

Abstract
The clinical manifestations of Graves’ ophthalmopathy (GO) stem from a combination of increased orbital fat and extraocular muscle volume within the orbital space. Fibroblasts residing within these tissues are thought to be targets of autoimmune attack in the disease. Thyrotropin receptor (TSHr) mRNA and functional protein have been demonstrated in orbital fibroblasts from both normal individuals and GO patients, with higher levels present in the latter. Autoantibodies directed against TSHr or the insulin-like growth factor-1 (IGF-1) receptor have been implicated in GO pathogenesis. Evidence from our laboratory suggests that monoclonal TSHr autoantibodies (TRAbs) are potent stimulators of adipogenesis in GO orbital cells. Therefore, it is possible that circulating TRAbs in Graves’ patients both stimulate overproduction of thyroid hormones and increase orbital adipose tissue volume. Antibodies to the IGF-1 receptor appear to impact GO pathogenesis through recruitment and activation of T-cells and stimulation of hyaluronan production, processes that play key roles in the development of inflammation and increased orbital tissue swelling. Although originally thought to represent another causative agent, antibodies to extraocular muscles are now generally thought to be secondary to extraocular muscle inflammation and damage.
Introduction
Graves’ disease (GD) was named after the Irish physician Robert James Graves (1797–1853), who described the syndrome of hyperthyroidism, goiter, and exophthalmos. This autoimmune disease has an incidence of 1/1000 women per year and represents the most common form of hyperthyroidism. The overproduction of thyroid hormones by thyroid follicular cells in GD is mediated by autoantibodies directed against the thyroid-stimulating hormone receptor (TSHr). Graves’ ophthalmopathy (GO; also known as thyroid-associated ophthalmopathy or thyroid eye disease) is clinically evident in 25–50% of patients with GD (1). While the majority of patients experience only mild ocular symptoms, 3–5% of patients with GO suffer from severe disease (2). The spectrum of eye manifestations ranges from lid lag and retraction to proptosis, ophthalmoplegia, conjunctivitis, chemosis, and corneal ulceration, to loss of vision.
The clinical manifestations of GO stem from a combination of increased orbital fat and extraocular muscle volume within the orbital space. Because the bony orbit lacks compliance, anterior displacement of the contained tissues may result, leading to proptosis, or protrusion of the globe. The increased orbital pressure also causes impairment of venous and lymphatic outflow and congestive swelling of the periorbital tissues (3). Although orbital adipose tissue volume expansion predominates in some patients and increased extraocular muscle volume is prominent in others, most patients show a combination of both processes (Fig. 1).
FIG. 1.
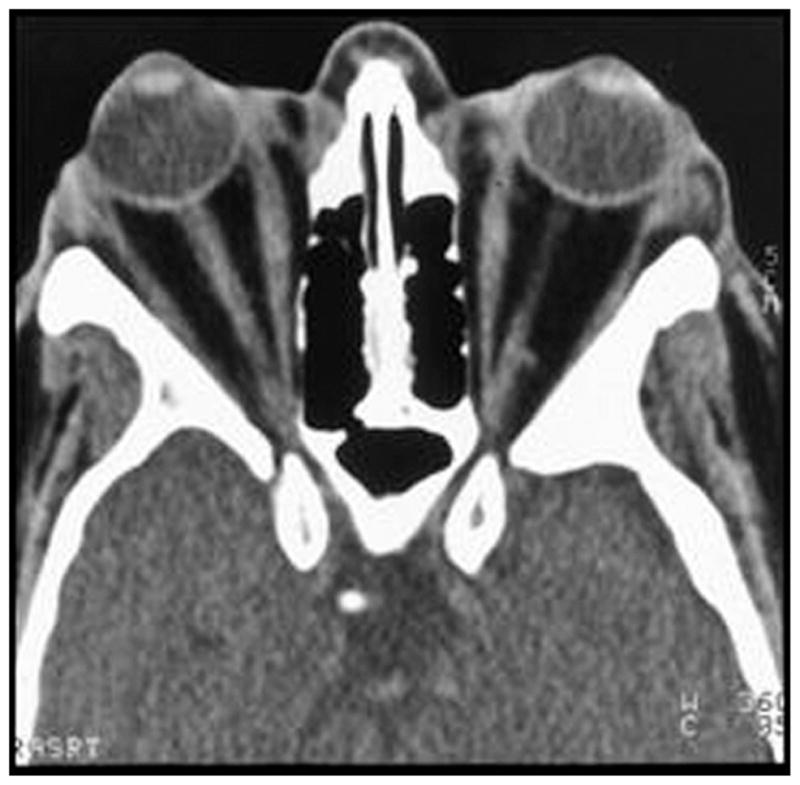
Computerized tomographic scan of the orbits of a patient with Graves’ ophthalmopathy showing enlargement of both the orbital fat and the extraocular muscles. The expanded orbital tissues cause forward displacement of the globe and impairment of venous and lymphatic outflow from the orbit.
Histochemical examination of orbital tissues in GO reveals a lymphocytic infiltration, consisting primarily of T lymphocytes, and the presence of inflammatory cytokines (4). Fibroblasts residing within the orbital connective/adipose tissue compartment and investing the extraocular muscle cells are thought to be targets of autoimmune attack in the disease. These multipotent cells are markedly heterogeneous and may be partially characterized according to their expression of the surface glycoprotein Thy-1 (5,6). Although its function as a receptor is unknown, this marker appears to distinguish distinct subgroups that differ in their responses to adipogenic stimuli and in their biosynthetic properties. The minority of cells derived from the orbital connective/adipose tissue compartment are Thy-1− and thus capable of adipogenesis. In contrast, those investing the extraocular muscles (and found within dermal tissues) uniformly display Thy-1 (Thy-1+) and do not undergo adipogenesis when similarly stimulated. This phenotypic heterogeneity in fibroblasts within the orbit may impact the clinical presentation of the disease as regard the relative contributions of adipose tissue and extraocular muscle expansion (6).
Fibroblast heterogeneity extends as well to cells derived from other anatomic sites; while orbital connective tissue fibroblasts treated with interferon-γ or leukoregulin synthesize high levels of hyaluronan, dermal fibroblasts produce only small quantities of this glycosaminoglycan (7,8). Furthermore, peroxisome proliferator–activated receptor-γ (PPAR-γ) agonists enhance differentiation of preadipocyte fibroblast from subcutaneous sites, while not exerting any effect on omental fibroblasts despite the PPAR-γ receptor being expressed at similar levels in fibroblasts from both sites (9). Although the mechanisms at play have yet to be clarified, these and other phenotypic differences between fibroblasts may help to explain why orbital adipose tissue is targeted in GO while other fat depots appear not to be impacted.
Involvement of Autoantibodies in GO Pathogenesis
TSHr autoantibodies
The close clinical association between onset of Graves’ hyperthyroidism and the development of GO suggests that these two conditions may share pathogenic mechanisms. Because autoantibodies directed against TSHr [TSHr autoantibodies (TRAbs)] are known to be responsible for the hyperthyroidism of GD, investigators have long sought evidence that TRAbs might be involved as well in GO pathogenesis. Clinical studies show that GO prevalence is increased in GD patients having the highest levels of TRAbs, and that euthyroid patients with GO generally have elevated TRAb levels (10,11). Furthermore, the clinical activity score, a composite based on signs of inflammation such as orbital pain, conjunctival erythema, and chemosis, is correlated with levels of both TSHr stimulatory and TSH-binding inhibitory TRAbs; a weaker, but also significant, correlation was found between levels of these antibodies and proptosis (12). In a large longitudinal study, TSH-binding inhibitory antibody levels were significantly higher in patients with severe disease than in patients with mild GO (13).
A prerequisite for the involvement of TSHr and TRAbs in GO would seem to be that the TSHr is expressed in affected orbital tissue. This has been convincingly shown in several studies demonstrating both TSHr mRNA and protein in orbital fibroblasts from normal individuals and patients with GO (14–16). Further supporting evidence implicating TSHr derives from correlations between TSHr expression in GO tissues and the activity or severity of the disease; TSHr mRNA levels are higher in orbital adipose tissue of GO patients than in orbital tissue from patients without eye disease (17). These findings suggest that increased TSHr expression may either be directly involved in the development of GO, or secondary to its development. Further, orbital tissues from patients with active GO have significantly higher levels of TSHr expression than do tissues from patients with inactive disease (18).
Studies suggest that extrathyroidal TSHr is functional and exhibits properties similar to those of the thyroidal receptor; TSH stimulation of TSHr in human orbital fibroblasts and abdominal preadipocytes leads to activation of p70 S6 kinase (p70 S6K), an enzyme recognized as a downstream target of TSHr in thyroid cells (19). In addition, the treatment of both normal and GO orbital fibroblast cultures with TSH results in increased cyclic adenosine monophosphate (AMP) production (20). This is most pronounced in cultures subjected to conditions that favor adipocyte differentiation, suggesting that mature fat cells express TSHr to a greater degree than do preadipocyte fibroblasts.
Orbital preadipocyte fibroblast cultures treated with the PPAR-γ agonist rosiglitazone, or cultured under adipogenic other conditions, undergo marked adipogenesis and show increased expression of TSHr and adipocyte-associated genes, including PPAR-γ, adiponectin, and leptin (21,22). Similarly, specimens of orbital adipose tissue from GO patients over-express PPAR-γ, adiponectin, leptin, and TSHr compared with normal orbital adipose tissue (22,23). These studies suggest that de novo adipogenesis is enhanced in the orbits of GO patients, and that the increase in levels of TSHr expression seen in GO orbital tissues is a consequence of this process. Studies from our laboratory have shown that monoclonal TRAbs are potent stimulators of adipogenesis in cultures of GO and normal orbital preadipocyte fibroblasts (24). Therefore, it is possible that circulating TRAbs in patients with GD might stimulate not only overproduction of thyroid hormones, but also the increase in orbital adipose tissue volume seen in patients with GO.
Mechanisms involved in enhanced orbital adipogenesis in GO were suggested by gene array studies showing markedly elevated levels of secreted frizzled-related protein-1 (sFRP-1) in GO orbital adipose tissues compared with normal orbital tissues (23). This protein is known to inhibit wingless-type (Wnt) signaling, itself a tonic inhibitor of adipogenesis. Whether TRAbs might increase sFRP-1 production within the orbit, and thus be responsible for the stimulation of adipogenesis in these tissues in GO, is the subject of ongoing investigation.
IGF-1 receptor autoantibodies
Involvement of the insulin-like growth factor-1 (IGF-1) receptor (IGF-1r) and autoantibodies to the receptor in the pathogenesis of GO was first suggested by investigators who demonstrated high-affinity IGF-1 binding sites on human orbital fibroblasts (25). In addition, these investigators found that immunoglobulin G (IgG) from the sera of patients with GD, with or without GO, inhibited labeled [125I] IGF-1 binding to these cells, whereas IgG from normal subjects showed no such effect. In more recent studies, IgG from patients with GD was shown to induce the expression in GO orbital fibroblasts of interleukin-16 (IL-16) and RANTES, factors playing roles in T cell migration and activation, while having no effect in orbital fibroblasts from normal individuals (26). The induction was inhibited by both IGF-1 and the IGF-1r–specific ligand, Des (1–3), suggesting that it is mediated through the IGF-1r and that IgG from Graves’ patients contains autoantibodies directed against this receptor. These same investigators also demonstrated that Graves’ IgG induces hyaluronan synthesis in GD orbital fibroblasts, and that these cells express higher levels of IGF-1r than do normal orbital fibroblasts (27). As IGF-1 is also known to be a potent stimulator of adipogenesis (Fig. 2), it may be that autoantibodies directed against IGF-1r in the sera of patients with GO act upon the diverse population of orbital fibroblasts to stimulate some to differentiate into adipocytes and others to increase production of hyaluronan. The chemoattractant effect that these autoantibodies exert through their stimulation of IL-6 and RANTES would increase recruitment of inflammatory cells into the orbit and perpetuate the autoimmune response.
FIG. 2.
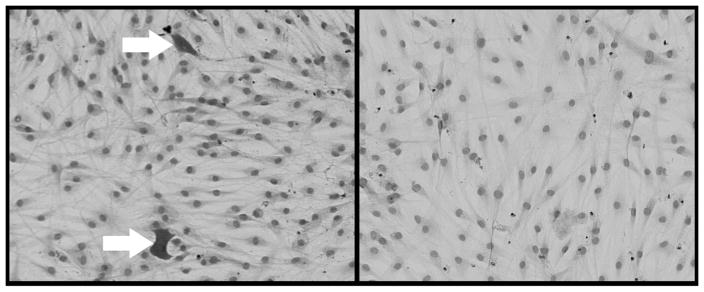
Human orbital fibroblasts stained with Oil Red O and hematoxylin showing the presence of mature fat cells (arrows) after 18-day exposure to adipogenic culture media containing insulin-like growth factor-1 (left; 10 nM), or cultured under standard conditions (right; 10× magnification).
Antibodies to extraocular muscle
The extraocular muscle bodies are generally enlarged in patients with GO and intimately involved in the disease process. Based on these observations, early studies focused on the potential role of extraocular muscle cells as target cells in GO, and of muscle antigens as autoantigens in the disease. Indeed, numerous studies showed that GO sera recognize various components of extraocular muscle, with reactivity to a 64 kDa protein in as high as 71% of patients with GO. This reactivity was shown to be present in only about 35% of sera from individuals having GD without GO, or Hashimoto’s thyroiditis (28–30). In these studies, the level of antibody activity appeared to correlate well with the presence of GO (29,30). The most prevalent autoantibodies were directed toward membranous and cytoplasmic antigens in extraocular muscles (31).
More recent characterization of muscle antigens has focused on two proteins termed “G2s” and “Fp” (32). G2s is strongly expressed in thyroid and extraocular muscle, but is also present in skeletal muscle, myocardium, and other organs. Fp (the “64 kDa protein”) is a flavoprotein subunit of mitochondrial succinate dehydrogenase found in various tissues, including skeletal muscle and liver. Because the two proteins are found intracellularly, it is unlikely that the autoantibodies directed against these antigens are primarily pathogenic. They rather appear to represent the sequela of muscle damage with subsequent release of antigens into the circulation. While not being specific to GO, antibodies directed against G2s and Fp are sensitive markers of eye muscle damage in GD patients. Their presence appears to be closely associated with the ocular myopathy subtype of GO and thus may have a predictive role in the development of extraocular muscle involvement in GD patients without overt GO (33).
Novel Approaches to Therapy
The commonly used medical treatments for GO are only moderately effective in halting or reversing the ocular symptoms, and their use may be limited by unwanted side effects. Current understanding of pathogenic mechanisms involved in the development of GO suggests several novel therapeutic targets whose role in GO therapy warrant further study (Fig. 3). Costimulation inhibitors that block early steps in T and B cell activation hold promise as agents that would inhibit both the production of autoantibodies by B cells and the secretion of inflammatory cytokines by T cells (34). This class of agents includes CTLA4-Ig (abatacept), a blocker of CD28 ligation that prevents delivery of the second costimulatory signal required for optimal T cell activation, and alefacept, an antibody to the CD2 marker that blocks effector memory T cells. In addition, evidence for the direct participation of TRAbs and IGF-1r autoantibodies in GO pathogenesis suggests that rituximab, an agent that blocks CD20 ligation and the early phases of B cell maturation, might prove beneficial in the disease (35). A trial of rituximab therapy in patients with refractory systemic lupus erythematosus found improved disease activity with a significant decline in the levels of anti-dsDNA antibody levels (36–38). A similarly beneficial effect in patients with rheumatoid arthritis, improving rheumatoid factor levels and disease parameters, was seen following this therapy (39). The few reports of the use of rituximab in GO patients hold promise and offer firm rationale for prospective, randomized, controlled trials of this agent for the treatment of GO (40,41). Another approach to GO therapy at the level of the involved cellular receptors would be to block receptor binding of TRAbs or IGF-1 autoantibodies utilizing specific monoclonal anti-IGF-1r or TSHr antibodies. Downstream effects of autoantibody binding might be modulated by small molecule inhibitors of tyrosine kinase to block IGF-1r activation, or fragments of antisense RNA (42).
FIG. 3.

Potential novel therapeutic targets in Graves’ ophthalmopathy. [1] Inhibition of T and B cell activation: costimulation inhibitors (CTLA4-Ig, alefacept). [2] Inhibition of B cell maturation, autoantibody production: rituximab. [3] Inhibition of autoantibody binding to insulin-like growth factor-1 receptor (IGF-1r) and thyroid-stimulating hormone receptor (TSHr): specific anti-IGF-1r or TSHr antibodies, inhibitors of IGF-1r tyrosine kinase, antisense RNA. [4] Inhibition of adipogenesis: peroxisome proliferator–activated receptor-γ antagonists. [5] Decrease inflammation: non-steroidal antiinflammatory drugs (NSAIDs) or anticytokine agents (infliximab, adalimumab, etanercept, and anakinra).
Further downstream in the pathogenic pathway, orbital preadipocyte differentiation into mature adipocytes might be inhibited. If this could be achieved within the orbit, the increase commonly seen in orbital adipose tissue volume might be prevented, limiting the development of proptosis and impairment of venous and lymphatic outflow. One approach might be to use an antagonist to PPAR-γ (such as GW9662) that blocks receptor ligation, preventing fat cell development. Conversely, because PPAR-γ agonists (thiazolidinediones) are known to stimulate adipogenesis in orbital fibroblast cultures (21), it has been suggested that these agents may be relatively contraindicated in GO patients; a patient with stable GO for 2 years was reported to have developed increased proptosis after initiation of rosiglitazone for type 2 diabetes mellitus (43). Finally, the inflammation characteristic of the disease, perhaps representing the most commonly debilitating of the symptoms, might be addressed using nonsteroidal antiinflammatory drugs (NSAIDs) or anticytokine agents, including antitumor necrosis factor-alpha (anti-TNFα) (infliximab, ad-alimumab, and etanercept) or anti-IL-1 receptor agents (anakinra), known to be effective in the treatment of rheumatoid arthritis (44).
Summary
Evidence to date implicates autoantibodies to both TSHr and IGF-1r in the development of the orbital changes of GO. The effects of TSHr ligation by TRAbs may be mediated through increased sFRP-1 production by orbital fibroblasts with inhibition of the Wnt signaling pathway, leading to increased adipogenesis. Antibodies to the IGF-1r appear to play role in the disease process through recruitment and activation of T cells and stimulation of hyaluronan production, processes that play key roles in the development of inflammation and increased orbital tissue swelling. Although originally thought to another causative agent, antibodies to extraocular muscles are now generally believed to be secondary to extraocular muscle inflammation and damage. However, there may be a role for measurement of these antibodies in monitoring or predicting ocular involvement in GO.
Despite advancing knowledge concerning GO pathogenesis, many questions remain. It is unknown why orbital fibroblasts expressing Thy-1 do not differentiate into adipocytes, while Thy-1− fibroblasts appear to be preadipocytes. In addition, reasons for site-specific fibroblast responses to immunologic or pharmacologic stimuli are unclear.
Current treatment strategies for medical management of GO are inadequate. These therapies are only moderately effective in halting or reversing disease symptoms, or are associated with untoward adverse effects. Better understanding of the pathogenesis of GO will lead to randomized, prospective clinical trials of novel agents that will ultimately benefit patients suffering from this debilitating disease.
References
- 1.Bahn RS, Heufelder AE. Pathogenesis of Graves’ ophthalmopathy. N Engl J Med. 1993;329:1468–1475. doi: 10.1056/NEJM199311113292007. [DOI] [PubMed] [Google Scholar]
- 2.Prabhakar BS, Bahn RS, Smith TJ. Current perspective on the pathogenesis of Graves’ disease and ophthalmopathy. Endocr Rev. 2003;24:802–835. doi: 10.1210/er.2002-0020. [DOI] [PubMed] [Google Scholar]
- 3.Garrity JA, Bahn RS. Pathogenesis of graves ophthalmopathy: implications for prediction, prevention, and treatment. Am J Ophthalmol. 2006;142:147–153. doi: 10.1016/j.ajo.2006.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weetman AP, Cohen S, Gatter KC, Fells P, Shine B. Immunohistochemical analysis of the retrobulbar tissues in Graves’ ophthalmopathy. Clin Exp Immunol. 1989;75:222–227. [PMC free article] [PubMed] [Google Scholar]
- 5.Sorisky A, Pardasani D, Gagnon A, Smith TJ. Evidence of adipocyte differentiation in human orbital fibroblasts in primary culture. J Clin Endocrinol Metab. 1996;81:3428–3431. doi: 10.1210/jcem.81.9.8784110. [DOI] [PubMed] [Google Scholar]
- 6.Smith TJ, Koumas L, Gagnon A, Bell A, Sempowski GD, Phipps RP, Sorisky A. Orbital fibroblast heterogeneity may determine the clinical presentation of thyroid-associated ophthalmopathy. J Clin Endocrinol Metab. 2002;87:385–392. doi: 10.1210/jcem.87.1.8164. [DOI] [PubMed] [Google Scholar]
- 7.Smith TJ, Bahn RS, Gorman CA, Cheavens M. Stimulation of glycosaminoglycan accumulation by interferon gamma in cultured human retroocular fibroblasts. J Clin Endocrinol Metab. 1991;72:1169–1171. doi: 10.1210/jcem-72-5-1169. [DOI] [PubMed] [Google Scholar]
- 8.Smith TJ, Wang HS, Evans CH. Leukoregulin is a potent inducer of hyaluronan synthesis in cultured human orbital fibroblasts. Am J Physiol: Cell Physiol. 1995;268:C382–C388. doi: 10.1152/ajpcell.1995.268.2.C382. [DOI] [PubMed] [Google Scholar]
- 9.Adams M, Montague CT, Prins JB, Holder JC, Smith SA, Sanders L, Digby JE, Sewter CP, Lazar MA, Chatterjee VK, O’Rahilly S. Activators of peroxisome proliferator-activated receptor gamma have depot-specific effects on human preadipocyte differentiation. J Clin Invest. 1997;100:3149–3153. doi: 10.1172/JCI119870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khoo DH, Eng PH, Ho SC, Tai ES, Morgenthaler NG, Seah LL, Fong KS, Chee SP, Choo CT, Aw SE. Graves’ ophthalmopathy in the absence of elevated free thyroxine and triiodothyronine levels: prevalence, natural history, and thyrotropin receptor antibody levels. Thyroid. 2000;10:1093–1100. doi: 10.1089/thy.2000.10.1093. [DOI] [PubMed] [Google Scholar]
- 11.Khoo DH, Ho SC, Seah LL, Fong KS, Tai ES, Chee SP, Eng PH, Aw SE, Fok AC. The combination of absent thyroid peroxidase antibodies and high thyroid-stimulating immunoglobulin levels in Graves’ disease identifies a group at markedly increased risk of ophthalmopathy. Thyroid. 1999;9:1175–1180. doi: 10.1089/thy.1999.9.1175. [DOI] [PubMed] [Google Scholar]
- 12.Gerding MN, van der Meer JW, Broenink M, Bakker O, Wiersinga WM, Prummel MF. Association of thyrotrophin receptor antibodies with the clinical features of Graves’ ophthalmopathy. Clin Endocrinol. 2000;52:267–271. doi: 10.1046/j.1365-2265.2000.00959.x. [DOI] [PubMed] [Google Scholar]
- 13.Eckstein AK, Plicht M, Lax H, Neuhauser M, Mann K, Lederbogen S, Heckmann C, Esser J, Morgenthaler NG. Thyrotropin receptor autoantibodies are independent risk factors for Graves’ ophthalmopathy and help to predict severity and outcome of the disease. J Clin Endocrinol Metab. 2006;91:3464–3470. doi: 10.1210/jc.2005-2813. [DOI] [PubMed] [Google Scholar]
- 14.Feliciello A, Porcellini A, Ciullo I, Bonavolonta G, Avvedimento EV, Fenzi G. Expression of thyrotropin-receptor mRNA in healthy and Graves’ disease retro-orbital tissue. Lancet. 1993;342:337–338. doi: 10.1016/0140-6736(93)91475-2. [DOI] [PubMed] [Google Scholar]
- 15.Heufelder AE, Dutton CM, Sarkar G, Donovan KA, Bahn RS. Detection of TSH receptor RNA in cultured fibroblasts from patients with Graves’ ophthalmopathy and pretibial dermopathy. Thyroid. 1993;3:297–300. doi: 10.1089/thy.1993.3.297. [DOI] [PubMed] [Google Scholar]
- 16.Starkey K, Janezic A, Jones G, Jordan N, Baker G, Ludgate M. Adipose thyrotrophin receptor expression is elevated in Graves’ and thyroid eye diseases ex vivo and indicates adipogenesis in progress in vivo. J Mol Endocrinol. 2003;30:369–380. doi: 10.1677/jme.0.0300369. [DOI] [PubMed] [Google Scholar]
- 17.Bahn RS, Dutton CM, Natt N, Joba W, Spitzweg C, Heufelder AE. Thyrotropin receptor expression in Graves’ orbital adipose/connective tissues: potential autoantigen in Graves’ ophthalmopathy. J Clin Endocrinol Metab. 1998;83:998–1002. doi: 10.1210/jcem.83.3.4676. [DOI] [PubMed] [Google Scholar]
- 18.Wakelkamp IM, Bakker O, Baldeschi L, Wiersinga WM, Prummel MF. TSH-R expression and cytokine profile in orbital tissue of active vs. inactive Graves’ ophthalmopathy patients. Clin Endocrinol. 2003;58:280–287. doi: 10.1046/j.1365-2265.2003.01708.x. [DOI] [PubMed] [Google Scholar]
- 19.Bell A, Gagnon A, Grunder L, Parikh SJ, Smith TJ, Sorisky A. Functional TSH receptor in human abdominal pre-adipocytes and orbital fibroblasts. American Journal of Physiology: Cell Physiol. 2000;279:C335–C340. doi: 10.1152/ajpcell.2000.279.2.C335. [DOI] [PubMed] [Google Scholar]
- 20.Valyasevi RW, Erickson DZ, Harteneck DA, Dutton CM, Heufelder AE, Jyonouchi SC, Bahn RS. Differentiation of human orbital preadipocyte fibroblasts induces expression of functional thyrotropin receptor. J Clin Endocrinol Metab. 1999;84:2557–2562. doi: 10.1210/jcem.84.7.5838. [DOI] [PubMed] [Google Scholar]
- 21.Valyasevi RW, Harteneck DA, Dutton CM, Bahn RS. Stimulation of adipogenesis, peroxisome proliferator-activated receptor-gamma (PPARgamma), and thyrotropin receptor by PPARgamma agonist in human orbital pre-adipocyte fibroblasts. J Clin Endocrinol Metab. 2002;87:2352–2358. doi: 10.1210/jcem.87.5.8472. [DOI] [PubMed] [Google Scholar]
- 22.Kumar S, Coenen MJ, Scherer PE, Bahn RS. Evidence for enhanced adipogenesis in the orbits of patients with Graves’ ophthalmopathy. J Clin Endocrinol Metab. 2004;89:930–935. doi: 10.1210/jc.2003-031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Leontovich A, Coenen MJ, Bahn RS. Gene expression profiling of orbital adipose tissue from patients with Graves’ ophthalmopathy: a potential role for secreted frizzled-related protein-1 in orbital adipogenesis. J Clin Endocrinol Metab. 2005;90:4730–4735. doi: 10.1210/jc.2004-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stan MN, Coenen MJ, Bahn RS. Adipogenesis is stimulated by thyrotropin receptor autoantibodies and TSH in Graves’ orbital preadipocytes. Thyroid. 2006;16:859–860. [Google Scholar]
- 25.Weightman DR, Perros P, Sherif IH, Kendall-Taylor P. Autoantibodies to IGF-1 binding sites in thyroid associated ophthalmopathy. Autoimmunity. 1993;16:251–257. doi: 10.3109/08916939309014643. [DOI] [PubMed] [Google Scholar]
- 26.Pritchard J, Han R, Horst N, Cruikshank WW, Smith TJ. Immunoglobulin activation of T cell chemoattractant expression in fibroblasts from patients with Graves’ disease is mediated through the insulin-like growth factor I receptor pathway. J Immunol. 2003;170:6348–6354. doi: 10.4049/jimmunol.170.12.6348. [DOI] [PubMed] [Google Scholar]
- 27.Smith TJ, Hoa N. Immunoglobulins from patients with Graves’ disease induce hyaluronan synthesis in their orbital fibroblasts through the self-antigen, insulin-like growth factor-I receptor. J Clin Endocrinol Metab. 2004;89:5076–5080. doi: 10.1210/jc.2004-0716. [DOI] [PubMed] [Google Scholar]
- 28.Kodama K, Sikorska H, Bandy-Dafoe P, Bayly R, Wall JR. Demonstration of a circulating autoantibody against a soluble eye-muscle antigen in Graves’ ophthalmopathy. Lancet. 1982;2:1353–1356. doi: 10.1016/s0140-6736(82)91267-3. [DOI] [PubMed] [Google Scholar]
- 29.Hiromatsu Y, Cadarso L, Salvi M, Wall JR. Significance of cytotoxic eye muscle antibodies in patients with thyroid-associated ophthalmopathy. Autoimmunity. 1990;5:205–213. doi: 10.3109/08916939009002979. [DOI] [PubMed] [Google Scholar]
- 30.Hiromatsu Y, Sato M, Tanaka K, Shoji S, Nonaka K, Chinami M, Fukazawa H. Significance of anti-eye muscle antibody in patients with thyroid-associated ophthalmopathy by quantitative western blot. Autoimmunity. 1992;14:9–16. doi: 10.3109/08916939309077351. [DOI] [PubMed] [Google Scholar]
- 31.Kiljanski JI, Peele K, Stachura I, Pickeral J, Stolarski C, Kennerdell JS, Wall JR. Antibodies against striated muscle, connective tissue and nuclear antigens in patients with thyroid-associated ophthalmopathy: should Graves’ disease be considered a collagen disorder? J Endocrinol Invest. 1997;20:585–591. doi: 10.1007/BF03346914. [DOI] [PubMed] [Google Scholar]
- 32.Mizokami T, Salvi M, Wall JR. Eye muscle antibodies in Graves’ ophthalmopathy: pathogenic or secondary epiphenomenon? J Endocrinol Invest. 2004;27:221–229. doi: 10.1007/BF03345270. [DOI] [PubMed] [Google Scholar]
- 33.De Bellis A, Perrino S, Coronella C, Sansone D, Ruocco G, Tirelli G, Di Martino S, Conte M, Bellastella G, Wall JR, Bellastella A, Bizzarro A. Extraocular muscle antibodies and the occurrence of ophthalmopathy in Graves’ disease. Clin Endocrinol. 2004;60:694–698. doi: 10.1111/j.1365-2265.2004.02036.x. [DOI] [PubMed] [Google Scholar]
- 34.Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S, Russell A, Dougados M, Emery P, Nuamah IF, Williams GR, Becker JC, Hagerty DT, Moreland LW. Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA4Ig. N Engl J Med. 2003;349:1907–1915. doi: 10.1056/NEJMoa035075. [DOI] [PubMed] [Google Scholar]
- 35.Maloney DG, Grillo-Lopez AJ, White CA, Bodkin D, Schilder RJ, Neidhart JA, Janakiraman N, Foon KA, Liles TM, Dallaire BK, Wey K, Royston I, Davis T, Levy R. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood. 1997;90:2188–2195. [PubMed] [Google Scholar]
- 36.Leandro MJ, Cambridge G, Edwards JC, Ehrenstein MR, Isenberg DA. B-cell depletion in the treatment of patients with systemic lupus erythematosus: a longitudinal analysis of 24 patients. Rheumatology. 2005;44:1542–1545. doi: 10.1093/rheumatology/kei080. [DOI] [PubMed] [Google Scholar]
- 37.Cambridge G, Leandro MJ, Teodorescu M, Manson J, Rahman A, Isenberg DA, Edwards JC. B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006;54:3612–3622. doi: 10.1002/art.22211. [DOI] [PubMed] [Google Scholar]
- 38.Ng KP, Leandro MJ, Edwards JC, Ehrenstein MR, Cambridge G, Isenberg DA. Repeated B cell depletion in treatment of refractory systemic lupus erythematosus. Ann Rheum Dis. 2006;65:942–945. doi: 10.1136/ard.2005.044487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cambridge G, Stohl W, Leandro MJ, Migone TS, Hilbert DM, Edwards JC. Circulating levels of B lymphocyte stimulator in patients with rheumatoid arthritis following rituximab treatment: relationships with B cell depletion, circulating antibodies, and clinical relapse. Arthritis Rheum. 2006;54:723–732. doi: 10.1002/art.21650. [DOI] [PubMed] [Google Scholar]
- 40.El Fassi D, Nielsen CH, Hasselbalch HC, Hegedus L. Treatment-resistant severe, active Graves’ ophthalmopathy successfully treated with B lymphocyte depletion. Thyroid. 2006;16:709–710. doi: 10.1089/thy.2006.16.709. [DOI] [PubMed] [Google Scholar]
- 41.Salvi M, Vannucchi G, Campi I, Curro N, Dazzi D, Simonetta S, Bonara P, Rossi S, Sina C, Guastella C, Ratiglia R, Beck-Peccoz P. Treatment of Graves’ disease and associated ophthalmopathy with the anti-CD20 monoclonal antibody rituximab: an open study. Eur J Endocrinol. 2007;156:33–40. doi: 10.1530/eje.1.02325. [DOI] [PubMed] [Google Scholar]
- 42.Surmacz E. Growth factor receptors as therapeutic targets: strategies to inhibit the insulin-like growth factor I receptor. Oncogene. 2003;22:6589–6597. doi: 10.1038/sj.onc.1206772. [DOI] [PubMed] [Google Scholar]
- 43.Starkey K, Heufelder A, Baker G, Joba W, Evans M, Davies S, Ludgate M. Peroxisome proliferator-activated receptor-gamma in thyroid eye disease: contraindication for thiazolidinedione use? J Clin Endocrinol Metab. 2003;88:55–59. doi: 10.1210/jc.2002-020987. [DOI] [PubMed] [Google Scholar]
- 44.Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]