

Abstract
Alzheimer’s disease (AD) is the major cause of dementia in the United States. At the cellular level, the brains of AD patients are characterized by extracellular dense plaques and intracellular neurofibrillary tangles whose major components are the β-amyloid peptide and tau, respectively. The β-amyloid peptide is a cleavage product of the amyloid precursor protein (APP); mutations in APP have been correlated with a small number of cases of familial Alzheimer’s disease. APP is the canonical member of the APP family, whose functions remain unclear. The nematode Caenorhabditis elegans, one of the premier genetic workhorses, is being used in a variety of ways to address the functions of APP and determine how the β-amyloid peptide and tau can induce toxicity. First, the function of the C. elegans APP-related gene, apl-1, is being examined. Although different organisms may use APP and related proteins, such as APL-1, in different functional contexts, the pathways in which they function and the molecules with which they interact are usually conserved. Second, components of the γ-secretase complex and their respective functions are being revealed through genetic analyses in C. elegans. Third, to address questions of toxicity, onset of degeneration, and protective mechanisms, different human β-amyloid peptide and tau variants are being introduced into C. elegans and the resultant transgenic lines examined. Here, we summarize how a simple system such as C. elegans can be used as a model to understand APP function and suppression of β-amyloid peptide and tau toxicity in higher organisms.
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder affecting over 5 million Americans and over 26 million people worldwide (Association 2008; Brookmeyer et al. 2007). As the US population lives longer, the prevalence of AD will increase and become more of a health concern and financial burden (Association 2008; Hebert et al. 2003). Thus far, AD is incurable and its etiology is unknown.
Inheritance of AD
One of the major risk factors for AD is family history and genetic predisposition (Association 2008). No mutation has been linked to sporadic AD, which is late in onset (>65 years) and accounts for most AD cases. In contrast, several mutations have been linked to familial AD (FAD), which has an early onset (<40 years) (Campion et al. 1999; Chartier-Harlin et al. 1991; Murrell et al. 1991) and can be seen in patients as young as 25 (Miklossy et al. 2003). The brains of AD patients are characterized by the accumulation of dense plaques and neurofibrillary tangles (Kidd 1964; Krigman et al. 1965; Luse and Smith 1964; Terry et al. 1964). The major components of the dense plaques is the β-amyloid peptide (Aβ; (Glenner and Wong 1984; Masters et al. 1985), which is a cleavage product of the amyloid precursor protein (APP; Kang et al. 1987); the major component of the neurofibrillary tangles is hyperphosphorylated tau, a microtubule-associated protein (MAP; Goedert et al. 1989). Mutations in the genes encoding APP (Chartier-Harlin et al. 1991; Goate et al. 1991; Murrell et al. 1991; Hardy 2009a) and presenilins (PSEN1 and PSEN2; (Levy-Lahad et al. 1995a, b; Rogaev et al. 1995; Sherrington et al. 1995; Hardy 2009b, c), which are proteases that are part of the γ-secretase complex responsible for cleaving APP (Kimberly et al. 2003;Li et al. 2000), have been correlated to FAD. Moreover, a duplication of the APP locus can lead to FAD (Cabrejo et al. 2006; Rovelet-Lecrux et al. 2006; Sleegers et al. 2006). These data suggest that disruption of APP metabolism is one of the causative factors in the disease.
The APP gene is alternatively spliced to give rise to a single-pass transmembrane domain protein (Kang et al. 1987; Ponte et al. 1988; Tanzi et al. 1988; Yoshikai et al. 1990). APP is cleaved through two major proteolytic pathways: the α-or β-secretase pathways (Fig. 1; for reviews, see (Gralle and Ferreira 2007; Nunan and Small 2000; Selkoe 1999). Initial cleavage by the α- or β-secretase releases an extracellular fragment sAPPα or sAPPβ, respectively. Subsequent cleavage in the β-secretase pathway by γ-secretase leads to the production of Aβ and release of a cytoplasmic fragment (AICD). In contrast, because α-secretase cleaves within the Aβ sequence, the subsequent γ-secretase cleavage releases the cytoplasmic AICD fragment, but does not produce Aβ. Hence, the β/γ-secretase pathway is likely favored in the pathogenesis of AD (for reviews, see Nunan and Small 2000; Selkoe 1999). Presenilin 1 (PSEN1) and 2 (PSEN2) are part of the γ-secretase complex that releases Aβ (Kimberly et al. 2003; Li et al. 2000). The specificity of the secretase cleavage sites can vary. Normally, 90% of the derived Aβ consists of 40 amino acids (=Aβ40) and the other 10% consists of 42 and 43 amino acids (=Aβ42 and Aβ43, respectively; Haass and Selkoe 1993). Aβ42 and Aβ43 are highly fibrillogenic and readily aggregate (Haass and Selkoe 1993). Deposition of Aβ, and particularly Aβ42, is presumed to be neurotoxic (Pike et al. 1993; Yankner et al. 1990).
Fig. 1.
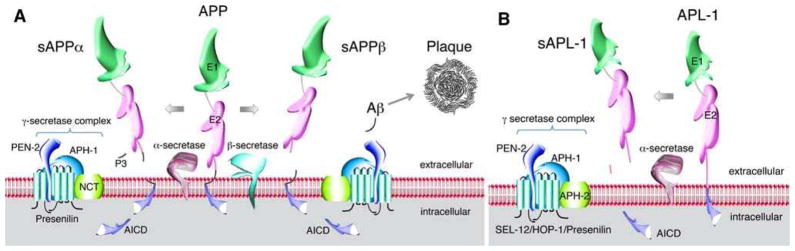
Schematic of the processing pathways of human amyloid precursor protein (APP; a) and its C. elegans ortholog APL-1 (b). a) APP undergoes two processing pathways, α/γ-secretase or β/γ-secretase, to produce sAPPα/AICD or sAPPβ/AICD, respectively. Only the β/γ-secretase pathway produces the amyloid peptide (Aβ). α-Secretase corresponds to ADAM17/TACE, β-secretase to BACE. b) In C. elegans, APL-1 undergoes at least one processing pathway to produce sAPL-1 and presumably AICD. α-Secretase may correspond to SUP-17, ADAM10, or ADM-4 ADAM17/TACE. No β-secretase activity has been described in C. elegans. NCT/APH-2 nicastrin; conserved domains: cysteine-rich E1 and acidic residue-rich E2 domain, AICD APP or APL-1 intracellular cytoplasmic domain.
Understanding the role of APP: use of a mouse model system
A powerful approach to elucidate the in vivo function of a protein is to inactivate the gene and observe the defects caused in the organism. Inactivation of APP in Mus musculus revealed several deficits, such as reduced brain and body weight, a malformation of forebrain commissures, a deficit in grip strength, an alteration in circadian locomotor activity, a hypersensitivity to seizures, and impairments in spatial learning and long-term potentiation (Li et al. 1996; Magara et al. 1999; Perez et al. 1997; Steinbach et al. 1998; Tremml et al. 1998; Zheng et al. 1995). Overexpression of APP in mice also resulted in several phenotypes, which varied in severity, presumably because of varying levels of APP expression. These phenotypes include lethality, neophobia, impaired spatial alteration, reactive gliosis, and an increase in the number of synaptophysin and GAP-43 immunoreactive presynaptic terminals (Hsiao et al. 1995; Mucke et al. 1994).
APP is the canonical member of the APP family of proteins that includes APLP1 and APLP2, which share high sequence similarity to APP within the extracellular and cytoplasmic domains, but do not contain Aβ (Slunt et al. 1994; Sprecher et al. 1993; Wasco et al. 1992, 1993a, b). The different members of the APP family probably originated from one ancestral gene, which was duplicated and translocated. Over time, the duplicated genes evolved to have slightly different, but still overlapping functions. Mice in which APLP1 is inactivated show a postnatal growth defect (Heber et al. 2000), whereas mice in which APLP2 is inactivated appear wild type (Von Koch et al. 1997). However, APLP1-APLP2 and APLP2-APP, but not APLP1-APP, double knockouts show early postnatal lethality (Heber et al. 2000). Furthermore, in addition to postnatal lethality, the triple APP-APLP1-APLP2 knockout mice also show a smoothened brain resembling human lissencephaly type II and neuronal ectopias (Herms et al. 2004). These experiments highlight an essential role for the APP family proteins in development and viability.
Understanding the role of APP: use of the C. elegans model system
The nematode Caenorhabditis elegans contains only one APP-related gene, apl-1 (Daigle and Li 1993). Because genetic manipulations are easier and faster in C. elegans than in a mammalian system, C. elegans presents an attractive alternative model to examine APP function and its mechanisms of action. APL-1 is also a single-pass transmembrane domain protein and shares many conserved domains with the mammalian APP family members; however, like APLP1 and APLP2, APL-1 does not show any sequence similarity in the Aβ peptide region (Fig. 1). Nevertheless, a lack of sequence similarity does not exclude the possibility of APL-1 having an “Aβ peptide equivalent.” The Drosophila APP-related protein (Appl) also lacks sequence similarity to the mammalian Aβ peptide region (Rosen et al. 1989), but contains an Aβ peptide equivalent that upon cleavage can form neurotoxic plaques (Carmine-Simmen et al. 2009). Animals carrying mutations in Drosophila appl are viable (Luo et al. 1992), but unhealthy (D. Kretzschmar, personal communication).
Similar to the APP gene family, apl-1 has an essential function. Knockout of apl-1 causes 100% lethality during early larval development (Table 1; Hornsten et al. 2007). The onset of apl-1 lethality in C. elegans is comparable developmentally to the postnatal lethality seen in the APP family mouse knockouts. In wild-type C. elegans, each larval stage is punctuated by a molt, when the old cuticle is sloughed off and replaced by a new cuticle. apl-1 mutants synthesize a new cuticle, but have difficulty shedding their old cuticle; hence, apl-1 mutants die during the transition from the first to second larval stage (L1–L2). Several other phenotypes were seen, some at low penetrance, including arrest as L1 larvae and/or severe morphogenetic defects, and some at high penetrance, such as the presence of large vacuoles in hypodermal cells. All phenotypes were rescued by germline transformation with a genomic fragment containing the apl-1 region as well as an APL-1 translational fusion with green fluorescent protein (GFP) at the 3′end (APL-1::GFP; Table 1). The lethality was not rescued with a genomic fragment containing a premature stop codon in the coding region. Hence, APL-1 is an essential protein for postembryonic development. The goal of examining apl-1 in C. elegans is to gain insight into the functional domains and pathways of APL-1 and translate these findings to mammals.
Table 1.
Knockouts of C. elegans orthologues of human genes implicated in AD
| Human | Caenorhabditis elegans | ||||
|---|---|---|---|---|---|
| Role | Gene | Gene | Knockout (null) alleles | Phenotypes of null alleles | References |
| Amyloid Precursor Protein Family | |||||
| APP | APP/APLP1/APLP2 | apl-1 | yn10, yn23, yn28, yn29, yn30, yn31, yn32 | larval lethal; molting defect; vacuoles; morphological defects | (Hornsten et al. 2007) |
| Processing Enzymes of APP | |||||
| α-secretase | |||||
| ADAM10 | sup-17 | n1306, n1315, n1316, n1318, n1319am, n1320 | lethal | (Tax et al. 1997) | |
| ADAM17/TACE | adm-4 | ok265 | wild type; functional redundancy between SUP-17 and ADM-4 | (Jarriault and GreenwalD 2005) | |
| β-secretase | |||||
| BACE1 | no endogenous b-secretase activity that cleaves human APP found in transgenic C. elegans | (Link 2006) | |||
| γ-secretase complex | |||||
| Presenilins | PSEN1 or 2 | sel-12 | ar171, ty11 | disrupted vulva morphogenesis; egg laying defective | (Cinar et al. 2001; Levitan and Greenwald 1995) |
| PSEN1 or 2 | hop-1 | ar179 | functionally redundant with sel-12 | (Wen et al. 2000) | |
| APH-1 | APH-1 | aph-1 | ep140, ep169, ep170, ep216, ep411, ep413, zu123, or28 | no anterior pharynx; maternal effect embryonic lethal; hypodermis fails to enclose body; egg laying defective; APH-2 localized to cytoplasm rather than cell surface | (Francis et al. 2002; Goutte et al. 2002) |
| Nicastrin | APH-2 | aph-2 | zu181 | no anterior pharynx; maternal effect embryonic lethal | (Goutte et al. 2000) |
| PEN-2 | PEN-2 | pen-2 | ep219, ep220 ep221, ep336, ep412, ep423 | no anterior pharynx; maternal effect embryonic lethal; hypodermis fails to enclose body; egg laying defective | (Francis et al. 2002) |
| Physical Interactors with APP | |||||
| Fe65 | FE65 | feh-1 | gb561 | embryonic/larval lethal and larval arrest | (Napolitano et al. 2008; Zambrano et al. 2002) |
| Mena | MENA | unc-34 | e951, gm104, gm114 | uncoordinated; axon guidance defect; reduced brood size | (Withee et al. 2004) |
| Tau and Suppressors of Tau Pathogenesis | |||||
| Tau | TAU | ptl-1 | ok621 | incompletely penetrant embryonic lethal; escapers have mechanosensory defect | (Gordon et al. 2008) |
| sut-1 | bk79 | suppresses tau pathogenesis | (Kraemer and Schellenberg 2007) | ||
| MSUT-2 | MSUT-2 | sut-2 | bk741 | suppresses tau pathogenesis | (Guthrie et al. 2009) |
Not included: ADAM9 and APOE4, since no orthologues identified in C. elegans
Functional domains within APL-1
In mammals, Fe65 binds to the APP cytoplasmic tail (Sabo et al. 2001). FEH-1, the C. elegans ortholog of Fe65, also binds directly to the cytoplasmic tail of APL-1 (Zambrano et al. 2002). Knockdown of apl-1 or feh-1 by RNAi causes hyperactive pharyngeal pumping (Zambrano et al. 2002); knockdown of feh-1 also causes an incompletely penetrant embryonic lethality or L1 arrest (Zambrano et al. 2002). These findings suggest that APL-1 acts as a receptor, which transduces a signal through FEH-1 during postembryonic development. However, germline transformation of the transmembrane and cytoplasmic domains of APL-1 did not rescue the apl-1 loss-of-function lethality, although this finding does not preclude that APL-1 signals through the cytoplasmic domain for other functions. In contrast, the extracellular domain of APL-1 was able to rescue the loss-of-function apl-1 lethality (Hornsten et al. 2007). Furthermore, apl-1(yn5) is a viable deletion allele that produces only the extracellular domain of APL-1 (APL-1EXT), indicating that only the extracellular part of APL-1 is necessary and sufficient for viability (Hornsten et al. 2007). APL-1EXT corresponds to the entire APL-1 extracellular domain and is not further cleaved by α-secretase (Hornsten et al. 2007). These findings lead to a new model whereby APL-1 is a multifunctional protein, and in one of its functions, APL-1 is cleaved and the extracellular fragment (sAPL-1) acts as a ligand. Similarly, a knockin of only the extracellular domain of mammalian APP (sAPPα) was able to rescue the phenotypes of an APP knockout mouse (Ring et al. 2007) and the lethality of APP-APLP2 double knockout mice (U. Muller, personal communication). Knockdown of apl-1 by RNAi in wild-type animals shortened body size (Niwa et al. 2008), which is consistent with the reduced body size seen in APP knockout mice (Tremml et al. 1998). Hence, these results highlight how findings in the C. elegans model can be translated back to mammals.
To identify functional domains within sAPL-1, different domains were tested for their ability to rescue the apl-1 loss-of-function lethality. The extracellular domain of the APP family contains two conserved domains, a cysteine-rich E1 domain and an E2 domain rich in acidic residues (Wang and Ha 2004; Zheng and Koo 2006); the E1 and E2 domains do not appear to share any structural similarities. Surprisingly, the presence of either the E1 or E2 domain was sufficient to rescue the apl-1 loss-of-function lethality (Hornsten et al. 2007). The apparent functional redundancy between the two domains is unexpected, but suggests that the extracellular APL-1 fragment interacts through multiple domains.
Functional localization of APL-1
Similar to the ubiquitously expressed human APP (Ponte et al. 1988; Tanzi et al. 1988), apl-1 is expressed during all developmental stages and in many cell types, such as muscles, glial cells, hypodermal cells, and neurons (Fig. 2; Hornsten et al. 2007). To determine in which cell-types apl-1 expression is sufficient for viability, APL-1 expression was driven by different cell-type specific promoters. Because the apl-1 loss-of-function lethality is due to a molting defect, APL-1 expression in hypodermis cells, which produce the cuticle during molting, was expected to rescue the lethality; however, no rescue was detected. In contrast, pan-neuronal APL-1 expression was able to rescue the apl-1 loss-of-function lethality. Furthermore, pan-neuronal expression of only the extracellular domain of APL-1 was also sufficient to rescue lethality, suggesting that after APL-1 is cleaved, sAPL-1 is released from neurons to promote molting (Hornsten et al. 2007). These results raise several questions, including the fate of the extracellular sAPL-1 fragment, the identity of its binding partners, and whether sAPL-1 can be released from a subset of neurons for rescue.
Fig. 2.
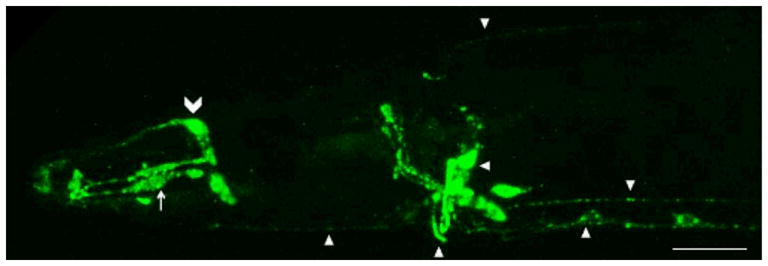
Expression of an APL-1::GFP translational fusion in C. elegans. APL-1::GFP is seen in multiple cell types, including neurons and processes (arrowheads), glial sheath cells (arrow), and muscle cells (chevron). Anterior head region shown, ventral side is down. Scale bar = 10 μm
The secretases in C. elegans
Although APL-1 has not been directly tested for cleavage by different secretases, C. elegans has several orthologues to the mammalian secretases. The secretases will be discussed briefly here, although in the reverse order in which they act. As mentioned above, APP undergoes two cleavages to release Aβ. The second cleavage is mediated by the γ-secretase complex, which consists of at least four proteins: presenilins (PSEN1 and PSEN2), APH-1, Nicastrin/APH-2, and PEN-2 (Kimberly et al. 2003). Mutations in the presenilins were first correlated to FAD in 1995, but their cellular functions were initially unclear (Levy-Lahad et al. 1995a; Rogaev et al. 1995; Sherrington et al. 1995). A few months after these initial reports, the C. elegans ortholog SEL-12 PSEN was identified as a suppressor of LIN-12 Notch signaling (Levitan and Greenwald 1995). This work was significant to the AD field in two ways. First, it provided the first insights into the function of presenilins: presenilins were mediators of Notch signaling, which was well characterized for its critical role in cell-fate decisions during development (reviewed in Greenwald 2005). Second, it raised the possibility that presenilins mediated the activity of multiple proteins in addition to APP and Notch; hence, therapeutic strategies to disrupt presenilins would disrupt not only APP and Notch, but other proteins as well.
Some clues as to how SEL-12 PSEN was acting were made from analysis of sel-12 PSEN suppression of different lin-12 Notch alleles. Gain-of-function mutations in lin-12 Notch cause production of ectopic pseudovulvae (Greenwald et al. 1983), whose formation can be suppressed by decreased sel-12 PSEN activity (Levitan and Greenwald 1995); however, when LIN-12 Notch was constitutively activated by removal of its extracellular domain, decreased sel-12 PSEN activity no longer suppressed formation of pseudovulvae, suggesting that sel-12 PSEN acts either directly on the LIN-12 Notch receptor or upstream in the Notch pathway (Levitan and Greenwald 1998). Loss of sel-12 PSEN caused defects in vulval morphogenesis, leading to an egg-laying defect (Levitan and Greenwald 1995) and deficits in thermal memory (Wittenburg et al. 2000). Germline transformation of sel-12 PSEN mutants with human presenilins restored the egg-laying defect to wild type, indicating that the human and C. elegans genes are functionally homologous (Levitan et al. 1996). Subsequent screening in C. elegans revealed that, like the mammalian systems, C. elegans has multiple presenilins: SEL-12, HOP-1 (Li and Greenwald 1997; Westlund et al. 1999), and SPE-4 (L’Hernault and Arduengo 1992). SPE-4 is required only for spermatogenesis (Arduengo et al. 1998) and loss of sel-12 PSEN cannot be rescued by spe-4 (Eimer 2003). In contrast, loss of sel-12 PSEN can be rescued by hop-1 and knockout of sel-12 and hop-1 in a double mutant caused a synthetic lethality, indicating that the two genes are not only functionally homologous, but have functional overlap (Li and Greenwald 1997; Westlund et al. 1999).
By cleverly tagging each transmembrane domain with a lacZ reporter tag, the Greenwald group demonstrated that SEL-12 PSEN undergoes an obligatory endoproteolysis after the sixth transmembrane domain (Li and Greenwald 1996), similar to the obligatory endoproteolysis of mammalian presenilins (Thinakaran et al. 1996). Mutations in sel-12 PSEN or human presenilins that lead to its own miscleavage increased the levels of Aβ42 in human cell cultures (Okochi et al. 2000). Four years after the initial identification of the presenilins, several groups, including a C. elegans and Drosophila group, proposed that the presenilins were the proteases within the γ-secretase complex (De Strooper et al. 1999; Struhl and Greenwald 1999; Wolfe et al. 1999; Ye et al. 1999).
Multiple lines of evidence, however, indicated that γ-secretase was a complex of proteins; mammalian PSEN1, for instance, co-fractionated in a high molecular weight complex (Li et al. 2000). Further immunoextraction of proteins tightly associated with PSEN1 yielded nicastrin, a glycosylated transmembrane protein that binds PSEN1/2 and APP, suggesting that nicastrin’s role was to target a substrate to the γ-secretase complex (Yu et al. 2000). Loss of aph-2/nicastrin in C. elegans resulted in maternal effect embryonic lethality, similar to the phenotype seen in glp-1 Notch mutants (Goutte et al. 2000; Levitan et al. 2001). Given the high molecular weight complex that co-frac-tionated with PSEN1, other components in addition to nicastrin had to be present. The power of the genetic approaches available in C. elegans again contributed to the identification of these other components. Genetic screens for enhancers of sel-12 PSEN phenotypes in C. elegans pulled out APH-1 and PEN-2 (Francis et al. 2002; Goutte et al. 2000), whose mammalian orthologues were the major missing components within the γ-secretase complex. Loss of aph-1 or pen-2 conferred maternal effect embryonic lethality, similar to the phenotypes seen in glp-1 Notch and aph-2/nicastrin mutants (Francis et al. 2002; Goutte et al. 2000). APH-1, a seven transmembrane domain protein, and PEN-2, a two transmembrane domain protein, bind PSENs and nicastrin to facilitate assembly and maturation of the γ-secretase complex (Francis et al. 2002; Goutte et al. 2000, 2002; Gu et al. 2003; Levitan et al. 2001). An additional gene identified in a genetic screen for suppression of sel-12 PSEN activity was sel-10, which encodes a protein of the CDC4/CUL-1 E2-E3 ubiquitin ligase family (Hubbard et al. 1997). SEL-10 physically interacts with SEL-12 PSEN, presumably to target it for degradation (Wu et al. 1998). Similarly, the human homolog SEL-10 physically interacts with and facilitates ubiquitination of human PSEN1 and affects Ab42 production in mammalian cells (Li et al. 2002).
Two α-secretases, sup-17 ADAM10 and adm-4 ADAM17/TACE, have been identified in C. elegans (Jarriault and Greenwald 2005; Tax et al. 1997). These two proteases act redundantly to process LIN-12 Notch (Jarriault and Greenwald 2005) and presumably APL-1. Loss of sup-17 ADAM10 results in lethality, which can be rescued by germline transformation with either sup-17 ADAM10 or adm-4 ADAM17/TACE (Jarriault and Greenwald 2005; Tax et al. 1997; Wen et al. 1997). Thus far, no β-secretase activity that can cleave human APP has been identified in C. elegans (Link 2006).
Regulation of APL-1 in C. elegans
Regulation of APL-1 expression has only been explored for the time point of the L4 larval-to-adult transition. In wild-type animals, the seams cells, a specialized type of hypodermal cells, serve as stem cells to generate new hypodermal cells during the different molts; however, during the L4 larval-to-adult transition, the seam cells undergo terminal differentiation and join the hypodermal syncytium (Sulston and White 1980). The L4 larval-to-adult transition is regulated by the heterochronic genes hbl-1, lin-41, and lin-42 (Abrahante et al. 2003; Fay et al. 1999; Jeon et al. 1999; Slack et al. 2000). Loss of these heterochronic genes caused adult fates to be executed precociously and an incompletely penetrant molting defect from L4 to adult (Abrahante et al. 2003; Jeon et al. 1999; Slack et al. 2000); in contrast, overexpression of these genes caused a reiteration of larval cell fates (Abrahante et al. 2003; Slack et al. 2000). Hence, expression of these heterochronic genes must be down-regulated for entry into the adult stage. This down-regulation occurs in part through negative regulation by let-7, a heterochronic microRNA that has been shown to suppress human cancers (Kumar et al. 2008). hbl-1 negatively regulates let-7, such that expression of let-7 does not occur before L3 (Roush and Slack 2009). mir-48 and mir-84, heterochronic microRNAs of the let-7 family, are responsible for controlling the L2–L3 larval transition partially by negatively regulating hbl-1 (Abbott et al. 2005). Loss-of-function let-7 mutations cause a supernumerary fifth molt during the L4 to adult transition, resulting in the production of extra seam cells, and an adult lethality due to vulval bursting, presumably due to the extra hypodermal cells (Reinhart et al. 2000).
During the L4 larval-to-adult transition, apl-1 expression appears in seam cells (Niwa et al. 2008). Knockdown of apl-1 by RNAi reduced apl-1 levels to 40% of wild type and suppressed the vulval bursting and additional seam cells in let-7 mutants (Niwa et al. 2008). These data suggest that APL-1 is regulated by let-7 at the L4 larval-to-adult transition to allow proper molting. If apl-1 acts downstream of the let-7 targets hbl-1, lin-41, and lin-42, then loss of apl-1 should enhance the molting phenotype of hbl-1, lin-41, and lin-42 mutants. Knockdown of apl-1 by RNAi indeed enhanced the L4 to adult molting defects in the hbl-1, lin-41, and lin-42 single mutants, suggesting that apl-1 acts downstream and is a potential target of these heterochronic genes (Niwa et al. 2008). A cold-sensitive mutation of mir-48 causes a low penetrance supernumerary molt at the young adult stage, presumably because hbl-1 is not down-regulated (Reinhart et al. 2000). The penetrance of this phenotype is enhanced in a non-temperature dependent manner in a mir-84 loss-of-function background and the mir-48; mir-84 double mutant shows two cuticles (Abbott et al. 2005). Knockdown of apl-1 in mir-48; mir-84 double mutants is sufficient to suppress the supernumerary molt and formation of a double cuticle (Niwa et al. 2008). These results indicate that APL-1 expression is temporally regulated by heterochronic microRNAs and regulators during the L4 larval-to-adult transition.
Overexpression of APL-1 induces several phenotypes
A duplication of the APP locus is correlated with FAD (Cabrejo et al. 2006; Rovelet-Lecrux et al. 2006; Sleegers et al. 2006), suggesting that increased Aβ levels contributes to the FAD pathology. However, overexpression of the APP extracellular fragment, hyper-signaling through the APP cytoplasmic domain, or toxicity of the APP cytoplasmic fragment may also contribute to the FAD pathology. For instance, the extracellular domain of human APP (sAPP) can bind to the non-canonical DR6 death receptor to induce cell and axonal degeneration in the absence of trophic factors (Nikolaev et al. 2009). In addition, transgenic mice overexpressing an FAD version of human APP with an additional mutation (D664A) in a caspase cleavage site to inhibit the release of the cytoplasmic peptide (APP-C31) show reduced behavioral defects, despite increased Aβ deposits compared to the FAD version alone (Galvan et al., 2008), implicating an important role for the cytoplasmic tail in FAD pathogenesis. Hence, the effects of APL-1 overexpression were examined in C. elegans through the use of transgenic animals (Table 1). When generating transgenic animals, microinjected or co-injected DNA appear as extrachromosomal arrays, which are inherited by progeny at a low frequency because the arrays can be lost during cell division. The arrays, which are generally multiple tandem copies of the single or co-injected DNA, can be integrated into the genome, which allow their Mendelian transmission (Mello and Fire 1995). To determine whether APL-1 overexpression causes any phenotypes, several transgenic lines in which different apl-1 transgenes were present as arrays or integrated into the genome in an otherwise wild-type background were examined. By Western blot analysis, the transgenic lines expressed APL-1 or APL-1::GFP at levels from 15-to 180-fold higher than wild type (Hornsten et al. 2007).
The overexpression lines had defects in brood size, movement, and viability (Table 1); the severity of these defects was strongly correlated with the level of APL-1 overexpression (Hornsten et al. 2007). Wild-type animals generally lay between 250 and 300 eggs (Byerly et al. 1976). Animals overexpressing APL-1 laid significantly fewer eggs than wild type (Hornsten et al. 2007). Because apl-1(yn5) APL-1EXT animals also showed a decreased number of progeny, brood size may be decreased by elevated levels of sAPL-1, perhaps because of interference with cell–cell interactions or adhesion defects that disrupt morphogenesis and/or gonadal development (Hornsten et al. 2007). All apl-1 transgenic overexpression strains also showed significantly reduced swimming and crawling rates compared to wild type (Table 1). Increasing levels of APL-1, therefore, inhibit movement, perhaps by interfering with motor neuron functions.
The overexpression line that had the highest levels of APL-1 (~180-fold higher than wild type), ynIs79 APL-1::GFP, exhibited the most severe phenotypes, including an incompletely penetrant (70%) larval lethality (Table 1; Hornsten et al. 2007). One other overexpression line, ynIs86 APL-1, also showed lethality, but at a much lower rate (5.5%). ynIs79 APL-1::GFP overexpression animals appear morphologically wild type at hatching. At variable times during L1, ynIs79 APL-1::GFP animals became translucent and large gaps became evident between organs. These phenotypes are consistent with disruptions in cell adhesion whereby elevated levels of APL-1 interfere with normal adhesion contacts between cells. Alternatively, APL-1 overexpression could cause defects in osmoregulation. Mutations in clr-1, which encodes a phosphatase involved in regulating osmotic pressure (Kokel et al. 1998), also cause a translucent phenotype (Hedgecock et al. 1990). Decreased sel-12 PSEN activity partially suppressed the apl-1 overexpression lethality, suggesting that the interactions between the presenilins and the APP family are conserved between worms and mammals (Hornsten et al. 2007).
Loss-and gain-of-function apl-1 lethality are not mediated by caspases
Mammalian APP can regulate apoptosis (for review, see Chen 2004) as well as be cleaved by different caspases (Barnes et al. 1998; Gervais et al. 1999). The apl-1 loss-of-function and overexpression-induced lethality were examined for activation of an apoptotic or necrotic cell death pathway. ced-3 encodes a caspase that is essential for execution of apoptosis in C. elegans (Horvitz 1999) and crt-1 encodes calreticulin, which is essential for execution of necrotic cell deaths (Xu et al. 2001) in C. elegans. Neither loss of ced-3 caspase nor loss of crt-1 calreticulin activity rescued the apl-1 loss-of-function lethality, indicating that this lethality is not due to ectopic activation of either cell death pathway. Similarly, the APL-1 overexpression lethality of ynIs79 APL-1::GFP is not due to activation of the ced-3 apoptotic pathway (Hornsten et al. 2007). Whether the apl-1 lethality is mediated by an autophagic pathway is unknown.
The amyloid cascade hypothesis: C. elegans models to induce degeneration
Hardy and Allsop first proposed the amyloid cascade hypothesis in 1991 to explain the pathology seen in AD (Hardy and Allsop 1991). Briefly, they postulated that imbalances in APP metabolism lead to increased levels of Aβ, and in particular Aβ42, which can then aggregate into plaques. Formation of these plaques can in turn lead to the other pathologies, such as neurofibrillary tangles and neurodegeneration, seen in AD. C. elegans has been used in several ways to test the amyloid cascade hypothesis (Table 2).
Table 2.
Overexpression of C. elegans proteins implicated in AD
| C. elegans protein | C. elegans promoter | Expression in C. elegans | Fold over-expression | Rescuing ability & phenotypes | Transgene name/(plasmid) | References |
|---|---|---|---|---|---|---|
| Endogenous expression of APL-1 | ||||||
| APL-1 | apl-1 | head and tail neurons, ventral cord, hypodermis and supporting cells, vulva muscles | 125x | rescues apl-1 null lethality; low level (5.5%) L1 lethality; reduced brood size; sluggish | ynIs86 | (Hornsten et al. 2007) |
| APL-1::GFP | 180x | rescues apl-1 null lethality; high level (70%) L1 lethality and morphological defects; cell and organ detachment; reduced brood size; sluggish | ynIs79 | (Hornsten et al. 2007) | ||
| APL-1 extracellular domain | rescues apl-1 null lethality; slowed development; reduced brood size; sluggish | ynIs71, ynEx106, ynIs106A | (Hornsten et al. 2007) | |||
| APL-1ΔGoa | rescues apl-1 null lethality | (Hornsten et al. 2007) | ||||
| APL-1ΔE1b | rescues apl-1 null lethality | (Hornsten et al. 2007) | ||||
| APL-1ΔE2c | rescues apl-1 null lethality | (Hornsten et al. 2007) | ||||
| APL-1ΔE1-E2d | no rescue of apl-1 null lethality | (Hornsten et al. 2007) | ||||
| Neuronal expression of APL-1 | ||||||
| APL-1 | snb-1 | constitutively in all neurons; pharynx; arcade cells; distal tip cell; vulval muscle; spermatheca; gonad sheath cells; body wall muscle; hypodermis; seam cellse | 71x and 17x respectively | rescues apl-1 null lethality; reduced brood size; sluggishness | ynIs12, ynIs13 | (Hornsten et al. 2007) |
| APL-1 extracellular domainf | rescues apl-1 null lethality | ynEx166 | (Hornsten et al. 2007) | |||
| APL-1::GFP | rab-3 | constitutively in all neurons | rescues apl-1 null lethality | (Hornsten et al. 2007) | ||
| Expression of Proteins in the γ-secretase complexg | ||||||
| SEL-12 | sel-12 | constitutively in most cell types, except intestine | rescues sel-12 null phenotypes | byIs100, byIs101, SEL-12 | (Levitan et al. 1996; Wittenburg et al. 2000) | |
| SEL-12 | ttx-3 | only in AIY neuron | rescues sel-12 null phenotypes | (pBY478) | (Wittenburg et al. 2000) | |
| SEL-12 | egl-13 | Pi cell, neurons, bodywall muscles, intestine | partially rescues egg laying defect and Pi cell fate of sel-12 null worms | (cHNC2) | (Cinar et al. 2001) | |
| HOP-1 | sel-12 | constitutively in most cell types, except intestine | rescues sel-12 null phenotypes | HOP-1 | (Li and Greenwald 1997) | |
| APH-1 | sel-12 | constitutively in most cell types, except intestine | rescues egg laying defect of aph-1 null worms (Note: no rescue under aph-1 endogenous promoter) | Ce3aph-1 | (Francis et al. 2002) | |
| PEN-2 | pen-2 | neurons, muscles, intestine, vulva | rescues egg laying defect of pen-2 null worms | pen-2 genomic | (Francis et al. 2002) | |
| PEN-2 | sel-12 | constitutively in most cell types, except intestine | rescues egg laying defect of pen-2 null worms | Ce pen-2 | (Francis et al. 2002) | |
| Endogenous expression of the α-secretase | ||||||
| ADM-4 | adm-4 | pharynx, intestine, tail | rescues sterility of sup-17; adm-4 double mutants | arEx399, arEx400 | (Jarriault and Greenwald 2005) | |
APL-1ΔGo: deletion of Go-binding sequence
APL-1ΔE1: deletion of E1 domain
APL-1ΔE2: deletion of E2 domain
APL-1ΔE1-E2: deletion of E1 through E2 domains
Expression pattern of Psnb-1::GFP (BC11116; Hunt-Newbury et al. 2007)
APL-1EXT: the entire extracellular domain of APL-1. APL-1EXT is not further cleaved and is slightly larger than sAPL-1
No rescue of maternal effect lethal phenotype for aph-1, aph-2, pen-2, probably due to co-suppression of their endogenous locus in the germline, a general germline effect described in Dernburg et al. (2000)
Mutations correlated with FAD favor production of Aβ42 and Aβ43 and this increased Aβ42 production presumably leads to a higher accumulation of Aβ into neurotoxic extracellular plaques (Lorenzo and Yankner 1996). In mice, introduction of Aβ42 leads to neurodegeneration and a severe decrease in lifespan (Laferla et al. 1995). To determine whether Aβ plaques are toxic in C. elegans, human Aβ42 was introduced into muscle cells by germline transformation (Table 2; Link 1995). The resultant transgenic animals showed a progressive, irreversible paralysis (Link 1995). Staining these transgenic worms with a human Aβ42 specific antibody (mAbG8) revealed an accumulation of Aβ42 deposits, which reacted with Congo red, thioflavin S, and X-34, all markers of Aβ42 deposits (Fay et al. 1998; Link 1995). Ultrastructural examination of the Aβ42 deposits, however, revealed that the deposits were located in the cytoplasm of the muscle cells (Link et al. 2001) rather than as extracellular deposits or plaques as seen in AD brains (Selkoe 2001); the ultrastructural images were insufficient to resolve whether the Aβ42 deposits were in the cytoplasm or, more likely, within intracellular inclusions. This intracellular localization was somewhat surprising given that the human Ab42 construct was made with an artificial signal sequence that should have led to the extracellular release of Aβ42. The artificial signal sequence is functional, although cleavage occurs after the signal sequence so that the Aβ sequence corresponds to amino acids 3–42 rather than 1–42; the Aβ3-42 species appears more toxic than Aβ1-42 species (McColl et al. 2009). Collectively, these results demonstrated that after production, Aβ42 is targeted either by the endoplasmic reticulum (ER) quality control system and retrotranslocated for degradation (Link 2006) and/or sequestered into intracellular inclusions by autophagic vesicles (Florez-McClure et al. 2007). Aβ42 co-immunoprecipitated with several chaperone-related proteins, including two HSP70 ER chaperone proteins, in C. elegans (Fonte et al. 2002). Intracellular Aβ42 accumulation seems to be crucial in AD pathogenesis (Gouras et al. 2000; Laferla et al. 1997), and some Aβ42 transgenic mouse models that display AD behavioral phenotypes also show intracellular Aβ42 accumulation rather than extracellular plaque formation (Chui et al. 1999; Kuo et al. 2001; Laferla et al. 1995; Li et al. 1999; Oddo et al. 2003b; Wirths et al. 2001). Hence, intracellular Aβ42 accumulation could be a first step in AD pathogenesis, and the C. elegans human Aβ42 transgenic animals are an excellent model to examine intracellular Aβ42 toxicity and metabolism.
Investigating Aβ42 toxicity in C. elegans
The amyloid cascade hypothesis implicitly implies that Aβ42 plaques and not soluble forms of Aβ42 are the toxic species in vivo. The human Aβ42 transgenic worms offer several advantages to investigating the intracellular toxicity of Aβ species in vivo. To determine which residues are relevant to fibril formation in vivo, transgenic lines containing different human Aβ42 variants that do not form fibrils and plaques in vitro were generated in C. elegans and examined for their fibril formation and intracellular deposits (Table 2; Fay et al. 1998). Transgenic worms expressing human Aβ42 variants containing Leu17Pro and Met35Cys substitutions or an artificial single chain Aβ42 dimer formed no thioflavin S-reactive deposits, although some of the strains had Aβ42 variant levels comparable to lines expressing wild-type Aβ42 (Fay et al. 1998). Hence, Aβ42 residues Leu17 and Met35 are required for fibril formation and Aβ42 deposits in vivo as well. To determine whether presence of intracellular Aβ42 deposits is correlated with paralysis, an inducible system for human Aβ42 expression in muscles was developed (Link et al. 2003). Within several hours of Aβ42 induction, animals showed paralysis without any detectable Aβ42 deposits (Drake et al. 2003). Hence, Aβ42 deposits are not required to induce the Aβ42 paralysis phenotype in C. elegans, suggesting that either intracellular Aβ42 deposits are not the toxic species or a small Aβ42 aggregation not visible as a deposit is also toxic. Consistent with these findings, the transgenic AD model mouse (Tg2576) displayed morphological, behavioral and memory deficits months before Aβ42 plaque deposition was apparent (Jacobsen et al. 2006). Furthermore, plaque prevalence does not strictly correlate with dementia in AD (Davies et al. 1988; Mann et al. 1992; Schmitt et al. 2000).
How can Aβ42 be toxic in C. elegans even in the absence of Aβ42 deposits? One possibility is that Aβ42 induces cells to undergo oxidative stress. Transgenic worms expressing human Aβ42, but not the Aβ42 Met35Cys variant, showed increased levels of protein oxidation (Yatin et al. 1999). A closer look at the temporal formation of plaques and the oxidative stress response revealed that oxidation of proteins precedes presence of Aβ42 deposits (Drake et al. 2003), suggesting that soluble Aβ42 or small aggregates of Aβ42, rather than deposits, may induce oxidative stress in vivo, thereby causing Aβ42 toxicity. Similarly, rat hippocampal cell cultures exposed to exogenous Aβ42, but not the Aβ42 Met35Nle variant, have increased levels of protein carbonyls and neurotoxicity, presumably due to increased protein oxidation (Yatin et al. 1999). A second possibility for Aβ42 toxicity is that intracellular Aβ42 inclusions are disrupting the ER-assisted degradation or autophagic degradation pathways (Florez-McClure et al. 2007). Hence, possible mechanisms for Aβ42 toxicity in C. elegans are oxidative stress and/or disruption of ERAD or autophagy pathways (see also below).
Cellular response to human Aβ42 expression in C. elegans
Assuming that some form of Aβ42 is toxic or disruptive to cells, then a defense mechanism that protects the cells must be initially mounted; such a mechanism is likely conserved among all animals. To identify this Aβ42 detoxification pathway in C. elegans, two approaches were undertaken. First, as mentioned above, proteins that co-immunoprecipitated with Aβ42 were characterized by MALTI-TOF to reveal two HSP70-related proteins, three HSP-16 αB-crystallin-related proteins, and a putative negative regulator of HSP70, 3R05F9.10, all of which are involved in chaperone activity (Fonte et al.2002). RNAi knockdown of the putative negative regulator of HSP70 (R05F9.10) partially suppressed the paralysis of worms expressing human Aβ42 in muscles, suggesting that the transgenic animals up-regulate heat shock proteins to cope with the Aβ42 toxicity (Fonte et al. 2002). Furthermore, HSP-16 co-localizes with Aβ42 and binds it directly in vitro (Fonte et al. 2002, 2008). Inducing Aβ42 expression leads to a strong transcriptional increase of HSP-16 (Fonte et al. 2002) and targeted over-expression of HSP-16 partially suppresses Aβ42 toxicity (Fonte et al. 2008). The second approach to identify components of a degradation pathway was a microarray analysis to find genes that are either strongly up-or down-regulated by induced human Aβ42 expression in C. elegans. Genes that were up-regulated, as further verified by RT-PCR, included the two HSP-16 chaperone proteins and two proteins with homology to the TNFAIP1 family, which are induced by the tumor necrosis factor α (Link et al. 2003). Interestingly, subsequent RT-PCR of CRYAB, a mammalian ortholog of HSP-16, and TNFAIPI showed their up-regulation in AD brain tissues, suggesting a common Aβ42 detoxification pathway in worms and mammals (Link et al. 2003).
Clearance of Aβ42 in C. elegans
The finding of a potential Aβ42 detoxification pathway leads to the question of how Aβ42 is cleared from cells in C. elegans. One general mechanism could be through autophagy, whereby misfolded, excess, or used proteins or other cellular products are engulfed into a double membrane to form autophagic vesicles, which fuse with lysosomes where the engulfed components are degraded. In AD brains, there is an abnormal accumulation of autophagic vesicles, particularly in the dystrophic neurites of neocortical and hippocampal neurons (Nixon et al. 2005; Yu et al. 2005). In mouse models of AD, autophagic vacuoles contain APP, Aβ, and the APP cytoplasmic terminal fragment CTFβ after β-secretase cleavage (Yu et al. 2004). Similarly, inducible expression of human Aβ42 in muscles leads to an accumulation of autophagic vesicles in C. elegans (Florez-Mcclure et al. 2007). To determine whether autophagic vesicle formation causes paralysis, autophagic vesicle formation was blocked by RNAi knockdown of bec-1, which encodes an APG6/VPS30 protein that is part of the complex that localizes proteins to autophagic vesicles, and atg-7, which encodes an E1 ubiquitin-activating-like enzyme orthologous to the autophagic budding yeast protein Apg7p. Such knockdown in Aβ42 transgenic animals had no effect on the levels of Aβ42 or paralysis rate. In contrast, knockdown of lysosomal component genes, such as aspartyl proteases (asp-2, asp-4, asp-5, and asp-6), lysosomal-associated membrane proteins (lmp-1 and lmp-2), and vacuolar proton-translocating ATPase (vha-15), by RNAi enhanced the paralysis rate and caused higher Aβ42 levels. Hence, Aβ42 toxicity in C. elegans may result from impaired lysosomal degradation due to defective lysosomal formation and/or vesicular acidification (Florez-Mcclure et al. 2007).
Interestingly, a mutation in daf-2, which encodes an insulin/IGF-1 receptor, decreases paralysis and levels of Aβ42 peptide, but not Aβ42 mRNA levels, in C. elegans with inducible expression of human Aβ42 in muscles (Florez-Mcclure et al. 2007). These animals also had an increased number of lysosomes and few or no autophagic vesicles. While these data may suggest the presence of an autophagy-independent route for Aβ42 degradation in daf-2 IGF-1/Insulin Receptor mutants, bec-1 APG6/VPS30 is needed for the suppression of the paralysis and for deceased Aβ42 peptide levels (Florez-Mcclure et al. 2007). Hence, Aβ42 is targeted to lysosomes in part by autophagic vesicles.
Age-dependent onset of proteotoxicity
Mutations in daf-2 insulin/IGF-1 receptor extend lifespan, such that animals live twice as long as wild type (Kenyon et al. 1993). DAF-2 insulin/IGF-1 receptor is a negative regulator of the FOXO transcription factor DAF-16 (Lin et al. 1997; Ogg et al. 1997), which activates genes involved in longevity, stress resistance, protein folding, and detoxification (Barsyte et al. 2001; Honda and Honda 1999; Murphy et al. 2003; Walker et al. 2001). The lifespan extension in daf-2 insulin/IGF-1 receptor mutants also depends on the presence of the heat shock factor 1 (HSF-1) transcription factor (Hsu et al. 2003). However, although both DAF-16 and HSF-1 are necessary, they are not sufficient for the extended longevity in daf-2 insulin/IGF-1 receptor mutants (Hsu et al. 2003; Lin et al. 2001) and overexpression of DAF-16 or HSF-1 does not mimic the extreme longevity of daf-2 insulin/IGF-1 receptor mutants (Henderson and Johnson 2001; Hsu et al. 2003). Interestingly, reducing daf-2 insulin/IGF-1 receptor activity not only dramatically increases lifespan, but also makes these worms much healthier (Kenyon et al. 1993). Indeed, knockdown of daf-2 insulin/IGF-1 receptor by RNAi in worms expressing human Aβ42 in muscles, not only decreases paralysis, but also dramatically increases their lifespan (Cohen et al. 2006). These effects are dependent on DAF-16 and HSF-1 and do not affect overall mRNA or protein levels of human Aβ42 (Cohen et al. 2006). Given that the Aβ42 deposits are intracellular, these results suggest that the delay in Aβ42 toxicity in daf-2 knockdown animals is due to Aβ42 being removed more efficiently either through clearance or detoxification (Cohen et al. 2006). A possible future approach to AD could be a pharmacological treatment via the insulin signaling pathway. Some AD patients have altered insulin signaling, which manifests in a brain specific diabetes (Steen et al. 2005).
Pharmacological approach to ameliorate Aβ42 toxicity in C. elegans
Several drugs, particularly ones that decrease reactive oxygen species (ROS) generated by oxidative stress, have been examined for their effects in decreasing toxicity in the transgenic worms expressing human Aβ42 in muscles. EGb761, an extract from the Ginkgo biloba that is sold as a dietary supplement, is reported to have several beneficial properties, such as neuroprotection (anti-apoptotic effects in cell culture; Smith et al. 2002), inhibition of Aβ aggregation (Luo et al. 2002), lifespan extension in rats (Winter 1998), cognitive enhancer in mice (Winter 1991), and stabilization of symptoms and improvement of cognitive performance in AD patients (Curtis-Prior et al. 1999; Le Bars et al. 1997;Le Bars et al. 2000; Mix and Crews 2002; Oken et al. 1998). EGb761 was also found to increase lifespan and slow the rate of paralysis in human Aβ42 transgenic worms (Wu and Luo 2005; Wu et al. 2006). EGb761 decreases toxic ROS levels induced by Aβ42 in C. elegans muscles and inhibits Aβ42 oligomerization and deposits (Smith and Luo 2003; Wu et al. 2006). However, human Aβ42 transgenic worms treated with ascorbic acid, an antioxidant, or ginkgolide GB or GC, two components within EGb761, had low levels of ROS or Aβ42 oligomers, respectively, but high rates of Aβ42-induced paralysis (Wu et al. 2006). Hence, whether levels of ROS or Aβ42 oligomers underlie the increased lifespan and slower rate of paralysis with EGb761 treatment is unclear. Other chemicals that protect against ROS levels are isoflavones (Mahn et al.2005). Glycitein, an isoflavone isolated from soy beans, also suppressed the Aβ42-induced toxic hydrogen peroxide levels, attenuated the paralysis rate, and decreased numbers of Aβ42 deposits in transgenic Aβ42 worms (Gutierrez-Zepeda et al. 2005), indicating that in some instances decreasing ROS levels can decrease Aβ42 toxicity.
Reserpine is an FDA approved psychopharmalogical and antihypertensive drug (NDA#009838; Bleuler and Stoll 1955; Vakil 1949) that downregulates biogenic amines through inhibition of the vesicular monoamine transporter (VMAT; Metzger et al. 2002). Surprisingly, in human Aβ42 transgenic worms, reserpine increased lifespan and slowed the rate of paralysis without affecting overall Aβ mRNA and protein levels, similar to knockdown of daf-2 insulin/IGF-1 receptor by RNAi (Arya et al. 2009). However, in contrast to knockdown of daf-2 insulin/IGF-1 receptor, lifespan extension by reserpine is not dependent on daf-16 (Srivastava et al. 2008). Whether the protective efficacy of reserpine is mediated through the insulin signaling pathway is not known.
Drugs that inhibit presenilin activity have also been tested in C. elegans. The compound BMS AG6B was first discovered in a cell-based screen for compounds that alter Aβ ratios. Applying BMS AG6B on C. elegans induced a specific inhibition of sel-12 PSEN function and thus revealed the mechanism of action for the compound (reviewed in Carroll and Fitzgerald 2003).
Tau in C. elegans
Because mutations correlated with FAD were biased toward the amyloid cascade hypothesis, much of AD research has been focused on APP and Aβ. However, a second postmortem criterion in AD is the presence of intracellular neurofibrillary tangles, whose major component is hyperphosphorylated tau, a MAP (Brion et al. 1991; Delacourte and Defossez 1986; Grundke-Iqbal et al. 1986; Kosik et al. 1986). There are several reasons for the neglect of the tau pathology. First, neurofibrillary tangles are not uniquely seen in AD, but also in other neurodegenerative diseases, such as frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17; Hulette et al. 1999; Mirra et al. 1999; Spillantini et al. 1998). Second, there are no FAD cases correlated with mutations in tau (Kwon 2008). Third, Aβ42 deposition precedes and can induce hyperphosphorylation of tau (Lewis et al. 2001; Oddo et al. 2003a; Zheng et al. 2002). Nevertheless, the transgenic mouse model that phenocopies the most pathologies seen in AD is a triple transgenic mouse, whose transgenes carry mutations in APP, presenilin (PSEN1), and tau. These triple transgenic mice of mutant APPSwe, PSEN1M146V, and tauP301L display plaques and tangles and show behavioral deficits that are characteristic of AD (Oddo et al. 2003b).
Tau-related protein in C. elegans
Although no tau homolog is present in C. elegans, a ‘protein with tau-like repeats’ (PTL-1) has been identified (Table 3; Goedert et al. 1996; Mcdermott et al. 1996). PTL-1 shares 50% sequence identity within the microtubule tandem repeats with mammalian tau, MAP2, and MAP4 (Goedert et al. 1996; Mcdermott et al. 1996). PTL-1 is expressed in the embryonic epidermis, intestinal muscle, mechanosensory neurons, a number of head neurons, and motor neurons in the ventral nerve cord (Goedert et al. 1996), binds to microtubules, and facilitates microtubule assembly in ptl-1 transfected COS cells (Goedert et al. 1996). Inactivation of ptl-1 caused an incompletely penetrant embryonic lethality; escaping animals showed a reduced mechanosensory response and enhanced defects in animals also carrying mutations in mec-12 and mec-7, which encode alpha and beta tubulin subunits, respectively (Gordon et al. 2008). These results indicate that PLT-1 is involved in two main processes: elongation during development and mechanosensation in larval and adult animals; both roles may require PTL-1 to provide structural support for microtubules.
Table 3.
AD toxicity models of human proteins expressed in C. elegans
| Human protein/peptide | C. elegans promoter | Expression in C. elegans | Phenotypes | Strain/transgene name/(plasmid) | References |
|---|---|---|---|---|---|
| Expression of the human beta amyloid peptide | |||||
| Aβ1-42 (wild type) | unc-54 | constitutively in muscles | age-dependent progressive paralysis; forms amyloid deposits; increased oxidative stress | CL2005, CL2006, CL1019, CL1118, CL1119, CL1120, CL1121, CL2120 | (Fay et al. 1998; Link 1995; Link et al. 2001; Yatin et al. 1999) |
| Dimer Aβ1-42 | no formation of amyloid deposits | CL2109, CL3109 | (Fay et al. 1998; Link et al. 2001) | ||
| Met35Cys Aβ1-42 | no formation of amyloid deposits; no increase in oxidative stress | CL3115 | (Fay et al. 1998; Yatin et al. 1999) | ||
| Aβ1-42 (with long 3′UTR) | myo-3 | inducible Aβ1-42 in body wall muscles | rapid paralysis; oxidative stress precedes amyloid deposition; autophagosome accumulation | CL4176 | (Drake et al. 2003; Florez-McClure et al. 2007; Link et al. 2003) |
| Aβ1-42 | snb-1 | inducible Aβ1-42 in all neurons | normal movement; forms amyloid deposits; reduced chemotaxis towards Benzaldehyde; hypersensitive to serotonin | CL2241, CL2355 | (Link 2006; Wu et al. 2006) |
| Expression of Components of the γ-Secretase Complex | |||||
| PSEN1 | sel-12 | constitutively in most cell types, except intestine | rescues sel-12 null phenotypes | PS1, (pBY146) | (Levitan et al. 1996; Wittenburg et al. 2000) |
| Mutant PSEN1 variants | fails to rescue sel-12 null phenotypes | PS1ΔE9, PS1M146L, PS1H163R, PS1L266V, PS1A286E, PS1C410Y, A246(pBY147) | (Levitan et al. 1996; Wittenburg et al. 2000) | ||
| PSEN2 | rescues sel-12 null phenotypes | PS2 | (Levitan et al. 1996) | ||
| Nicastrin | rescues egg-laying defect of aph-2 null | hNCT FL | (Levitan et al. 2001) | ||
| Mutant Nicastrin variants | partially rescues egg-laying defect of aph-2 null | DYIGS, AAIGS, Δ340, Δ 69, EC | (Levitan et al. 2001) | ||
| APH-1 | human APH-1 is unable to rescue egg-laying defect of aph-1 null worms; human APH-1 can partially rescue egg-laying defect of aph-1 null only in mixture with Hpen-2, Haph-1a, Haph1b and HPSEN1 | Haph-1a, Haph1b | (Francis et al. 2002) | ||
| PEN-2 | Partially rescues egg-laying defect of pen-2 null (with long 3′ UTR) only in mixture with Hpen-2, Haph-1a, Haph1b and HPSEN1 | Hpen-2 | (Francis et al. 2002) | ||
| Expression of Human Tau and Variants | |||||
| Tau (4R1N isoform, most abundant form in human brain) | aex-3 | constitutively in all neurons | age-dependent progressive uncoordination and accumulation of insoluble tau; neurodegeneration; reduced lifespan compared to non- transgenic worms | N-1, N-2 | (Kraemer et al. 2003) |
| V337M Tau (4R1N) (FTDP-17 mutation) | aex-3 | constitutively in all neurons | stronger age-dependent progressive uncoordination and accumulation of insoluble tau; neurodegeneration; reduced lifespan compared to non-transgenic worms | 337M-1,3337M-2 | (Kraemer et al. 2003) |
| P301L tau (4R1N) (FTDP-17 mutation) | aex-3 | constitutively in all neurons | strong age-dependent progressive uncoordination and accumulation of insoluble tau; neurodegeneration; reduced lifespan compared to non-transgenic worms | 301L-1, 301L-2 | (Kraemer et al. 2003) |
| Tau WT4R (wild type) | mec-7 | touch neurons (ALML/R, AVM, PLML/R, PVM); weak in FLP, PVD, BDU | age-dependent progressive impairment in touch response; neurodegeneration; little tau accumulation in PLM neuron | tmIs82, tmIs83, tmIs84, tmIs85, tmIs171 | (Miyasaka et al. 2005) |
| Tau WT R (wild type) | mec-7 | touch neurons (ALML/R, AVM, PLML/R, PVM); weak in FLP, PVD, BDU | age-dependent progressive impairment in touch response, neurodegeneration | tmIs110, tmIs173 | (Miyasaka et al. 2005) |
| P301L tau (FTDP-17 mutation) | mec-7 | touch neurons (ALML/R, AVM, PLML/R, PVM); weak in FLP, PVD, BDU | strong age-dependent progressive impairment in touch response; neurodegeneration; strong tau accumulation in PLM neuron (as wild type tau WT4R) | tmIs81, tmIs178, tmIs179 | (Miyasaka et al. 2005) |
| R406W tau (FTDP-17 mutation) | mec-7 | touch neurons (ALML/R, AVM, PLML/R, PVM); weak in FLP, PVD, BDU | strong age-dependent progressive impairment in touch response; neurodegeneration; strong tau accumulation in PLM neuron (as wild-type tau WT4R) | tmIs146, tmIs147, tmIs148, tmIs149 | (Miyasaka et al. 2005) |
| Tau352 (=fetal, 352aa isoform) wildtype | rgef-1 | constitutively in all neurons | age-dependent progressive uncoordination; neurodegeneration | VH255, VH1016, VH1018 | (Brandt et al. 2009) |
| Tau352 PHP (pseudo- hyperphosphorylated) | rgef-1 | constitutively inall neurons | strong-age dependent progressive uncoordination; neurodegeneration | VH254, VH1014, VH1015, | (Brandt et al. 2009) |
| Tau352 Ala (10 Ser/Thr phosphorlyation sites substituted with Ala) | rgef-1 | constitutively in all neurons | early onset of age-dependent progressive uncoordination and reduced lifespan compared towild- type tau352 | VH418, VH421 | (Brandt et al. 2009) |
Human tau transgenic models in C. elegans
Several transgenic human tau models have been generated in C. elegans (Table 2). These include transgenic lines overexpressing wild-type human fetal or adult tau, both of which can be phosphorylated by C. elegans, as well as transgenic lines overexpressing human tau containing mutations that cause hyperphosphorylation, correspond to mutations seen in FTDP-17, or correspond to the tau mutation in the triple transgenic mice APPswe, PSEN1M146V, and tauP301L (Brandt et al. 2009; Kraemer et al. 2003; Miyasaka et al. 2005). Transgenic lines that overexpress wild-type or mutant tau in neurons show two major phenotypes, which differ in severity and progression depending on whether fetal or adult tau was expressed and on the levels of tau overexpression; in addition, the mutant tau lines showed more severe phenotypes than the wild-type tau lines. First, the transgenic animals were uncoordinated and showed reduced motility (Brandt et al. 2009; Kraemer et al. 2003), and in the mutant adult tau transgenic lines, this uncoordination became progressively worse with age (Kraemer et al. 2003). Second, the transgenic mutant tau animals showed axonal degeneration whereby gaps and discontinuities appeared in the axonal bundles of the dorsal and/or ventral nerve cords (Brandt et al. 2009; Kraemer et al. 2003); by electron microscopy analysis, axons were dilated, vacuolated, and irregular (Kraemer et al. 2003). In transgenic lines where tau is overexpressed in the mechanosensory neurons, animals lost their sense of touch and this defect became progressively worse with age; the cell bodies of the mechanosensory neurons were swollen, axons were thinner, kinked, and tortuous, and microtubules were broken (Miyasaka et al. 2005). Although all tau over-expressing lines produced insoluble tau, phenotypes were detected even in the absence of insoluble tau. Thus, over-expression of tau either over-stabilizes microtubules or hyperphosphorylated tau is toxic (Brandt et al. 2009). These sets of data show that wild-type human tau is sufficient to induce neurodegeneration in a multicellular organism and its toxicity increases with age.
Mutants that suppress the tau pathology highlight genes that may act in the pathway that mediates tau toxicity. Two suppressors of tau toxicity, sut-1 and sut-2, have been isolated in C. elegans using a motility assay. sut-1 encodes an RNA and snRNP-binding protein (MacMorris et al. 2007), which is localized predominantly in the nucleus (Kraemer and Schellenberg 2007). In sut-1 mutants, tau-induced toxicity, as assessed by decreased motility, is partially suppressed and levels of tau are decreased (Kraemer and Schellenberg 2007). SUT-1 physically binds to UNC-34/Mena/Enabled, presumably in the cytoplasmic compartment in C. elegans (Kraemer and Schellenberg 2007). unc-34/Mena/Enabled-sut-1 double mutants show enhanced tau-induced toxicity and show severely decreased motility (Kraemer and Schellenberg 2007), suggesting that unc-34 is either needed for the sut-1 suppression or acts in a parallel pathway to affect motility. In mammals, Mena binds Fe65 (Ermekova et al. 1997) and forms a complex with APP (Sabo et al. 2001). Whether SUT-1, UNC-34/Mena/Enabled, FEH-1/Fe65, and APL-1 form a complex and how loss of sut-1 mediates detoxification of tau in C. elegans is unknown, but could involve shuttling of APP and/or tau to different compartments. sut-2 encodes a novel subtype of a CCCH zinc finger (Guthrie et al. 2009; Kraemer and Schellenberg 2007), which is similar to the zinc fingers contained within Nab2 in yeast and ZC3H14 in humans (Kelly et al. 2007); these zinc fingers bind polyadenosine RNA with high affinity (Kelly et al. 2007). SUT-2 interacts with ZYG-12, a HOOK cytoskeletal linker protein necessary for the attachment between centrosomes and the nucleus (Guthrie et al. 2009), suggesting that SUT-2 may be involved in tau movement. The role of the SUT proteins in suppressing tau toxicity, however, is still unclear.
Conclusion
The model organism C. elegans offers many approaches in understanding the role of genes implicated in AD. First, because the function of APP and related proteins are still unclear, C. elegans can be used to probe basic functions and different functional domains of an APP-related protein. The ease of performing genetic screens and generating transgenic strains allows researchers to answer questions that would be much more difficult and time-consuming to perform in a mammalian system. For instance, the in vivo relevance of the extracellular domain of an APP-related protein was determined first in C. elegans. By judicious disruption of different domains, the functional domains within the extracellular domain can be determined. In addition, forward genetic screens can be performed to identify genes in the apl-1 pathway; these genes may similarly function in an APP pathway. Second, over 50% of FAD cases have a mutation in the presenilins, which cleave APP. The in vivo function of the presenilins and two components of the γ-secretase complex were first identified in C. elegans and genetic screens can continue to identify relevant genes. Third, overexpression of APP and/or processing through the β-/γ-secretase pathway are linked to AD. Overexpression of C. elegans APL-1 is also toxic. Pathways to relieve this toxicity, such as APL-1/APP clearance, may also be conserved. Fourth, artificial introduction of human Aβ in C. elegans causes toxicity and this toxicity can be used to probe which residues are critical for toxicity, deposition, and Aβ plaque formation in a multicellular organism. Overexpression of Aβ is linked to oxidative stress, and several genes and drugs that decrease oxidative stress and Aβ toxicity in C. elegans may similarly decrease Aβ toxicity in humans. For instance, recent research suggests that autophagy may be a mechanism to clear excess APP and/or Aβ. Lastly, the finding that human tau is hyperphosphorylated by endogenous C. elegans enzymes and the resulting neurodegeneration caused by overexpression of human tau opened a new approach to find suppressors of tau pathogenesis. In summary, C. elegans is a valuable genetic model system that can provide insights into the function and metabolism of APP and tau and their contributions to AD.
Acknowledgments
We wish to thank Casey Brander for help with the figures and lab members for helpful discussions. This work was supported by grants from the Alzheimer’s Association, National Institutes Health, and National Science Foundation (CL) and a National Institutes of Health RCMI grant to City College.
References
- Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, et al. The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev Cell. 2005;9(3):403–414. doi: 10.1016/j.devcel.2005.07.009. S1534-5807(05)00289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahante JE, Daul AL, Li M, Volk ML, Tennessen JM, Miller EA, et al. The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev Cell. 2003;4(5):625–637. doi: 10.1016/s1534-5807(03)00127-8. S1534580703001278. [DOI] [PubMed] [Google Scholar]
- Arduengo PM, Appleberry OK, Chuang P, L’Hernault SW. The presenilin protein family member SPE-4 localizes to an ER/Golgi derived organelle and is required for proper cytoplasmic partitioning during Caenorhabditis elegans spermatogenesis. J Cell Sci. 1998;111:3645–3654. doi: 10.1242/jcs.111.24.3645. [DOI] [PubMed] [Google Scholar]
- Arya U, Dwivedi H, Subramaniam JR. Reserpine ameliorates Abeta toxicity in the Alzheimer’s disease model in Caenorhabditis elegans. Exp Gerontol. 2009;44:462–466. doi: 10.1016/j.exger.2009.02.010. [DOI] [PubMed] [Google Scholar]
- Association. 2008 Alzheimer’s disease facts and figures. Alzheimers Dement. 2008;4(2):110–133. doi: 10.1016/j.jalz.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Barnes NY, Li L, Yoshikawa K, Schwartz LM, Oppenheim RW, Milligan CE. Increased production of amyloid precursor protein provides a substrate for caspase-3 in dying motoneurons. J Neurosci. 1998;18(15):5869–5880. doi: 10.1523/JNEUROSCI.18-15-05869.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsyte D, Lovejoy DA, Lithgow GJ. Longevity and heavy metal resistance in daf-2 and age-1 long-lived mutants of Caenorhabditis elegans. FASEB J. 2001;15(3):627–634. doi: 10.1096/fj.99-0966com. [DOI] [PubMed] [Google Scholar]
- Bleuler M, Stoll WA. Clinical use of reserpine in psychiatry: comparison with chlorpromazine. Ann N Y Acad Sci. 1955;61(1):167. doi: 10.1111/j.1749-6632.1955.tb42463.x. [DOI] [PubMed] [Google Scholar]
- Brandt R, Gergou A, Wacker I, Fath T, Hutter H. A Caenorhabditis elegans model of tau hyperphosphorylation: induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol Aging. 2009;30:22–33. doi: 10.1016/j.neurobiolaging.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Brion JP, Hanger DP, Bruce MT, Couck AM, Flament-Durand J, Anderton BH. Tau in Alzheimer neurofibrillary tangles. N-and C-terminal regions are differentially associated with paired helical filaments and the location of a putative abnormal phosphorylation site. Biochem J. 1991;273(Pt 1):127–133. doi: 10.1042/bj2730127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007;3(3):186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- Byerly L, Cassada RC, Russell RL. The life cycle of the nematode Caenorhabditis elegans. I. Wild-type growth and reproduction. Dev Biol. 1976;51(1):23–33. doi: 10.1016/0012-1606(76)90119-6. 0012-1606(76)90119-6. [DOI] [PubMed] [Google Scholar]
- Cabrejo L, Guyant-Marechal L, Laquerriere A, Vercelletto M, De la Fourniere F, Thomas-Anterion C, et al. Phenotype associated with APP duplication in five families. Brain. 2006;129(Pt 11):2966–2976. doi: 10.1093/brain/awl237. [DOI] [PubMed] [Google Scholar]
- Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, et al. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet. 1999;65(3):664–670. doi: 10.1086/302553. S0002-9297(07)62317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmine-Simmen K, Proctor T, Tschape J, Poeck B, Triphan T, Strauss R, et al. Neurotoxic effects induced by the Drosophila amyloid-beta peptide suggest a conserved toxic function. Neurobiol Dis. 2009;33(2):274–281. doi: 10.1016/j.nbd.2008.10.014. S0969-9961(08) 00263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll PM, Fitzgerald K. Model organisms in drug discovery. Wiley; New York: 2003. [Google Scholar]
- Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, et al. Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature. 1991;353(6347):844–846. doi: 10.1038/353844a0. [DOI] [PubMed] [Google Scholar]
- Chen YZ. APP induces neuronal apoptosis through APP-BP1-mediated downregulation of beta-catenin. Apoptosis. 2004;9(4):415– 422. doi: 10.1023/B:APPT.0000031447.05354.9f. [DOI] [PubMed] [Google Scholar]
- Chui DH, Tanahashi H, Ozawa K, Ikeda S, Checler F, Ueda O, et al. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5(5):560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- Cinar HN, Sweet KL, Hosemann KE, Earley K, Newman AP. The SEL-12 presenilin mediates induction of the Caenorhabditis elegans uterine pi cell fate. Dev Biol. 2001;237:173–182. doi: 10.1006/dbio.2001.0374. [DOI] [PubMed] [Google Scholar]
- Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Curtis-Prior P, Vere D, Fray P. Therapeutic value of Ginkgo biloba in reducing symptoms of decline in mental function. J Pharm Pharmacol. 1999;51(5):535–541. doi: 10.1211/0022357991772817. [DOI] [PubMed] [Google Scholar]
- Daigle I, Li C. apl-1,a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci USA. 1993;90(24):12045–12049. doi: 10.1073/pnas.90.24.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies L, Wolska B, Hilbich C, Multhaup G, Martins R, Simms G, et al. A4 amyloid protein deposition and the diagnosis of Alzheimer’s disease: prevalence in aged brains determined by immunocytochemistry compared with conventional neuropathologic techniques. Neurology. 1988;38(11):1688–1693. doi: 10.1212/wnl.38.11.1688. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398(6727):518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Delacourte A, Defossez A. Alzheimer’s disease: tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J Neurol Sci. 1986;76(2–3):173–186. doi: 10.1016/0022-510x(86)90167-x. [DOI] [PubMed] [Google Scholar]
- Dernburg AF, Zalevsky J, Colaiacovo MP, Villeneuve AM. Transgene-mediated cosuppression in the C. elegans germ line. Genes Dev. 2000;14(13):1578–1583. [PMC free article] [PubMed] [Google Scholar]
- Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1–42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging. 2003;24(3):415–420. doi: 10.1016/s0197-4580(02)00225-7. [DOI] [PubMed] [Google Scholar]
- Eimer S. Dissertation. Ludwig-Maximilians-Universitaet Muenchen; Muenchen: 2003. Analysis and suppression of mutant presenilin sel-12 in Caenorhabditis elegans. [Google Scholar]
- Ermekova KS, Zambrano N, Linn H, Minopoli G, Gertler F, Russo T, et al. The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J Biol Chem. 1997;272(52):32869–32877. doi: 10.1074/jbc.272.52.32869. [DOI] [PubMed] [Google Scholar]
- Fay DS, Fluet A, Johnson CJ, Link CD. In vivo aggregation of beta-amyloid peptide variants. J Neurochem. 1998;71(4):1616–1625. doi: 10.1046/j.1471-4159.1998.71041616.x. [DOI] [PubMed] [Google Scholar]
- Fay DS, Stanley HM, Han M, Wood WB. A Caenorhabditis elegans homologue of hunchback is required for late stages of development but not early embryonic patterning. Dev Biol. 1999;205(2):240–253. doi: 10.1006/dbio.1998.9096. S0012-1606(98)99096-0. [DOI] [PubMed] [Google Scholar]
- Florez-McClure ML, Hohsfield LA, Fonte G, Bealor MT, Link CD. Decreased insulin-receptor signaling promotes the autophagic degradation of beta-amyloid peptide in C. elegans. Autophagy. 2007;3:569–580. doi: 10.4161/auto.4776. [DOI] [PubMed] [Google Scholar]
- Fonte V, Kapulkin V, Taft A, Fluet A, Friedman D, Link CD. Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc Natl Acad Sci USA. 2002;99:9439–9444. doi: 10.1073/pnas.152313999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonte V, Kipp DR, Yerg J, Merin D, Forrestal M, Wagner E, et al. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem. 2008;283(2):784–791. doi: 10.1074/jbc.M703339200. [DOI] [PubMed] [Google Scholar]
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, et al. aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell. 2002;3:85–97. doi: 10.1016/s1534-5807(02)00189-2. [DOI] [PubMed] [Google Scholar]
- Galvan V, Zhang J, Gorostiza OF, Banwait S, Huang W, Ataie M, et al. Long-term prevention of Alzheimer’s disease-like behavioral deficits in PDAPP mice carrying a mutation in Asp664. Behav Brain Res. 2008;191(2):246–255. doi: 10.1016/j.bbr.2008.03.035. S0166-4328(08)00180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97(3):395–406. doi: 10.1016/s0092-8674(00)80748-5. S0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122(3):1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349(6311):704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3(4):519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- Goedert M, Baur CP, Ahringer J, Jakes R, Hasegawa M, Spillantini MG, et al. PTL-1, a microtubule-associated protein with tau-like repeats from the nematode Caenorhabditis elegans. J Cell Sci. 1996;109:2661–2672. doi: 10.1242/jcs.109.11.2661. [DOI] [PubMed] [Google Scholar]
- Gordon P, Hingula L, Krasny ML, Swienckowski JL, Pokrywka NJ, Raley-Susman KM. The invertebrate microtubule-associ-ated protein PTL-1 functions in mechanosensation and development in Caenorhabditis elegans. Dev Genes Evol. 2008;218(10):541. doi: 10.1007/s00427-008-0250-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156(1):15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutte C, Hepler W, Mickey KM, Priess JR. aph-2 encodes a novel extracellular protein required for GLP-1-mediated signaling. Development. 2000;127(11):2481–2492. doi: 10.1242/dev.127.11.2481. [DOI] [PubMed] [Google Scholar]
- Goutte C, Tsunozaki M, Hale VA, Priess JR. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA. 2002;99:775–779. doi: 10.1073/pnas.022523499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralle M, Ferreira ST. Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog Neurobiol. 2007;82(1):11–32. doi: 10.1016/j.pneurobio.2007.02.001. S0301-0082(07)00016-0. [DOI] [PubMed] [Google Scholar]
- Greenwald I. Worm-Book, editor. The C. elegans Research Community. WormBook; 2005. LIN-12/Notch signaling in C. elegans. http://www.wormbook.org. [DOI] [Google Scholar]
- Greenwald IS, Sternberg PW, Horvitz HR. The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell. 1983;34(2):435–444. doi: 10.1016/0092-8674(83)90377-x. 0092-8674(83)90377-X. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83(13):4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Chen F, Sanjo N, Kawarai T, Hasegawa H, Duthie M, et al. APH-1 interacts with mature and immature forms of presenilins and nicastrin and may play a role in maturation of presenilin/nicastrin complexes. J Biol Chem. 2003;278:7374–7380. doi: 10.1074/jbc.M209499200. [DOI] [PubMed] [Google Scholar]
- Guthrie CR, Schellenberg GD, Kraemer BC. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum Mol Genet. 2009;18(10):1825–1838. doi: 10.1093/hmg/ddp099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Zepeda A, Santell R, Wu Z, Brown M, Wu Y, Khan I, et al. Soy isoflavone glycitein protects against beta amyloid-induced toxicity and oxidative stress in transgenic Caenorhabditis elegans. BMC Neurosci. 2005;6:54. doi: 10.1186/1471-2202-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell. 1993;75(6):1039–1042. doi: 10.1016/0092-8674(93)90312-e. [DOI] [PubMed] [Google Scholar]
- Hardy J. APP mutations table. 2009a http://www.alzforum.org/res/com/mut/app/table1.asp.
- Hardy J. [Accessed 14 Apr 2009];Presenilin-1 mutations table. 2009b http://www.alzforum.org/res/com/mut/pre/table1.asp.
- Hardy J. [Accessed 14 Apr 2009];Presenilin-2 mutations table. 2009c http://www.alzforum.org/res/com/mut/pre/table2.asp.
- Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12(10):383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20(21):7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60(8):1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Hall DH. The unc-5, unc-6, and unc-40 genes guide circumferential migrations of pioneer axons and mesodermal cells on the epidermis in C. elegans. Neuron. 1990;4(1):61–85. doi: 10.1016/0896-6273(90)90444-k. 0896-6273(90)90444-K. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, et al. Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J. 2004;23(20):4106–4115. doi: 10.1038/sj.emboj.7600390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. Faseb J. 1999;13(11):1385–1393. [PubMed] [Google Scholar]
- Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, Xu X, et al. APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci USA. 2007;104(6):1971–1976. doi: 10.1073/pnas.0603997104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvitz HR. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999;59(7 Suppl):1701s–1706s. [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- Hsu A-L, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300(5622):1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Hubbard EJ, Wu G, Kitajewski J, Greenwald I. sel-10, a negative regulator of lin-12 activity in Caenorhabditis elegans, encodes a member of the CDC4 family of proteins. Genes Dev. 1997;11(23):3182–3193. doi: 10.1101/gad.11.23.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulette CM, Pericak-Vance MA, Roses AD, Schmechel DE, Yamaoka LH, Gaskell PC, et al. Neuropathological features of frontotemporal dementia and Parkinsonism linked to chromosome 17q21-22 (FTDP-17): Duke family 1684. J Neuropathol Exp Neurol. 1999;58(8):859–866. doi: 10.1097/00005072-199908000-00008. [DOI] [PubMed] [Google Scholar]
- Hunt-Newbury R, Viveiros R, Johnsen R, Mah A, Anastas D, Fang L, et al. High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol. 2007;5(9):e237. doi: 10.1371/journal.pbio.0050237. 07-PLBI-RA-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, et al. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103(13):5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarriault S, Greenwald I. Evidence for functional redundancy between C. elegans ADAM proteins SUP-17/Kuzbanian and ADM-4/TACE. Dev Biol. 2005;287(1):1–10. doi: 10.1016/j.ydbio.2005.08.014. S0012-1606(05) 00542-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon M, Gardner HF, Miller EA, Deshler J, Rougvie AE. Similarity of the C. elegans developmental timing protein LIN-42 to circadian rhythm proteins. Science. 1999;286(5442):1141–1146. doi: 10.1126/science.286.5442.1141. [DOI] [PubMed] [Google Scholar]
- Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, et al. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325(6106):733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- Kelly SM, Pabit SA, Kitchen CM, Guo P, Marfatia KA, Murphy TJ, et al. Recognition of polyadenosine RNA by zinc finger proteins. Proc Natl Acad Sci USA. 2007;104(30):12306–12311. doi: 10.1073/pnas.0701244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366(6454):461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kidd M. Alzheimer’s disease—an electron microscopical study. Brain. 1964;87:307–320. doi: 10.1093/brain/87.2.307. [DOI] [PubMed] [Google Scholar]
- Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci USA. 2003;100(11):6382–6387. doi: 10.1073/pnas.1037392100. doi:10.1073/pnas. 1037392100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokel M, Borland CZ, DeLong L, Horvitz HR, Stern MJ. clr-1 encodes a receptor tyrosine phosphatase that negatively regulates an FGF receptor signaling pathway in Caenorhabditis elegans. Genes Dev. 1998;12(10):1425–1437. doi: 10.1101/gad.12.10.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA. 1986;83(11):4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer BC, Schellenberg GD. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum Mol Genet. 2007;16:1959–1971. doi: 10.1093/hmg/ddm143. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci USA. 2003;100:9980–9985. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krigman MR, Feldman RG, Bensch K. Alzheimer’s presenile dementia. A histochemical and electron microscopic study. Lab Invest. 1965;14:381–396. [PubMed] [Google Scholar]
- Kumar MS, Erkeland SJ, Pester RE, Chen CY, Ebert MS, Sharp PA, et al. Suppression of non-small cell lung tumor development by the let-7 microRNA family. Proc Natl Acad Sci USA. 2008;105(10):3903–3908. doi: 10.1073/pnas.0712321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Beach TG, Sue LI, Scott S, Layne KJ, Kokjohn TA, et al. The evolution of A beta peptide burden in the APP23 transgenic mice: implications for A beta deposition in Alzheimer disease. Mol Med. 2001;7(9):609–618. [PMC free article] [PubMed] [Google Scholar]
- Kwon J. Tau mutations table. 2008 http://www.alzforum.org/res/com/mut/tau/table1.asp.
- L’Hernault SW, Arduengo PM. Mutation of a putative sperm membrane protein in Caenorhabditis elegans prevents sperm differentiation but not its associated meiotic divisions. J Cell Biol. 1992;119:55–68. doi: 10.1083/jcb.119.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Tinkle BT, Bieberich CJ, Haudenschild CC, Jay G. The Alzheimer’s A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat Genet. 1995;9(1):21–30. doi: 10.1038/ng0195-21. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Troncoso JC, Strickland DK, Kawas CH, Jay G. Neuronal cell death in Alzheimer’s disease correlates with apoE uptake and intracellular Abeta stabilization. J Clin Invest. 1997;100(2):310–320. doi: 10.1172/JCI119536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bars PL, Katz MM, Berman N, Itil TM, Freedman AM, Schatzberg AF. A placebo-controlled, double-blind, randomized trial of an extract of Ginkgo biloba for dementia. North American EGb Study Group. Jama. 1997;278(16):1327–1332. doi: 10.1001/jama.278.16.1327. [DOI] [PubMed] [Google Scholar]
- Le Bars PL, Kieser M, Itil KZ. A 26-week analysis of a double-blind, placebo-controlled trial of the ginkgo biloba extract EGb 761 in dementia. Dement Geriatr Cogn Disord. 2000;11(4):230–237. doi: 10.1159/000017242. [DOI] [PubMed] [Google Scholar]
- Levitan D, Greenwald I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature. 1995;377(6547):351–354. doi: 10.1038/377351a0. [DOI] [PubMed] [Google Scholar]
- Levitan D, Greenwald I. Effects of SEL-12 presenilin on LIN-12 localization and function in Caenorhabditis elegans. Development. 1998;125(18):3599–3606. doi: 10.1242/dev.125.18.3599. [DOI] [PubMed] [Google Scholar]
- Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, et al. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1996;93:14940–14944. doi: 10.1073/pnas.93.25.14940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan D, Yu G, St George Hyslop P, Goutte C. APH-2/ nicastrin functions in LIN-12/Notch signaling in the Caenorhabditis elegans somatic gonad. Dev Biol. 2001;240:654–661. doi: 10.1006/dbio.2001.0486. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995a;269(5226):973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wijsman EM, Nemens E, Anderson L, Goddard KA, Weber JL, et al. A familial Alzheimer’s disease locus on chromosome 1. Science. 1995b;269(5226):970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Li X, Greenwald I. Membrane topology of the C. elegans SEL-12 presenilin. Neuron. 1996;17:1015–1021. doi: 10.1016/s0896-6273(00)80231-7. [DOI] [PubMed] [Google Scholar]
- Li X, Greenwald I. HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc Natl Acad Sci USA. 1997;94(22):12204–12209. doi: 10.1073/pnas.94.22.12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZW, Stark G, Gotz J, Rulicke T, Gschwind M, Huber G, et al. Generation of mice with a 200-kb amyloid precursor protein gene deletion by Cre recombinase-mediated site-specific recombination in embryonic stem cells. Proc Natl Acad Sci USA. 1996;93(12):6158–6162. doi: 10.1073/pnas.93.12.6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QX, Maynard C, Cappai R, McLean CA, Cherny RA, Lynch T, et al. Intracellular accumulation of detergent-soluble amyloidogenic A beta fragment of Alzheimer’s disease precursor protein in the hippocampus of aged transgenic mice. J Neurochem. 1999;72(6):2479–2487. doi: 10.1046/j.1471-4159.1999.0722479.x. [DOI] [PubMed] [Google Scholar]
- Li YM, Lai MT, Xu M, Huang Q, DiMuzio-Mower J, Sardana MK, et al. Presenilin 1 is linked with gamma-secretase activity in the detergent solubilized state. Proc Natl Acad Sci USA. 2000;97(11):6138–6143. doi: 10.1073/pnas.110126897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Pauley AM, Myers RL, Shuang R, Brashler JR, Yan R, et al. SEL-10 interacts with presenilin 1, facilitates its ubiquitination, and alters A-beta peptide production. J Neurochem. 2002;82(6):1540–1548. doi: 10.1046/j.1471-4159.2002.01105.x. [DOI] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/ forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278(5341):1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci USA. 1995;92:9368–9372. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link CD. C. elegans models of age-associated neurodegenerative diseases: lessons from transgenic worm models of Alzheimer’s disease. Exp Gerontol. 2006;41:1007–1013. doi: 10.1016/j.exger.2006.06.059. [DOI] [PubMed] [Google Scholar]
- Link CD, Johnson CJ, Fonte V, Paupard M, Hall DH, Styren S, et al. Visualization of fibrillar amyloid deposits in living, transgenic Caenorhabditis elegans animals using the sensitive amyloid dye, X-34. Neurobiol Aging. 2001;22:217–226. doi: 10.1016/s0197-4580(00)00237-2. [DOI] [PubMed] [Google Scholar]
- Link CD, Taft A, Kapulkin V, Duke K, Kim S, Fei Q, et al. Gene expression analysis in a transgenic Caenorhabditis elegans Alzheimer’s disease model. Neurobiol Aging. 2003;24:397–413. doi: 10.1016/s0197-4580(02)00224-5. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yankner BA. Amyloid fibril toxicity in Alzheimer’s disease and diabetes. Ann N Y Acad Sci. 1996;777:89–95. doi: 10.1111/j.1749-6632.1996.tb34406.x. [DOI] [PubMed] [Google Scholar]
- Luo L, Tully T, White K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron. 1992;9(4):595–605. doi: 10.1016/0896-6273(92)90024-8. [DOI] [PubMed] [Google Scholar]
- Luo Y, Smith JV, Paramasivam V, Burdick A, Curry KJ, Buford JP, et al. Inhibition of amyloid-beta aggregation and caspase-3 activation by the Ginkgo biloba extract EGb761. Proc Natl Acad Sci USA. 2002;99:12197–12202. doi: 10.1073/pnas.182425199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luse SA, Smith KR., Jr The ultrastructure of senile plaques. Am J Pathol. 1964;44(4):553–563. [PMC free article] [PubMed] [Google Scholar]
- MacMorris M, Kumar M, Lasda E, Larsen A, Kraemer B, Blumenthal T. A novel family of C. elegans snRNPs contains proteins associated with trans-splicing. RNA. 2007;13(4):511–520. doi: 10.1261/rna.426707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magara F, Muller U, Li ZW, Lipp HP, Weissmann C, Stagljar M, et al. Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the beta-amyloid-precursor protein. Proc Natl Acad Sci USA. 1999;96(8):4656–4661. doi: 10.1073/pnas.96.8.4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahn K, Borras C, Knock GA, Taylor P, Khan IY, Sugden D, et al. Dietary soy isoflavone induced increases in antioxidant and eNOS gene expression lead to improved endothelial function and reduced blood pressure in vivo. Faseb J. 2005;19(12):1755–1757. doi: 10.1096/fj.05-4008fje. [DOI] [PubMed] [Google Scholar]
- Mann DM, Jones D, South PW, Snowden JS, Neary D. Deposition of amyloid beta protein in non-Alzheimer dementias: evidence for a neuronal origin of parenchymal deposits of beta protein in neurodegenerative disease. Acta Neuropathol. 1992;83(4):415–419. doi: 10.1007/BF00713534. [DOI] [PubMed] [Google Scholar]
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82(12):4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl G, Roberts BR, Gunn AP, Perez KA, Tew DJ, Masters CL, et al. The Caernorhabditis elegans Abeta1-42 model of Alzheimer’s disease predominantly expresses Abeta3-42. J Biol Chem. 2009;284(34):22697–22702. doi: 10.1074/jbc.C109.028514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott JB, Aamodt S, Aamodt E. ptl-1, a Caenorhabditis elegans gene whose products are homologous to the tau micro-tubule-associated proteins. Biochemistry. 1996;35(29):9415–9423. doi: 10.1021/bi952646n. [DOI] [PubMed] [Google Scholar]
- Mello C, Fire A. DNA transformation. Methods Cell Biol. 1995;48:451–482. [PubMed] [Google Scholar]
- Metzger RR, Brown JM, Sandoval V, Rau KS, Elwan MA, Miller GW, et al. Inhibitory effect of reserpine on dopamine transporter function. Eur J Pharmacol. 2002;456(1–3):39–43. doi: 10.1016/s0014-2999(02)02647-x. [DOI] [PubMed] [Google Scholar]
- Miklossy J, Taddei K, Suva D, Verdile G, Fonte J, Fisher C, et al. Two novel presenilin-1 mutations (Y256S and Q222H) are associated with early-onset Alzheimer’s disease. Neurobiol Aging. 2003;24(5):655–662. doi: 10.1016/s0197-4580(02)00192-6. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Murrell JR, Gearing M, Spillantini MG, Goedert M, Crowther RA, et al. Tau pathology in a family with dementia and a P301L mutation in tau. J Neuropathol Exp Neurol. 1999;58(4):335–345. doi: 10.1097/00005072-199904000-00004. [DOI] [PubMed] [Google Scholar]
- Mix JA, Crews WD., Jr A double-blind, placebo-controlled, randomized trial of Ginkgo biloba extract EGb 761 in a sample of cognitively intact older adults: neuropsychological findings. Hum Psychopharmacol. 2002;17(6):267–277. doi: 10.1002/hup.412. [DOI] [PubMed] [Google Scholar]
- Miyasaka T, Ding Z, Gengyo-Ando K, Oue M, Yamaguchi H, Mitani S, et al. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol Dis. 2005;20(2):372–383. doi: 10.1016/j.nbd.2005.03.017. S0969-9961(05)00079-3. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, et al. Synaptotrophic effects of human amyloid beta protein precursors in the cortex of transgenic mice. Brain Res. 1994;666(2):151–167. doi: 10.1016/0006-8993(94)90767-6. 0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, et al. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424(6946):277–283. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Murrell J, Farlow M, Ghetti B, Benson MD. A mutation in the amyloid precursor protein associated with hereditary Alzheimer’s disease. Science. 1991;254(5028):97–99. doi: 10.1126/science.1925564. [DOI] [PubMed] [Google Scholar]
- Napolitano F, D’Angelo F, Bimonte M, Perrina V, D’Ambrosio C, Scaloni A, et al. A differential proteomic approach reveals an evolutionary conserved regulation of Nme proteins by Fe65 in C. elegans and mouse. Neurochem Res. 2008;33(12):2547–2555. doi: 10.1007/s11064-008-9683-z. [DOI] [PubMed] [Google Scholar]
- Nikolaev A, Mclaughlin T, O’Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Niwa R, Zhou F, Li C, Slack FJ. The expression of the Alzheimer’s amyloid precursor protein-like gene is regulated by developmental timing microRNAs and their targets in Caenorhabditis elegans. Dev Biol. 2008;315(2):418–425. doi: 10.1016/j.ydbio.2007.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Nunan J, Small DH. Regulation of APP cleavage by alpha-, beta-and gamma-secretases. FEBS Lett. 2000;483(1):6–10. doi: 10.1016/s0014-5793(00)02076-7. S0014-5793(00)02076-7. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging. 2003a;24(8):1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003b;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, et al. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389(6654):994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Oken BS, Storzbach DM, Kaye JA. The efficacy of Ginkgo biloba on cognitive function in Alzheimer disease. Arch Neurol. 1998;55(11):1409–1415. doi: 10.1001/archneur.55.11.1409. [DOI] [PubMed] [Google Scholar]
- Okochi M, Eimer S, Bottcher A, Baumeister R, Romig H, Walter J, et al. A loss of function mutant of the presenilin homologue SEL-12 undergoes aberrant endoproteolysis in Caenorhabditis elegans and increases abeta 42 generation in human cells. J Biol Chem. 2000;275(52):40925–40932. doi: 10.1074/jbc.M005254200. [DOI] [PubMed] [Google Scholar]
- Perez RG, Zheng H, Van der Ploeg LH, Koo EH. The beta-amyloid precursor protein of Alzheimer’s disease enhances neuron viability and modulates neuronal polarity. J Neurosci. 1997;17(24):9407–9414. doi: 10.1523/JNEUROSCI.17-24-09407.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13(4):1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponte P, Gonzalez-DeWhitt P, Schilling J, Miller J, Hsu D, Greenberg B, et al. A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature. 1988;331(6156):525–527. doi: 10.1038/331525a0. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403(6772):901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Ring S, Weyer SW, Kilian SB, Waldron E, Pietrzik CU, Filippov MA, et al. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J Neurosci. 2007;27(29):7817–7826. doi: 10.1523/JNEUROSCI.1026-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376(6543):775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Martin-Morris L, Luo LQ, White K. A Drosophila gene encoding a protein resembling the human beta-amyloid protein precursor. Proc Natl Acad Sci USA. 1989;86(7):2478–2482. doi: 10.1073/pnas.86.7.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roush SF, Slack FJ. Transcription of the C. elegans let-7 microRNA is temporally regulated by one of its targets, hbl-1. Dev Biol. 2009 doi: 10.1016/j.ydbio.2009.07.012. S0012-1606(09)01062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerriere A, Vital A, et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38(1):24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153(7):1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt FA, Davis DG, Wekstein DR, Smith CD, Ashford JW, Markesbery WR. “Preclinical” AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55(3):370–376. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399(suppl 6738):A23–A31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN- 29 transcription factor. Mol Cell. 2000;5(4):659–669. doi: 10.1016/s1097-2765(00)80245-2. S1097- 2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
- Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, et al. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain. 2006;129(Pt 11):2977–2983. doi: 10.1093/brain/awl203. [DOI] [PubMed] [Google Scholar]
- Slunt HH, Thinakaran G, Von Koch C, Lo AC, Tanzi RE, Sisodia SS. Expression of a ubiquitous, cross-reactive homologue of the mouse beta-amyloid precursor protein (APP) J Biol Chem. 1994;269(4):2637–2644. [PubMed] [Google Scholar]
- Smith JV, Luo Y. Elevation of oxidative free radicals in Alzheimer’s disease models can be attenuated by Ginkgo biloba extract EGb 761. J Alzheimers Dis. 2003;5(4):287–300. doi: 10.3233/jad-2003-5404. [DOI] [PubMed] [Google Scholar]
- Smith JV, Burdick AJ, Golik P, Khan I, Wallace D, Luo Y. Anti-apoptotic properties of Ginkgo biloba extract EGb 761 in differentiated PC12 cells. Cell Mol Biol (Noisy-le-grand) 2002;48(6):699–707. [PubMed] [Google Scholar]
- Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8(2):387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher CA, Grant FJ, Grimm G, O’Hara PJ, Norris F, Norris K, et al. Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry. 1993;32(17):4481–4486. doi: 10.1021/bi00068a002. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Arya U, SoundaraRajan T, Dwivedi H, Kumar S, Subramaniam JR. Reserpine can confer stress tolerance and lifespan extension in the nematode C. elegans. Biogerontology. 2008;9(5):309–316. doi: 10.1007/s10522-008-9139-5. [DOI] [PubMed] [Google Scholar]
- Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—is this type 3 diabetes? J Alzheimers Dis. 2005;7(1):63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- Steinbach JP, Muller U, Leist M, Li ZW, Nicotera P, Aguzzi A. Hypersensitivity to seizures in beta-amyloid precursor protein deficient mice. Cell Death Differ. 1998;5(10):858–866. doi: 10.1038/sj.cdd.4400391. [DOI] [PubMed] [Google Scholar]
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398(6727):522– 525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Sulston JE, White JG. Regulation and cell autonomy during postembryonic development of Caenorhabditis elegans. Dev Biol. 1980;78:577–597. doi: 10.1016/0012-1606(80)90353-x. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer’s disease. Nature. 1988;331(6156):528–530. doi: 10.1038/331528a0. [DOI] [PubMed] [Google Scholar]
- Tax FE, Thomas JH, Ferguson EL, Horvitz HR. Identification and characterization of genes that interact with lin-12 in Caenorhabditis elegans. Genetics. 1997;147(4):1675–1695. doi: 10.1093/genetics/147.4.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Gonatas NK, Weiss M. Ultrastructural studies in Alzheimer’s presenile dementia. Am J Pathol. 1964;44:269–297. [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17(1):181–190. doi: 10.1016/s0896-6273(00)80291-3. S0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Tremml P, Lipp HP, Muller U, Ricceri L, Wolfer DP. Neurobehavioral development, adult openfield exploration and swimming navigation learning in mice with a modified beta-amyloid precursor protein gene. Behav Brain Res. 1998;95(1):65–76. doi: 10.1016/s0166-4328(97)00211-8. [DOI] [PubMed] [Google Scholar]
- Vakil RJ. A clinical trial of Rauwolfia serpentina in essential hypertension. Br Heart J. 1949;11(4):350–355. doi: 10.1136/hrt.11.4.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, et al. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18(6):661–669. doi: 10.1016/s0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- Walker GA, White TM, McColl G, Jenkins NL, Babich S, Candido EP, et al. Heat shock protein accumulation is upregulated in a long-lived mutant of Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci. 2001;56:B281–B287. doi: 10.1093/gerona/56.7.b281. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F. Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA. 1992;89:10758–10762. doi: 10.1073/pnas.89.22.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, et al. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat Genet. 1993a;5(1):95–100. doi: 10.1038/ng0993-95. [DOI] [PubMed] [Google Scholar]
- Wasco W, Peppercorn J, Tanzi RE. Search for the genes responsible for familial Alzheimer’s disease. Ann N Y Acad Sci. 1993b;695:203–208. doi: 10.1111/j.1749-6632.1993.tb23053.x. [DOI] [PubMed] [Google Scholar]
- Wen C, Metzstein MM, Greenwald I. SUP-17, a Caenorhabditis elegans ADAM protein related to Drosophila KUZBANIAN, and its role in LIN-12/NOTCH signalling. Development. 1997;124(23):4759–4767. doi: 10.1242/dev.124.23.4759. [DOI] [PubMed] [Google Scholar]
- Wen C, Levitan D, Li X, Greenwald I. spr-2, a suppressor of the egg-laying defect caused by loss of sel-12 presenilin in Caenorhabditis elegans, is a member of the SET protein subfamily. Proc Natl Acad Sci USA. 2000;97:14524–14529. doi: 10.1073/pnas.011446498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westlund B, Parry D, Clover R, Basson M, Johnson CD. Reverse genetic analysis of Caenorhabditis elegans presenilins reveals redundant but unequal roles for sel-12 and hop-1 in Notch-pathway signaling. Proc Natl Acad Sci USA. 1999;96(5):2497–2502. doi: 10.1073/pnas.96.5.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter E. Effects of an extract of Ginkgo biloba on learning and memory in mice. Pharmacol Biochem Behav. 1991;38(1):109–114. doi: 10.1016/0091-3057(91)90597-u. [DOI] [PubMed] [Google Scholar]
- Winter JC. The effects of an extract of Ginkgo biloba, EGb 761, on cognitive behavior and longevity in the rat. Physiol Behav. 1998;63(3):425–433. doi: 10.1016/s0031-9384(97)00464-2. [DOI] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, et al. Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and prese-nilin-1 double-transgenic mice. Neurosci Lett. 2001;306(1–2):116–120. doi: 10.1016/s0304-3940(01)01876-6. S0304-3940(01)01876-6. [DOI] [PubMed] [Google Scholar]
- Withee J, Galligan B, Hawkins N, Garriga G. Caenorhabditis elegans WASP and Ena/VASP proteins play compensatory roles in morphogenesis and neuronal cell migration. Genetics. 2004;167(3):1165–1176. doi: 10.1534/genetics.103.025676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenburg N, Eimer S, Lakowski B, Röhrig S, Rudolph C, Baumeister R. Presenilin is required for proper morphology and function of neurons in C. elegans. Nature. 2000;406:306–309. doi: 10.1038/35018575. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398(6727):513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Wu Y, Luo Y. Transgenic C. elegans as a model in Alzheimer’s research. Curr Alzheimer Res. 2005;2(1):37–45. doi: 10.2174/1567205052772768. [DOI] [PubMed] [Google Scholar]
- Wu G, Hubbard EJ, Kitajewski JK, Greenwald I. Evidence for functional and physical association between Caenorhabditis elegans SEL-10, a Cdc4p-related protein, and SEL-12 presenilin. Proc Natl Acad Sci USA. 1998;95:15787–15791. doi: 10.1073/pnas.95.26.15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wu Z, Butko P, Christen Y, Lambert MP, Klein WL, et al. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci. 2006;26:13102–13113. doi: 10.1523/JNEUROSCI.3448-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Tavernarakis N, Driscoll M. Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron. 2001;31(6):957–971. doi: 10.1016/s0896-6273(01)00432-9. S0896-6273(01)00432-9. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250(4978):279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- Yatin SM, Varadarajan S, Link CD, Butterfield DA. In vitro and in vivo oxidative stress associated with Alzheimer’s amyloid beta-peptide (1–42) Neurobiol Aging. 1999;20:325–330. doi: 10.1016/s0197-4580(99)00056-1. (discussion 339–342) [DOI] [PubMed] [Google Scholar]
- Ye Y, Lukinova N, Fortini ME. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature. 1999;398(6727):525–529. doi: 10.1038/19096. [DOI] [PubMed] [Google Scholar]
- Yoshikai S, Sasaki H, Doh-ura K, Furuya H, Sakaki Y. Genomic organization of the human amyloid beta-protein precursor gene. Gene. 1990;87(2):257–263. doi: 10.1016/0378-1119(90)90310-n. [DOI] [PubMed] [Google Scholar]
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature. 2000;407:48–54. doi: 10.1038/35024009. [DOI] [PubMed] [Google Scholar]
- Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, et al. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer’s disease. Int J Biochem Cell Biol. 2004;36(12):2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, et al. Macroautophagy-a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171(1):87–98. doi: 10.1083/jcb.200505082. jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano N, Bimonte M, Arbucci S, Gianni D, Russo T, Bazzicalupo P. feh-1 and apl-1, the Caenorhabditis elegans orthologues of mammalian Fe65 and beta-amyloid precursor protein genes, are involved in the same pathway that controls nematode pharyngeal pumping. J Cell Sci. 2002;115(Pt 7):1411–1422. doi: 10.1242/jcs.115.7.1411. [DOI] [PubMed] [Google Scholar]
- Zheng H, Koo E. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:5. doi: 10.1186/1750-1326-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81(4):525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115(1):201–211. doi: 10.1016/s0306-4522(02)00404-9. S0306452202004049. [DOI] [PubMed] [Google Scholar]