

Summary
Living cells have evolved sophisticated signaling networks allowing them to respond to a wide array of external stimuli. Microfluidic devices, facilitating the analysis of signaling networks through precise definition of the cellular microenvironment often lack the capacity of delivering multiple combinations of different signaling cues, thus limiting the throughput of the analysis. To address this limitation, we developed a microfabricated platform combining microfluidic definition of the cell medium composition with dielectrophoretic definition of cell positions and protein microarray-based presentation of diverse signaling inputs. Ligands combined at different concentrations were spotted along with an extracellular matrix protein onto a glass substratum in alignment with an electrode array. This substratum was combined with a polydimethylsiloxane chip for microfluidic control of the soluble medium components, in alignment with the electrode and protein arrays. Endothelial cells were captured by dielectrophoretic force, allowed to attach and spread on the protein spots; and the signaling output of the NF-κB pathway in response to diverse combinations of IGF1 and TNF was investigated, in the absence and presence of variable dose of the pathway inhibitor. The results suggested that cells can be potently activated by immobilized TNF, with IGF1 having a modulating effect, the response that could be abolished to different degrees by the inhibitor. This study demonstrates considerable potential of combining precise cell patterning and liquid medium control with protein microarray technology for complex cell signaling studies in a high-throughput manner.
Introduction
Living cells are faced with complex combinations of environmental signals of diverse nature and types. For instance, the extracellular signaling cues capable of specifying alternative cell behaviors, can be a part of or localized to the extracellular matrix (ECM), be freely diffusible or embedded into the membranes of the neighboring cells. Correct interpretation of the complex totality of these cues can be critical to the cell survival and functioning. However, our understanding of these cell signaling functions is still limited, hampered in part by imperfection of the standard cell biology tools. In particular, few cell-compatible devices exist that can enable simultaneous highly controlled, high-throughput presentation of diverse extracellular stimuli, while also allowing a detailed assessment of the ensuing cell responses.
Arguably, the recent advances in development of microfabrication technology and its microfluidic applications can help address the challenge of rapid and detailed assaying of cell function in general, and cell signaling in particular.1–3 However, even this technology has primarily been used to design experiments specifically addressing a certain aspect of cell function only, e.g., the response to a single specific extracellular cue rather than more realistic combinations of these cues in the presence of well controlled initial cell and media conditions.
Novel tools addressing the above mentioned experimental limitations and thus facilitating the analysis of cell responses to complex combinations of stimuli and conditions might emerge from combination of several techniques, each permitting control of a certain aspect of cell function or a type of external stimuli. For instance, recently it was demonstrated how precisely defined complex ECM protein gradients could be combined with independently controlled gradients of a soluble cue, determining diverse patterns of growth cone migration.4 However, this technology does not allow one to precisely dictate the initial cell positions or use more than a few distinct chemical cues. This limitation can be overcome by locally presenting cells with multiple cues immobilized on the cell adhesion substratum through adoption of protein printing technology, ordinarily used for building protein-protein interaction arrays utilized in high-throughput proteomics analyses. For instance, as shown in a recent study, one can print arrays of ECM molecules and incubate live cells on the resulting protein spots.5 However, a precise definition of the initial cell positioning (other than due to the natural adhesion of cells to the printed ECM molecules) and continuous precise control of extracellular cell medium were lacking in this analysis. Another study extended the repertoire of proteins that can be presented to cells in the form of printed spots to antibodies specific to T-cell-surface proteins and cytokines secreted by T-cells.6 This allowed the investigators to study the cytokines secreted by few cells into their local micro-environment. However, in this case too, the initial positioning of cells was relatively ill-defined and the proteins used were not actively controlling cell function. Cells can be actively captured and positioned using dielectrophoretic (DEP) forces, due to cell polarization in inhomogeneous electric fields.7, 8 We recently showed that one can combine microfluidic control of cell environment with precise DEP-based cell capture to analyze interactions between endothelial cells or endothelial-tumor cells, on the scale of heterotypic and homotypic cell pairs.9 However, this methodology did not allow for high throughput and precise analysis of cell responses to multiple cell cues.
In this report, we demonstrate how microfluidic, dielectrophoretic and protein microarray technologies can be integrated for the first time into one functional cell-compatible device, allowing simultaneous control of the initial cell positioning, presentation of multiple cell cues and control of extracellular medium. Furthermore, we show that the printed proteins can include functional forms of cytokines and growth factors, allowing one to mimic natural capture of these factors by ECM and their presentation to cells in immobilized forms. This analysis extends the repertoire of the molecular factors that can be presented to cells using microarray printing techniques. Finally, we demonstrate that the microfluidic control can permit simultaneous determination of the action of different combination of soluble and surface-bound functional, biologically active molecules.
Materials and Methods
Cell culture and reagents
Immortalized human umbilical vein cells (iHUVEC) were a generous gift from the late Dr. Folkman, Massachusetts General Hospital). Cells were cultured in EGM-2 medium (basal medium plus a supplemental kit from Lonza) supplemented with 5% heat-inhibited fetal bovine serum (FBS). They were incubated at 37 °C and humidified 5% CO2/air atmosphere prior and during analysis. Human TNFα (PHC3015), IGF-1 and IKK inhibitor Sc514 were purchased from Invitrogen, Chemicon International and Calbiochem, respectively.
Design and fabrication of the cell patterning and culturing device
The fabrication procedure of microfluidic and dielectrophoretic parts of the device, schematically illustrated in Fig. 1A, was similar the design and fabrication reported previously for a combined microfludic-dielectrophoretic device for co-culture of heterotypic or homotypic cell pairs.9 Briefly, Indium Tin Oxide (ITO)-coated glass slides (Structure Probe, Inc.) were used to form an array of transparent ITO electrodes on glass substrata. Four electrode arrays were connected to 2 base bars for external electric connection. There were 240 electrodes pairs total in 4 channels (60 electrodes pairs per channel in 4 columns).
Figure 1.

A schematic flow chart of the experimental platform. Electrode array was formed by etching the Indium-Tin-Oxide (ITO) layer on a glass slide. The slide was then derivatized with N-hydroxysuccinimide (NHS) to enable protein immobilization (A). The center of each printed protein spot coincided with the mid-point between the tips of the two paired triangular electrodes. After protein array printing, a PDMS microchannel chip was bonded onto the slide by gentle bonding to form cell culture channels containing the protein spots aligned with electrodes for cell capture and culture in the presence of immobilized signaling ligands (B, C).
Differently from the design in reference 9, the glass substratum was then derivatized with N-hydroxysuccinimide (NHS) for subsequent protein printing. Protocol 9 from reference 10 was followed with minor modifications. NHS, Succinic Anhydride, 3-Aminopropyltriethoxysilane, N,N-Diisopropylethylamine and Dimethylformamide were purchased from Sigma-Aldrich and O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate was purchased from Advanced ChemTech. Piranha solution suggested in reference 10 was not used because it attacks ITO electrodes.
The passive diffusion gradient generating microfluidic channel network was based on the design previously described in 11 and modified for bonding to the ITO electrode containing slide, as described in 9. Briefly, the chip microfluidic network was fabricated by casting Polydimethylsiloxane (PDMS) onto a photoresist-based mold. The mold was created by spin-coating the photoresist SU8-2025 (Microchem, Inc). The SU-8 layer was then patterned with transparency mask and developed to form the microchannel network mold (Fig. 1B). The fluidic channels for cell culturing were 1.2 mm wide and 4 mm long, with different heights for side channels and test channels, 120 and 20 μm respectively, following the general design described. Shallow test channels will reduce the possibility of cross-flow during gradient formation. The method for fabrication of this dual-height microfluidic devices can be found in our previous publications.11, 12 These channels were then aligned with the electrode arrays and printed protein spots as shown in Fig. 1C and described below.
Protein preparation and printing
Solutions of designated protein mixtures were prepared in a 384-well plate (Genetix, New Milton, Hampshire), with 10 μl solution volumes in each well. To prevent the solutions within the multi-well plate and the printed spots from drying out, 12.5% glycerol was added. Oregon Green® 488 conjugate human type IV collagen (Invitrogen) at a concentration of 25 μg/ml was added to assist cell adhesion during cell culturing and to visualize the protein spots. The protein solutions were then spotted onto the NHS functionalized slide with electrode patterns by Capital Bio Personal Arrayer TM 16 (Capital Bio, San Diego, CA) or VersArray ChipWriter (Bio-Rad, Hercules, CA) with the centers of round spots carefully aligned with the midpoints between the paired triangular electrode tips (see Figs. 2A and 2C). To assure the alignment of the protein spots and the electrode array, a few spots of glycerol solutions were printed first. The offset of the spots and the electrodes was measured by visualization with microscopy. The glycerol spots were then washed away by sterile water and the position of the printing pin was adjusted according to the offset. The diameter of the printed spots was approximately 120 μm, corresponding to approximately 0.5 nl of the protein solution. The PDMS microfluidic chip with microchannel network was then carefully aligned with the glass slide, positioning the combined protein spot-ITO electrode network within the cell-culturing channels (Figs. 1 and 2B). The combined device was then bonded for 2 hours at 37 °C, to generate a liquid-tight seal and allow sufficient time for protein conjugation to NHS. Unconjugated protein was washed away by flowing NHS blocking buffer (50 mM ethanolamine in borate buffer, pH 8.0) using the microfluidic network in the chip. Cells were seeded into the chip and patterned precisely onto the printed protein spots by dielectropheretic force generated by non-uniform electric field defined by the triangular electrode array, as described in reference 9 and below (Figs. 2D and 3). After initial cell attachment, the DEP buffer was then exchanged with cell culturing EGM-2 medium by diffusion, as described in reference 9 and below.
Figure 2.

Combining dielectrophoresis-assisted microfluidics with protein micro-arraying for cell signaling studies. A: Four columns of Alexa 488 conjugated goat-anti-mouse antibody printed onto the glass substratum part of the device, with concentration increasing from left to right, 0.001, 0.01, 0.1 and 1 mg/ml, respectively; B: A photograph of a fully assembled functional device, highlighting the electrical wiring access to the on-glass ITO electrodes (parts not readily visible indicated in white broken lines), the PDMS microfluidic chips with channels filled with a food dye and inlets - annotated; C: Demonstration of the alignment of a part of the in-chip protein array (green spots indicating the printed antibody as in (A)) with the triangular electrode array inside one of the fluidic channels; D: One Oregon Green 488 conjugated collagen IV array spot (25 μg/ml coating concentration) with several iHUVECs captured and cultured on it. Cells were stained within the chip to indicate the location of the nucleus (stained with DAPI, blue) and anti-p65 antibody (red).
Figure 3.

Cell patterning onto the printed protein array spots within a device using DEP. The protein spotted, the cells and staining are as in Fig. 2D. A: DEP cell patterning of single cells, cell pairs and a small group of cells around each electrode pair. B: Protein spots (green) with a single cell, cell pair and a small group of cells. The flow rate and electric field controlling DEP were decreased in (B) to allow an increased number of cells to be captured.
In-chip cell patterning and culturing
Cell patterning and culturing process were essentially as reported in reference 9. Briefly, the microfluidic channels were pre-filled with solution made of 90% DEP medium (0.3 Osm sucrose solution with 1% fetal bovine serum and 20 mM HEPES, pH 7.4) and 10% HUVEC culture medium (EGM-2). Cells were detached from cell-culture flasks and resuspended in 200 μl of DEP medium at the density of 5.0×105 cells/ml,8 then introduced into the device through the cell-seeding ports (see Fig. 2B). After the cells entered the channels, an AC electrical potential of 4 Vpp at 3 MHz generated by a function generator (33220A, Agilent) was applied to the electrodes. The flow of this medium continued until every electrode tip captured a cell(s). Then the flow was reversed and the solution was changed to 90% DEP medium/10% culturing medium to wash away weakly adhered cells. The flow and electrical supply were then stopped and cells were allowed to attach in this solution for approximately 40 minutes at 37 °C. Cells were then provided with the culture medium through the feeding ports (see Fig. 2B), which replaced the DEP medium by diffusion, without perturbing cell positioning. Cells were then cultured at 37 °C and humidified 5% CO2/air atmosphere on the stage of a Zeiss inverted microscope (Axiovert 200M), allowing observation of cell spreading and positioning.
In-chip immunostaining
After incubation, cells were stained to analyze cells’ responses to the patterned protein combinations. Rabbit polyclonal anti-p65 antibody C-20 and secondary goat anti-rabbit Alexa Fluor 594 conjugated antibody (A11012) were purchased from Santa Cruz and Invitrogen, respectively. For in-chip immunostaining of the NF-κB (the p65 subunit), cells incubated in chip over time periods indicated in the text and figure legends were washed with ice-cold PBS and fixed by incubation in 4% paraformaldehyde for 20 min. Cells were then permeabilized with 0.1% Triton X-100 (in PBS + Ca/Mg) for 5 min. After blocking with 10% goat serum/PBS, the device was incubated with anti-p65 antibody at 1:100 dilution in 10% goat serum/PBS for one hour followed by the secondary antibody at 1:200 dilution in 10% goat serum/PBS for another hour. Hoechst staining was used to identify cells’ nuclei. The defined cell nucleus area was used for quantitative analysis of the nuclear NF-κB staining intensity and thus NF-κB translocation. All solutions were introduced and removed through the cell seeding ports. Phase-contrast and/or fluorescence images of fixed cells were taken using a cascade CCD camera (Photometrics) attached to the Zeiss microscope, using 40x oil-immersed lens.
Results
Development of the platform
We sought to develop an experimental platform that would combine the advantages of DEP-based cell patterning and microfluidic control embodied in the device described previously9 with protein array printing technology for precise and large-scale presentation of immobilized signaling ligands. The main features of the pervious design are a combination of an arraying of pairs of triangular DEP electrodes with a diffusion based media control in microfluidic device layer. In this study, we combined this basic design with protein array printing, so that the centers of the circular printed protein spots would be positioned at the geometric mid-points between the tips of the ITO electrodes in each electrode pair (Fig. 1). This design was expected to allow precisely controlled initial capture of cells at the central zones of printed protein spots in an arrayed form, allowing for formation of multiple spatially separated cell groups of controlled cell numbers and positioning, coincident with that of the locally defined printed ligand combinations. However, we immediately faced the potentially prohibitive challenge that the conditions usually employed for bonding the microfluidic and cell substratum parts of the device (particularly high ‘baking’ temperature) might promote protein denaturation. Thus it might be impossible to first print the protein spot arrays on the cell adhesion substratum and then seal the device with the microfluidic PDMS layer. Although a previously described technique employing vacuum-generated pressure-based device bonding might have allowed circumventing this problem, we found it to be not robust, convenient and reliable, within the framework of our fluidic network design.13 We therefore attempted instead to moderate the conditions required for formation of a watertight seal between the microfluidic and glass cell substratum parts. After extensive optimization, we found that bonding of the two device parts at 37 °C (the temperature not leading to protein denaturation) for 2 hours was sufficient for this purpose and ensured robust experimentation.
In printing protein microarrays, we followed the general procedures used for arrayed protein spotting,14, 15 relying on preparing of combinations of proteins mixtures in a 384-well plate and printing them with a standard contact micro-arrayer. To insure protein or dye immobilization, the ITO-electrode-glass substratum was pre-derivatized with NHS allowing formation of stable covalent amide groups with the available primary amino-groups of the immobilized proteins.10 We found that ITO electrodes did not interfere with NHS derivatization or protein arraying steps allowing precise co-localization of electrode pairs and protein spots.
Arrays of diverse protein combinations and concentrations and targeted cell capture
One major advantage of the device was the capability to test a serial of dilutions of a given protein or diverse combinations of several proteins simultaneously. To test the efficiency of protein immobilization, Alexa Fluor 594 conjugated goat-anti-mouse secondary antibody was used for printing. After immobilization and removal of the unbound protein, the chip was scanned with GenePix 4000B microarray scanner (Axon, Sunnyvale, CA). The fluorescent signal intensities of individual printed spots were extracted by GenePix 4.0 and evaluated. We found that, for the protein tested, the protein concentration of the printing solution and the fluorescence intensity of the arrayed spots, which represent the amount of the immobilized fluorescent antibody, displayed a linear saturating dependence in the 10–500 μg/ml range (Fig. 2A and Supp. Fig. 1A). Thus, in the following experiments, we limited the proteins to the 0–50 μg/ml range for printing.
Next we demonstrated that a functional device could indeed be fabricated using the methodology described (Fig. 2B). Furthermore, we could achieve a highly precise alignment of proteins array spots and the electrode arrays, generating 240 protein spot-DEP electrode combinations divided evenly among 4 microfluidic channels (Figs. 2A and 2C). We further tested whether two proteins could be successfully co-printed in a well defined mixtures, so that the ratio between the protein concentrations could be varied for different spots. We successfully printed four different concentrations varying over 3 orders of magnitude (1, 0.1, 0.01, 0.001 mg/ml) of the goat-anti-mouse secondary antibody conjugated with Alexa 488 (green) mixed with a constant high concentration (1 mg/ml) of this antibody conjugated with Alexa 594 (red). The results showed that the profile of the Alexa 488 fluorescence intensity was well preserved on the slide following protein adsorption vs. the initial concentration in the printing solution (Suppl. Fig. 1B), suggesting that a protein of interest present at a low concentration can be robustly immobilized on the slide even if printed in combination with another protein present at a much higher concentration, i.e., no protein competition for the available surfaces takes place, at the concentrations tested.
We next examined whether living cells could be successfully captured and cultured on the printed protein spots using DEP and microfluidic control. Several cell lines have been tested (See Supplementary Materials). We could successfully capture and culture cells in the locations specified by the electrodes and thus on the proteins spots (e.g., as shown in Fig. 2D). By varying the flow rate through the micro-electrode and protein spot containing channels, the gap between electrodes, and the electric field applied to the electrodes, we could control with high precision the number of cells captured per spot (Fig. 3), from single cells captured at the tip of one of the electrodes, to two cells captured at the tips of both electrodes, to progressively higher cells captured within the space between electrodes. This capability could not be achieved in situations when DEP control of cell capture was not exercised, thus illustrating the important role of the DEP based capture of cells. In the following analysis we focused on the experimental manipulation of the immortalized human umbilical vein endothelial cells (iHUVECs).
Printed TNFα and IGF1 synergistically stimulate the NF-κB pathway
We next investigated the functionality of the device by testing whether live cells captured onto the printed protein spots could be stimulated by active signaling ligands contained within these spots. We chose to analyze the response of the NF-κB signaling pathway to stimulation by a cytokine and a growth factor. Transcription factor NF-κB plays a role in many physiological functions, including inflammation, cancer development and immunity.16 Within the context of the innate immune response, this pathway is canonically stimulated by several cytokines, including the tumor necrosis factor-α (TNFα), interleukin-1 (IL-1), as well as components of the bacterial cell wall, lypo-polysaccharides (LPS).17 Growth factors also can stimulate this pathway in certain cell types.18 Following the pathway stimulation, NF-κB translocates into the nucleus, activating a plethora of genes. This translocation usually peaks within an hour, and is followed by a gradual decline to a sustained intermediate level, given the presence of a sustained stimulus.19 Furthermore, many of the diffusible growth factors or cytokines, including TNFα and IGF1 can undergo high affinity binding to proteins constituting ECM, and thus encounter their cognate receptors in such, immobilized form. It might thus be of interest to study whether immobilized signaling ligands can potently stimulate cells cultured within the described device.
To establish the control for our analysis, we first investigated the NF-κB activation in iHUVECs exposed to TNFα or a growth factor dissolved in the cell medium. We found that these cells displayed the expected initial temporal response to 10 ng/ml TNFα (Fig. 4A), achieving the peak response within 1 hr., but the response was relatively sustained thereafter, gradually adapting over the 3 hours time period. As some other growth factors, the insulin-like growth factor-1 (IGF1) might also stimulate NF-κB pathway, likely through activation of Akt, which can engage in cross-talk with the NF-κB pathway through modulation of the upstream kinase, IKK.20 We found that the activation of the pathway by 10 ng/ml of IGF1 was moderate and transient, peaking at 15 min, and returning to the baseline at 1 hour post-stimulation (Fig. 4A).
Figure 4.

Stimulation of the NF-κB pathway by printed TNFα and IGF1 in iHUVECs. A: Time courses of NF-κB stimulation in iHUVECs by a 10 ng/ml TNFα (filled circles) and 10 ng/ml IGF1 added to the cell medium. The fluorescence intensity values represent in arbitrary units the amount of NF-κB in cell nuclei measured by immunostaining using an anti-p65 antibody (approximately 60 cells were measured for each data point). Error bars represent the standard error of the mean; B: Stimulation of NF-κB by TNFα and IGF1 printed onto the glass cell adhesion substratum. In groups 1 to 4, the coating TNFα concentration changes 0, 2, 10 to 50 μg/ml, while within each group, the coating IGF1 concentration increases 0, 2, 10 to 50 μg/ml from left to right (approximately 30 cells were measured for each data point). Florescence intensities are as in (A). Error bars indicate the standard error of the mean. The insets show typical images of the p65 staining in cells captured on the protein spots corresponding to the indicated conditions (minimal and maximal coating TNFα and IGF1 concentrations). C. Stimulation of NF-κB by TNFα and IGF1 dissolved in the cell medium. From group 1 to 3, TNFα increased as 0,0.1 to 1 ng/ml, while within each group, IGF1 concentration increased from 0 (left) to 10 ng/ml (right).
We next printed spots of all combinations of four different concentrations of TNFα and IGF1 (16 combinations altogether) and analyzed NF-κB pathway activation by immunostaining of p65 (an NF-κB subunit) following 3 hours of cell incubation on the printed spots. In addition to containing different combinations of TNFα and IGF1, the protein solutions used for protein printing again contained constant concentrations of green-fluorescent human type IV collagen (25 μg/ml) to enable long-term cell adhesion to the printed protein spots and direct visualization of the spotted proteins. Each protein combination was printed in 15 replicate spots arranged in one column, utilizing the total of 240 spots (see Fig. 2). The concentrations in the printing solutions were 0, 2, 10 and 50 μg/ml for both TNFα and IGF1, yielding 16 distinct ligand combinations. We detected significant cell responses to printed TNFα on spots generated by the highest TNFα coating concentration (Fig. 4B), but not for lower signaling inputs. Nevertheless, the finding is of considerable importance, as it suggests that TNFα can be active even if surface-immobilized by covalent binding, and thus not being internalized by the cells, as is the case for soluble TNFα. Furthermore, the corresponding TNFα surface density was sufficient to generate a response close to the maximal when TNFα is presented in solution (Fig. 4A)), suggesting that the pathway could be effectively stimulated even though only the part of the cell surface facing the cell adhesion substratum was exposed to the signaling input, and thus only a fraction of TNFα receptors were engaged. Interestingly, we furthermore found that the cell response to TNFα at this ligand density was increasingly enhanced by progressively higher IGF1 levels. This was unexpected, given that IGF1 only had a transient effect on cells, when presented separately and in solution (Fig. 4A). As IGF1 failed to activate the NF-κB pathway at lower TNFα levels, our results suggested that IGF1 likely had only modulatory role at concentrations up to 10 ng/ml, adjusting the NF-κB response triggered by the canonical TNFα stimulation. This modulatory role of IGF1 was further confirmed by examination of cell responses to in TNFα and IGF1 as solutes in the cell media (Fig. 3C).
IκB kinase (IKK) inhibitor gradient affects signaling triggered by printed TNFα
The described experimental platform permits precise definition of the soluble components of the extracellular milieu. Thus the device may be used to examine cellular responses to simultaneously presented diverse combinations of soluble and surface-bound cues. One useful application of this capability lies in screening of libraries of soluble drugs that might be candidates for modulation of signaling responses. The NF-κB pathway is one of the more frequently targeted pathways in drug development, due to its importance in a multitude of important physiological and pathological processes. We therefore tested the suitability of the proposed device for a rapid determination of the dose response of the NF-κB pathway in iHUVECs to a drug targeting an upstream NF-κB activator, the IκB kinase (IKK). When presented to cells in Petri dishes, the drug, Sc514,17 progressively suppressed the nuclear NF-κB translocation in the 0–100 μM range following 3 hour stimulation by 10 ng/ml TNFα and 2 hour incubation with Sc514 (Fig. 5A). We took advantage of the ability to generate diffusion-based linear gradients within the device cross-channels containing cells, to expose iHUVECs cultured on TNFα spots (50 μg/ml coating concentration) for 1 hour followed by a subsequent 2 hours additional incubation in the 0–100 μM gradient of Sc514 (Fig. 5B). The results were fully consistent with the results of dose response experiments in Petri dishes (Fig. 5A) confirming IKK dependence of NF-κB activation by printed TNFα, and suggesting that the described device could indeed be used for complex combinatorial stimulation of cells by both surface-bound and soluble cues and drug screening, in a high-throughput manner.
Figure 5.
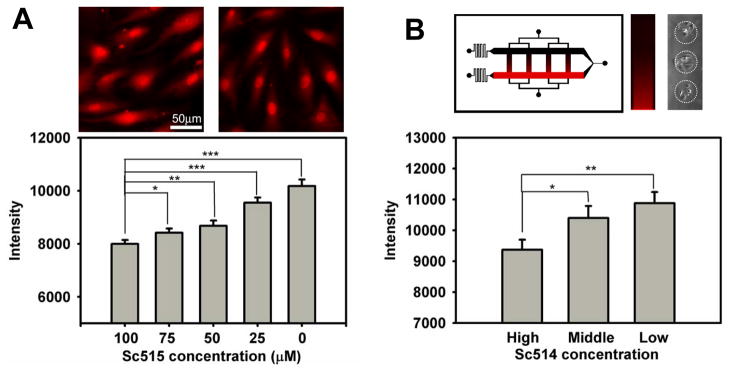
Inhibition of TNFα stimulated NF-κB activation by a mcirofluidically delivered IκB Kinase (IKK) inhibitor Sc-514. A: iHUVECs stimulated with 10 ng/ml TNFα added to the medium for 1 hour and exposed to different concentrations of Sc-514 into the medium for 2 hours. Approximately 80 cells were measured for each data point. Error bars represent the standard error of the mean. Insets show typical images corresponding to 100 (left) and 0 (right) μM of Sc514; B: iHUVECs stimulated by printed TNFα (50 μg/ml coating concentration) for 1 hour were exposed to a Sc-514 gradient within a microfluidic channel for subsequent 2 hours, with concentrations ranging from 0 to 100 μM. The gradient visualized within the device and one of the channels using Alexa 594 and the typical cell groups captured on the protein spots are shown in the upper part of the panel. The results were binned according to the Sc-514 concentration ranges: ‘low’ (0–33 μM), ‘medium’ (33–66 μM) and ‘high’ (66–100 μM). Approximately 50 cells were measured for each data point, with error bars representing the standard error of the mean. In both (A) and (B) T-test significance is represented by one asterisk, P< 0.05 to 0.01; two asterisks, P< 0.01 to 0.001, three asterisks, P< 0.001.
Discussion and conclusions
Progressive advances in the analysis of the in vivo cell behavior paint a picture of daunting complexity. The signals impinging on most cells are combinatorially complex, variable in space and time, and present in diverse chemical and physical forms. This intricacy of the physiological cell environment is rarely reproduced in vitro. Here we describe a miniature experimental platform that combines several recent technological advances within a single integrated device, permitting highly flexible experimental design and high throughput analysis of cell responses to complex variations in the immediate cellular microenvironments.
The particular architecture of the ITO electrode arrays employed in the analysis enables not only precise positioning of cells in pre-designated areas co-localized with the printed protein spots, but also allows one to control the numbers of cells captured per electrode pair, and thus per protein spot (Fig. 3). This feature permits the controlled analysis of cell ensembles of different sizes, from a single cell to cell pairs patterned at specific cell-cell distances, to multi-cellular groups of combined sizes limited only by the dimensions of the printed protein spots. The analysis of isolated cells can report on the importance of the input from the controlled signal presentation afforded by the microfluidic channel network vs. the effects exerted by the neighboring cells, the analysis that can be further refined by the study of homotypic and heterotypic cell pairs.9 Furthermore, the analysis of cell-cell communication in cell pairs can be enhanced if cells are captured on spots containing proteins that are also modifiers of cell communication processes, such as antibodies targeting cellular secretome. Finally, controlled scaling up of cell number captured on a single printed protein spot can be used to increase the throughput of signaling analysis by augmenting the number of cells analyzed.
Our analysis also revealed considerable flexibility in immobilization of combinations of diverse proteins in a single protein spot. Indeed, we could successfully print a multitude of protein mixtures of a cytokine, a growth factor and a cytoskeleton protein that collectively enabled cell adhesion, spreading and combinatorial stimulation of a signaling pathway. The doses of different proteins could be independently modulated ensuring possibility of complex dose-dependent combinations of surface immobilized cues.
Using this device, we have shown for the first time that the NF-κB pathway could be stimulated at the maximal level by immobilized ligand incapable of being internalized by cells, with only a fraction of cell surface and cell receptors being exposed to the signaling input. This finding suggested that TNFα, frequently bound to the extracellular matrix in vivo, could still be an effective stimulant, if a cell comes into contact with it. Furthermore, simultaneous stimulation of iHUVEC cells with 16 combinations of TNFα and IGF1 suggested that, at the doses printed, IGF1 failed to stimulate the NF-κB pathway directly, but could modulate the action of TNFα, revealing an interesting potential for a synergistic action between the cytokine and the growth factor, both of which were immobilized on the same surface and not internalized by the cells. By using the IKK-specific inhibitor, we confirmed that the surface-bound TNFα indeed stimulated the cells in a canonical fashion, and that this signaling could be modified by as soluble drug delivered at different doses.
In addition to scaling up the number of locally presented signaling inputs within a single device, immobilization of soluble ligands might enable a more detailed and high throughput analysis of the effects of many cytokines, chemokines and growth factors known to naturally bind to ECM components.21, 22 These effects can be contrasted in the same device with the effects of soluble forms of these ligands, using the microfluidic component of the device. Alternatively, the microfluidic channels can be used to deliver, with required kinetics, various modifiers of cell signaling, capable of affecting the pathways stimulated by the immobilized ligands. The results presented here demonstrate that the described experimental platform can potentially be an effective tool for a rapid screening of the effects of multiple doses of existing or prospective drugs on living cells exposed to combinatorially complex local signaling micro-environments.
Supplementary Material
Acknowledgments
The authors are grateful to Jef Boeke and members of Levchenko and Zhu labs for many fruitful discussions. They also acknowledge Dr. Jing Cheng and Capital Bio for providing protein printing equipment. The research was supported by NIH grants GM072024 to L.A. and RR020839 to A.L. and H.Z., and the National Basic Research Program of China (Grant No. 2010CB529205) to S.T.
References
- 1.Underhill G, Bhatia S. Curr Opin Chem Biol. 2007;11(4):357–66. doi: 10.1016/j.cbpa.2007.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.El-Ali J, Sorger PK, Jensen KF. Nature. 2006;442:403–411. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- 3.Whitesides GM. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 4.Wang CJ, Li X, Lin B, Shim S, Ming G-l, Levchenko A. Lab Chip. 2008;8:227– 237. doi: 10.1039/b713945d. [DOI] [PubMed] [Google Scholar]
- 5.Flaim C, Chien S, Bhatia S. Nat Methods. 2005;2(2):119–25. doi: 10.1038/nmeth736. [DOI] [PubMed] [Google Scholar]
- 6.Zhu H, Stybayeva G, Macal M, Ramanculov E, George M, Dandekar S, Revzin A. Lab Chip. 2008;8(12):2197–205. doi: 10.1039/b810244a. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Huang Y, Gascoyne P, Becker F. IEEE Transactions on Industry Applications. 1997;33:660–669. doi: 10.1109/28.585856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gray D, Tan J, Voldman J, Chen C. Biosensors and Bioelectronics. 2004;19:1765–1774. doi: 10.1016/j.bios.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 9.Yin Z, Noren D, Wang J, Hang R, Levchenko A. Molecular Systems Biology. 2008;4:232. doi: 10.1038/msb.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lue YR, Yeo SD, Tan L, Uttamchandani M, Chen GY, Yao SQ. Novel Strategies in Protein Microarray Generation and Detection. In: Schena M, editor. Protein Microarrays. Jones and Bartlett; Sudbury, MA: 2005. [Google Scholar]
- 11.Paliwal S, Iglesias PA, Campbell K, Hilioti Z, Groisman A, Levchenko A. Nature. 2007;446(7131):46–51. doi: 10.1038/nature05561. [DOI] [PubMed] [Google Scholar]
- 12.Groisman A, Lobo C, Cho H, Campbell J, Dufour Y, Stevens A, Levchenko A. Nat Methods. 2005;2(9):685–9. doi: 10.1038/nmeth784. [DOI] [PubMed] [Google Scholar]
- 13.Schaff UY, Xing MMQ, Lin KK, Pan N, Jeon NL, Simon SI. Lab Chip. 2007;7:448– 456. doi: 10.1039/b617915k. [DOI] [PubMed] [Google Scholar]
- 14.Halla DA, Ptaceka J, Snyder M. Mechanisms of Ageing and Development. 2007;128(1):161–167. doi: 10.1016/j.mad.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu H, Snyder M. Curr Opin Chem Biol. 2003;7(1):55–63. doi: 10.1016/s1367-5931(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 16.Dixit V, Mak TW. Cell. 2002;111(5):615–619. doi: 10.1016/s0092-8674(02)01166-2. [DOI] [PubMed] [Google Scholar]
- 17.Werner SL, Barken D, Hoffmann A. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 18.Obata H, Biro S, Arima N, Kaieda H, Kihara T, Eto H, Miyata M, Tanaka H. Biochemical and Biophysical Research Communications. 1996;224(1):27–32. doi: 10.1006/bbrc.1996.0979. [DOI] [PubMed] [Google Scholar]
- 19.Cheong R, Levchenko A. Trends in Cell Biology. 2008;18(3):112–118. doi: 10.1016/j.tcb.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poulaki V, Joussen AM, Mitsiades N, Mitsiades CS, Iliaki EF, Adamis AP. American Journal of Pathology. 2004;165(2):457–469. doi: 10.1016/S0002-9440(10)63311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollier BG, Kricker JA, Lonkhuyzen DRV, Leavesley DI, Upton Z. Endocrinology. 2007;149(3):1075–1090. doi: 10.1210/en.2007-0740. [DOI] [PubMed] [Google Scholar]
- 22.Hutchings H, Ortega N, Plouët J. The FASEB Journal. 2003;17:1520–1522. doi: 10.1096/fj.02-0691fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.