimoto Y. Cancer risk among children of atomic bomb survivors. A review of RERF epidemiologic studies. Radiation Effects Research Foundation. JAMA. 1990;264:596–600. [PubMed] [Google Scholar]
- 82.Yoshimoto Y, Neel JV, Schull WJ, Kato H, Soda M, et al. Malignant tumors during the first 2 decades of life in the offspring of atomic bomb survivors. Am J Hum Genet. 1990;46:1041–52. [PMC free article] [PubMed] [Google Scholar]
- 83.Little MP, Wakeford R, Charles MW. An analysis of leukaemia, lymphoma and other malignancies together with certain categories of non-cancer mortality in the first generation offspring (F1) of the Japanese bomb survivors. J Radiol Prot. 1994;14:203–18. [Google Scholar]
- 84.Hawkins MM, Draper GJ, Smith RA. Cancer among 1,348 offspring of survivors of childhood cancer. Int J Cancer. 1989;43:975–8. doi: 10.1002/ijc.2910430604. [DOI] [PubMed] [Google Scholar]
- 85.Mulvihill JJ, Myers MH, Connelly RR, Byrne J, Austin DF, et al. Cancer in offspring of long-term survivors of childhood and adolescent cancer. Lancet. 1987;2:813–7. doi: 10.1016/s0140-6736(87)91012-9. [DOI] [PubMed] [Google Scholar]
- 86.Dubrova YE, Nesterov VN, Krouchinsky NG, Ostapenko VA, Neumann R, et al. Human minisatellite mutation rate after the Chernobyl accident. Nature. 1996;380:683–6. doi: 10.1038/380683a0. [DOI] [PubMed] [Google Scholar]
- 87.Kirk KM, Lyon MF. Induction of congenital malformations in the offspring of male mice treated with X-rays at pre-meiotic and post-meiotic stages. Mutat Res. 1984;125:75–85. doi: 10.1016/0027-5107(84)90034-4. [DOI] [PubMed] [Google Scholar]
- 88.Nomura T. Paternal exposure to radiation and offspring cancer in mice: reanalysis and new evidence. J Radiat Res. 1991;32(Suppl 2):64–72. doi: 10.1269/jrr.32.supplement2_64. [DOI] [PubMed] [Google Scholar]
- 89.Selby PB. New York: Plenum Press; 1990. Experimental induction of dominant mutations in mammals by ionizing radiations and chemicals. [Google Scholar]
- 90.Cox R. Transgeneration carcinogenesis: are there genes that break the rules? Radiat Prot Bull. 1992;129:15–23. [Google Scholar]
- 91.Cattanach BM, Patrick G, Papworth D, Goodhead DT, Hacker T, et al. Investigation of lung tumour induction in BALB/cJ mice following paternal X-irradiation. Int J Radiat Biol. 1995;67:607–15. doi: 10.1080/09553009514550721. [DOI] [PubMed] [Google Scholar]
- 92.Cosgrove GE, Selby PB, Upton AC, Mitchell TJ, Steele MH, et al. Lifespan and autopsy findings in the first-generation offspring of X-irradiated male mice. Mutat Res. 1993;319:71–9. doi: 10.1016/0165-1218(93)90032-9. [DOI] [PubMed] [Google Scholar]
- 93.Takahashi T, Watanabe H, Dohi K, Ito A. 252Cf relative biological effectiveness and inheritable effect of fission neutrons in mouse liver tumorigenesis. Cancer Res. 1992;52:1948–53. [PubMed] [Google Scholar]
- 94.Nomura T. X-ray-induced germ-line mutation leading to tumors. Its manifestation in mice given urethane post-natally. Mutat Res. 1983;121:59–65. doi: 10.1016/0165-7992(83)90087-8. [DOI] [PubMed] [Google Scholar]
- 95.Cattanach BM, Papworth D, Patrick G, Goodhead DT, Hacker T, et al. Investigation of lung tumour induction in C3H/HeH mice, with and without tumour promotion with urethane, following paternal X-irradiation. Mutat Res. 1998;403:1–12. doi: 10.1016/s0027-5107(97)00322-9. [DOI] [PubMed] [Google Scholar]
- 96.Vorobtsova IE, Kitaev EM. Urethane-induced lung adenomas in the first-generation progeny of irradiated male mice. Carcinogenesis. 1988;9:1931–4. doi: 10.1093/carcin/9.11.1931. [DOI] [PubMed] [Google Scholar]
- 97.Vorobtsova IE, Aliyakparova LM, Anisimov VN. Promotion of skin tumors by 12-O-tetradecanoylphorbol-13-acetate in two generations of descendants of male mice exposed to X-ray irradiation. Mutat Res. 1993;287:207–16. doi: 10.1016/0027-5107(93)90013-6. [DOI] [PubMed] [Google Scholar]
- 98.Lord BI, Woolford LB, Wang L, Stones VA, McDonald D, et al. Tumour induction by methyl-nitroso-urea following preconceptional paternal contamination with plutonium-239. Br J Cancer. 1998;78:301–11. doi: 10.1038/bjc.1998.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lord BI, Woolford LB, Wang L, McDonald D, Lorimore SA, et al. Induction of lympho-haemopoietic malignancy: impact of preconception paternal irradiation. Int J Radiat Biol. 1998;74:721–8. doi: 10.1080/095530098140998. [DOI] [PubMed] [Google Scholar]
- 100.Bonde JP, Hjøllund HI, Henrikson TB, Jensen TK, Spanò M, et al. Epidemiologic evidence on biological and environmental male factors in embryonic loss. In: Robaire B, Hales BF, editors. Advances in Male Mediated Developmental Toxicity. New York: Kluwer Academic/Plenum Pub; 2003. pp. 25–35. [DOI] [PubMed] [Google Scholar]
- 101.Ramlau-Hansen CH, Stoltenberg CD, Hougaard KS, Parner ET, Toft G, et al. Male-mediated infertility in sons of building painters and gardeners: a nationwide register-based follow-up study. Reprod Toxicol. 2012;34:522–8. doi: 10.1016/j.reprotox.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 102.Savitz DA, Sonnenfeld NL, Olshan AF. Review of epidemiologic studies of paternal occupational exposure and spontaneous abortion. Am J Ind Med. 1994;25:361–83. doi: 10.1002/ajim.4700250306. [DOI] [PubMed] [Google Scholar]
- 103.Logman JF, de Vries LE, Hemels ME, Khattak S, Einarson TR. Paternal organic solvent exposure and adverse pregnancy outcomes: a meta-analysis. Am J Ind Med. 2005;47:37–44. doi: 10.1002/ajim.20102. [DOI] [PubMed] [Google Scholar]
- 104.Friedler G. Paternal exposures: impact on reproductive and developmental outcome. An overview. Pharmacol Biochem Behav. 1996;55:691–700. doi: 10.1016/s0091-3057(96)00286-9. [DOI] [PubMed] [Google Scholar]
- 105.Sheridan E, Wright J, Small N, Corry PC, Oddie S, et al. Risk factors for congenital anomaly in a multiethnic birth cohort: an analysis of the Born in Bradford study. Lancet. 2013;382:1350–59. doi: 10.1016/S0140-6736(13)61132-0. [DOI] [PubMed] [Google Scholar]
- 106.Anway MD, Memon MA, Uzumcu M, Skinner MK. Transgenerational effect of the endocrine disruptor vinclozolin on male spermatogenesis. J Androl. 2006;27:868–79. doi: 10.2164/jandrol.106.000349. [DOI] [PMC free article] [PubMed] [Google Scholar]