

Abstract
Background
Alternative splicing is important for increasing the complexity of the human proteome from a limited genome. Previous studies have shown that for some autoantigens, there is differential immunogenicity among alternatively spliced isoforms.
Objectives
Herein, we tested the hypothesis that alternative splicing is a common feature for transcripts of autologous proteins that are autoantigens. The corollary hypothesis tested was that nonautoantigen transcripts have a lower frequency of alternative splicing.
Methods
The extent of alternative splicing within 45 randomly selected self-proteins associated with autoimmune diseases was compared with 9554 randomly selected proteins in the human genome by using bioinformatics analyses. Isoform-specific regions that resulted from alternative splicing were studied for their potential to be epitopes for antibodies or T-cell receptors.
Results
Alternative splicing occurred in 100% of the autoantigen transcripts. This was significantly higher than the approximately 42% rate of alternative splicing observed in the 9554 randomly selected human gene transcripts (P < .001). Within the isoform-specific regions of the autoantigens, 92% and 88% encoded MHC class I and class II–restricted T-cell antigen epitopes, respectively, and 70% encoded antibody binding domains. Furthermore, 80% of the autoantigen transcripts underwent noncanonical alternative splicing, which is also significantly higher than the less than 1% rate in randomly selected gene transcripts (P < .001).
Conclusion
These studies suggest that noncanonical alternative splicing may be an important mechanism for the generation of untolerized epitopes that may lead to autoimmunity.
Furthermore, the product of a transcript that does not undergo alternative splicing is unlikely to be a target antigen in autoimmunity.
Keywords: Autoantigens, alternative splicing, exons, isoforms, antigen epitopes, immunogenicity, immune tolerance, autoimmune diseases
The breaking of immune tolerance to self-antigens is a central tenet in the pathogenesis of autoimmunity. Despite significant progress, the mechanisms by which self-proteins break immune tolerance and become auto-antigens remain poorly defined. One possible mechanism is alternative splicing, a process that removes introns and alters exons to generate multiple isoforms from a single pre-mRNA transcript. It is estimated that the human genome encodes as many as 60,000 genes, and bio-informatic analyses indicate that approximately 42% of randomly selected gene transcripts undergo alternative splicing.1–4
Alternative splicing is the major mechanism by which a small number of human genes can encode the larger complexity of the human proteome, which is estimated to be between 9 × 104 and 1 × 106 proteins.5 The majority of alternative splicing (70% to 88%)6 affects the coding region of mRNA, often resulting in the expression of additional exons that encode 16 to 100 amino acids on average.1 Because the length of peptides needed for antibody binding, MHC class I binding, and MHC class II binding is 8 to 15 amino acids, additional exons introduced by alternative splicing are long enough to produce novel antigenic epitopes. Because immune responses to autoantigen epitopes are highly specific,7 alternative splicing of exons could provide the structural basis for expression of novel untolerized antigen epitopes with altered antigenic properties and hence create the potential to break existing immune tolerance.8,9
Previously, 19 autoantigens were identified as having alternatively spliced isoforms,10–25 suggesting that alternative splicing may indeed contribute to the regulated expression of autoantigens. In addition, a recent report showed that intrathymic expression of proteo-lipid protein (PLP) was largely restricted to the shorter splice variant DM20. Pathogenic autoimmune responses targeted the autoantigen domain encoded by an exon of proteolipid protein that was untolerized because of extrathymic expression in the central nervous system.22 Two additional studies similarly revealed that autoimmune responses to the insulinoma-associated tyrosine phosphatase-like protein (IA-2), which is exclusively expressed in pancreatic islets, were targeted to the epitopes encoded by exon 13 in the full-length transcripts. However, the rest of the autoantigen was tolerized by the differential expression of an alternatively spliced isoform that lacked the exon 13-encoded protein domain in thymus and spleen.21,26 These experimental studies clearly demonstrated that the differential expression of alternatively spliced variants of each autoantigen resulted in novel protein domains that were encoded by an extra exon in peripheral tissues but were not expressed or tolerized in the thymus, thereby leading to the loss of immune tolerance and elicitation of an autoimmune response to the self-antigen.27 We previously reported that upregulated expression of the novel tumor antigens CML66 and CML28/Rrp46p is associated with immunogenicity of these antigens. We also recently identified a novel mechanism by which an alternative promoter and splicing can regulate the immunogenicity of CML66.28
Posttranscriptional splicing can be canonical or non-canonical (Fig 1). Canonical splicing removes introns that have 5′GT and 3′AG consensus flanking sequences. In noncanonical splicing, the intron flanking sequences can be a variety of other nucleotide pairs. It is estimated that less than 1% of human genes undergo noncanonical splicing. However, in tumor antigen transcripts, such noncanonical splicing had been reported to occur more frequently. Because most of the tumor antigens identified so far are self-antigens, it is well accepted that antitumor immunity is closely related to autoimmunity. Thus, we hypothesized that autoantigen transcripts will also have increased noncanonical splicing.
FIG 1.
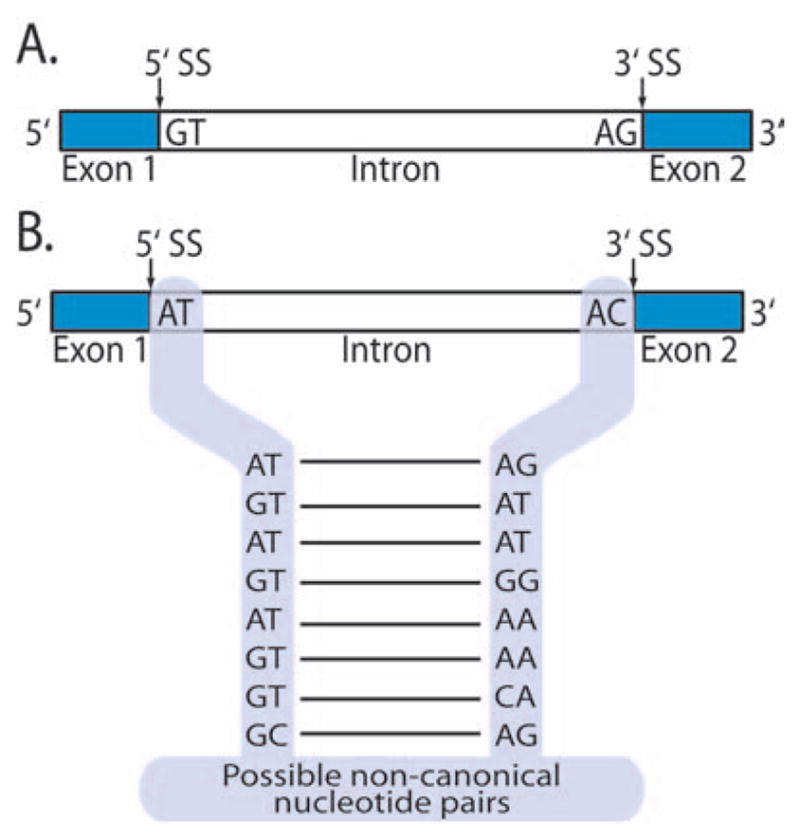
A, The 5′ and 3′ splice sites (SS) of introns typically occur at GT-AG flanking sequences. This type of splicing is termed canonical splicing, which accounts for >99% of splicing for randomly selected transcripts. B, Noncanonical splicing occurs when the intronic flanking sequences do not follow this GT-AG rule and accounts for <1% of splicing.
Herein, we investigated whether alternative splicing is a potential pathogenic mechanism limited to only a few autoantigens or whether it is a general phenomenon that occurs for most autoantigens. To test our hypothesis that a common characteristic of autoantigens is a high frequency of alternative splicing within their transcripts, we mined the GenBank databases. Bioinformatic analyses revealed that autoantigen transcripts do indeed exhibit a significantly greater frequency of alternative splicing than transcripts of nonautoantigens. Moreover, the occurrence of alternative splicing in the transcripts for all of the autoantigens studied suggests that this may be an important molecular mechanism in the pathogenesis of autoimmune diseases.
METHODS
Bioinformatic identification of alternatively spliced isoforms of autoantigens whose open reading frames are affected
The identification of novel alternatively spliced isoforms of autoantigens was performed by using the National Center for Biotechnological Information (NCBI) Web-based searching engines. First, the accession number of each published autoantigen was found by searching the GenBank database (http://www.ncbi.nlm.nih.gov/). Second, the LocusLink Web site (http://www.ncbi.nlm.nih.gov/LocusLink/index.html) was used to search the gene locus, the chromosome localization of the gene, and the exon/intron structure of the identified autoantigen mRNA transcripts. Third, the section Evidence Viewer under LocusLink was used to identify the mRNA-genomic alignments of the autoantigens and all of the alternative spliced isoforms (Table I). Therefore, our studies include the following splicing events: (1) a unique cassette exon that affects the open reading frame (ORF) of one isoform, but not that in another isoform, without causing a frame shift, thereby resulting in the expression of a novel protein substructure; (2) an alternative promoter/first exon that results in the novel N-terminal protein structure in one isoform but not in another isoform; (3) an alternative terminal exon that results in the extension of the C-terminus in one isoform, but not in another isoform; (4) junction regions newly formed via alternative splicing between the protein sequences encoded by 2 adjacent exons; and (5) terminal intron retention (unspliced intron) that results in C-terminal in frame extension with a novel protein structure or results in a truncated protein structure as a result of the introduction of a new stop codon.9 The expression sequence tag (EST) clones and the partial cDNA or mRNA sequences of the autoantigens were accepted as evidence of alternative spliced isoforms of the autoantigens (Table II), as described previously.3,4 Our classification of alternative spliced isoforms in Table I and Table II did not include the following sequences that do not affect the protein sequence of the autoantigens: (1) transcript sequences encoded solely in the intron region of the autoantigen genomic locus, without sharing any protein sequences (common exons) with the previously identified autoantigens; and (2) mRNA or cDNA sequences encoded by the minus-strand genomic DNA sequence of the same gene locus.
TABLE I.
Examples of alternatively spliced autoantigens
| Autoantigens | Isoform A§ | Isoform B§ | Frequency of HLA I† | Associated diseases |
|---|---|---|---|---|
| PM/Scl-75 | NM_005033 | U09215 | 17 | PM/Scl |
| PM/Scl-100 | NM_002685 | X66113 | 8 | PM/Scl |
| Ku70 | AK055786 | NM_001469 | 18 | PM/Scl |
| SSA/Ro-1‡ | U01882 | NM_003141 | 25 | SS, SLE |
| SSA/Ro-2‡ | AY205314 | NM_004600 | 11 | SS, SLE |
| Mi-2 | NM_001272 | U91543 | 6 | Polymyositis |
| Nuclear autoantigen sperm protein‡ | NM_152298 | NM_172164 | 97 | Vasectomy |
| NOR-90‡ | X56687 | NM_014233 | 11 | Scleroderma |
| Lamin A‡ | BC033088 | NM_005572 | 24 | WG |
| Nuclear mitotic apparatus protein 1‡ | BC004165 | NM_006185 | 407 | SS |
| CENP-A | BC000881 | NM_001809 | 11 | CREST |
| H+/K+ ATPase | NM_001676 | BC031609 | 3 | Autoimmune polyendocrine syndrome type II |
| BPAG1‡ | NM_001723 | NM_015548 | 1590 | Pemphigus |
| Phogrin‡ | NM_130843 | NM_002847 | 14 | DM type 1 |
| MBP‡ | M30047 | NM_002385 | 4 | EAE |
| DNA Topo2α‡ | AF285157 | NM_001067 | 9 | Morphea |
| RA33‡ | NM_002137 | NM_031243 | 3 | Rheumatoid arthritis |
| PCNA | XM_066450 | NM_002592 | 8/14¶ | SLE |
| Golgin-67‡ | NM_181076 | NM_015003 | 47 | SS, SLE |
| Proteinase 3 | X56132 | NM_002777 | 21 | WG |
| SmB/B′‡ | NM_003091 | X17568 | 0 | SLE, mixed connective tissue disease |
| CML-28 | NM_020158 | AF285785 | 13 | PM/Scl |
| α-Fodrin‡ | BC034956 | NM_003127 | 215 | SS |
| IA-2‡ | NA* | NM_002846 | 38 | DM type 1 |
| Thyroid peroxidase‡ | NM_175719 | NM_000547 | 28 | Autoimmune thyroiditis |
| SSB/La‡ | NM_003142 | X69804 | — | SS, SLE |
CREST, Calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia (a variant of scleroderma); SLE, systemic lupus erythematosus; SS, Sjögren syndrome; WG, Wegeners granulomatosus.
Reported in literature but not found in LocusLink.
HLA 2.1 scores of the unique segment of translated peptide for isoform (B) by using the SYFPEITHI epitope prediction website (≥9.4 is counted as significant).
Autoantigens with alternative-spliced isoforms, previously published: SSA/Ro-1,37 SSA/Ro-2,11 NOR-90,12 nuclear autoantigen sperm protein,16 lamin A,13 nuclear mitotic apparatus protein 1,18 BPAG1,15 phogrin,23 MBP,17 DNA topoisomerase 2α,19 RA33,25 Golgin-67,20 SmBB,10 α-fodrin,24 thyroid peroxidase,14 IA-2,21 and SSB/La.38
GenBank mRNA accession numbers. The shorter isoform is arbitrarily listed as isoform A. The untolerized isoform may be either the short or the long isoform.
PCNA isoform B has 2 isoform-specific regions.
TABLE II.
Autoantigens found in EST database listed in UniGene
| Autoantigens | Isoform A | Isoform B | Associated diseases |
|---|---|---|---|
| Jo-1 | NM_002109 | AK055917 | ASS |
| P450* | NM_000102 | AK094106 | Sjögren syndrome, SLE |
| U1–70 | NM_003089 | X06815 | SLE, MCTD |
| Gars† | NM_002047 | AK074524 | ASS |
| SRP | NM_003136 | U51920 | Polymyositis |
| Histone 2 | NM_003517 | NM_003516 | SLE‡ |
| Histone 4 | NM_175054 | NM_003545 | SLE‡ |
| SmD1 | NM_006938 | L36188 | SLE |
| Fibrillarin | NM_001436 | BC019609 | Scl |
| DNA Topo 1 | NM_003286 | NM_052963 | Scl |
| GAD65 | NM_000818 | BC039038 | DM type 1 |
| Lamin B1 | NM_005573 | BC012295 | SLE, autoimmune hepatitis |
| CENP-B | NM_001810 | BC021577 | CREST |
| CENP-C | NM_001812 | AV646089 | CREST |
| Ribosomal P2 | NM_001004 | BC007573 | SLE |
| U1-A | NM_004596 | BC000405 | SLE, MCTD |
| U1-C | NM_003093 | M18465 | SLE, MCTD |
| Ku-80 | NM_021141 | X57500 | SLE/overlap |
| MPO | BI028393 | NM_000250 | MPA§ |
ASS, Antisynthetase syndrome; CREST, calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia (a variant of scleroderma); MCTD, mixed connective tissue disease; Scl, scleroderma; SLE, systemic lupus erythematosus.
Cytochrome P450.
Glycyl-tRNA synthetase.
Especially in drug-induced lupus.
Microscopic angiitis.
Documented autoantigens
We analyzed the published human autoantigens experimentally identified in various systemic and organ-specific autoimmune diseases.29,30 The selection of these autoantigens for analyses was based on (1) their entries in the GenBank databases and (2) their documented association with common autoimmune diseases. To avoid sampling bias, selection of the autoantigens was made before the search for the information regarding splicing variations of the autoantigens.
Antigenic epitope analyses
To identify the antigenic structure within the protein domains encoded by the extra exons in the context of the alternatively spliced isoforms of the human autoantigens, we used the well-accepted Jameson-Wolf antigenic index31 to analyze the spliced isoform-specific antigenic structures for potential primary (linear or continuous) and secondary epitopes for antibody binding.30 In addition, we used 2 widely adapted Web site–based algorithms, the BioInformatics and Molecular Analysis Section algorithm at the National Institutes of Health Web site (http://bimas.dcrt.nih.gov/molbio/hla_bind/) and the SYFPEITHI algorithm (http://syfpeithi.bmi-heidelberg.com/Scripts/MHCServer.dll/EpPredict.htm), to analyze the MHC class I–restricted CD8+ and the MHC class II–restricted CD4+ T-cell antigenic epitopes.
Analysis for posttranslational modifications
To identify the potential sites for posttranslational modifications of isoform-specific regions, we used PROSITE (http://us.expasy.org/prosite/), a comprehensive database of protein families and domains, to help reliably identify which posttranslational modification sites (if any) a new protein sequence in the untolerized regions of the autoantigens has. Because the PROSITE database does not include all of the cleavage sites for granzyme B, a short Java-script program was written to detect all of the granzyme B cleavage sites.
Statistical estimation for the sample size
Our preliminary studies showed that the frequency of autoantigen transcripts undergoing alternative splicing was at least 30% higher than that observed for randomly selected genes (42%).3 Inferential hypothesis testing was based on 2 binomial proportions. Sample size determination and power calculations were based on an effect size of δ = 0.30, on the basis of the assumption that the alternative proportion of splice variants among autoantigens was p1 = 0.72 and that the null proportion was p0 = 0.42. A 1-sided test with an alternative hypothesis p1 > p0 was used assuming a Type I error probability of α = 0.05. Power, or the compliment of a Type II error probability (1-β), was calculated as a function of sample size by using the PASS 2000 package (Kaysville, Utah) for δ = 0.30. Power calculations indicate that for a sample size of 40 and effect size of 30%, at least 98% power can be attained for rejecting the null hypothesis when the alternative hypothesis is true. We conservatively used a sample size of 45 to ensure that at least 90% power was attained.
RESULTS
Alternative splicing is increased for autoantigens
For each of the 45 randomly selected autoantigens, we mined the GenBank databases and found that 26 have at least 2 isoforms that fulfilled our inclusion criteria, whereas the remaining 19 had no full-length alternatively spliced isoforms that met the criteria of the NCBI LocusLink. For these 19 autoantigens, we extended our search by blasting their sequences in the other NCBI nucleotide databases for evidence of alternative splicing. To our surprise, the sequence data revealed that all 19 autoantigens have convincing evidence of alternatively spliced isoforms that affected the ORF, which was verified by searching other NCBI databases, such as the Unigene Web site and the Aceview Web site. These EST databases were obtained from several genomic sequence–matched EST sequences, thus minimizing the potential artifacts in the EST cloning.32
Previous studies using data from the GenBank and EST databases showed that the rate of alternative splicing among randomly selected genes in the human genome was between 38%4 and 42%.3 Because those studies3,4 used the same GenBank databases, the same criteria for the definition of alternative splicing, and search strategies similar to those applied in this study, the data are all statistically comparable. To ensure that there were no sampling differences between the 2 previous studies,3,4 we performed a statistical analysis comparing their data and found that there was no statistical difference (P = .605) in the alternative splicing rate. To ensure that there were no sampling differences between the 2 previous studies3,4 and our study, we examined 50 randomly selected human genes and found that the alternative splicing rate among these 50 genes was 41% ± 10.5% (P > .05), indicating that our data are statistically comparable with theirs. In summary, our results showed that alternative splicing modulates the transcripts of all of the autoantigens examined, suggesting that alternative splicing modulates the transcripts of autoantigens at a significantly higher rate than that in randomly selected genes (P < .001).
Increased noncanonical splicing in autoantigen transcripts
Among the 45 autoantigens studied, 36 (80%) had evidence of noncanonical alternative splicing that did not conform to the GT-AG (Fig 1) rule in the consensus splicing junctions of exons and introns. Only 9 autoantigens (Centromere Protein C, GAD65, Histone 2, Histone 4, IA-2, Lamin B1, polymyositis/scleroderma overlap [PM/Scl]–75, Proteinase 3, and SmD1) exhibited canonical splicing. In contrast with this high rate of non-canonical splicing seen in autoantigens, less than 1% of randomly selected genes undergo noncanonical splicing.33 These data suggest that noncanonical spliceosomes may play an important role in the expression of proteins that have the potential to be autoantigens.33
Alternatively spliced isoform-specific regions encode potential sites for posttranslational modifications
Posttranslational modifications of autoantigens may contribute to their immunogenicity. Therefore, we hypothesized that alternatively spliced isoform-specific regions could encode untolerized posttranslational modification sites. We used PROSITE (Web-based search engine) to test this hypothesis. Of the 26 autoantigens, 77% encoded posttranslational modification sites in their isoform-specific regions, suggesting that these posttranslational modification sites could enhance the immunogenicity of the autoantigens.34
Alternatively spliced isoform-specific regions encode potential primary and secondary epitopes recognized by autoantibodies
Full-length isoforms are required for the analysis of isoform-specific regions using antigen index algorithms. Therefore, of the 45 autoantigens studied, only those listed in Table I could be analyzed. To test our hypothesis that alternatively spliced isoform-specific regions of autoantigens may encode epitopes recognized by autoantibodies, we used the Jameson-Wolf antigen index algorithm to evaluate the antigen index of each isoform-specific antigenic region.31 We used the 43 linear autoantigen epitopes that previously were experimentally defined30 as the reference epitopes. These 43 reference autoantigen epitopes had Jameson-Wolf antigen index scores that ranged from 1.56 to 4.36 (mean ± 2 SD = 2.96 ± 1.40), which served as the reference range (with a 95% CI) for antibody epitopes.
By using this criterion, of the 26 autoantigens with full-length isoforms (Table I), 18 (70%) of the auto-antigens encoded 1 or more antibody binding epitopes in their isoform-specific regions. In the isoform-specific regions of these 18 autoantigens, we found 92 potential primary and secondary epitope candidates for autoanti-body binding. These results demonstrate that the isoform-specific regions of autoantigens encode antigen epitopes that are eligible for autoantibody generation and binding.
Alternatively spliced isoform-specific regions encode potential MHC class I and class II–restricted T-cell antigen epitopes
We hypothesized that the alternatively spliced isoform-specific regions of autoantigens may encode MHC class I and/or MHC class II–restricted T-cell autoantigen epitopes. To obtain a statistical reference range for our prediction of MHC class I and II–restricted T-cell antigen epitope binding affinity, we used the SYFPEITHI algorithm to examine 56 HLA-A2.1–restricted CD8+ T-cell antigen epitopes and 18 HLA-DR4–restricted CD4+ T-cell antigen epitopes.35 The rationale for focusing on HLA-A2.1–restricted and HLA-DR4–restricted T-cell antigen epitopes to demonstrate potential T-cell antigen epitopes encoded by isoform-specific regions are that (1) they are among the best characterized human MHC alleles and have been widely used in experimental protocols for identification of antigenic epitopes, and (2) they are among the most representative human MHC alleles. The 56 HLA-A2.1 CD8+ T-cell antigen epitopes have predictive scores from 9.4 to 30.4 (mean ± 2 SD = 19.9 ± 10.5), and the 18 HLA-DR4–restricted CD4+ T-cell antigen epitopes have predictive scores from 7.0 to 31.4 (mean ± 2 SD = 19.2 ± 12.2), which can serve as valid criteria with 95% CI. These criteria suggest that if isoform-specific regions of the autoantigens have predictive scores within the ranges of 9.4 to 30.0 and 7.0 to 31.4, they would have a high probability of stably binding to HLA-A2.1 and HLA-DR4, respectively, and become immunodominant T-cell antigen epitopes for stimulation of CD8+ or CD4+ T cells. Of the 26 isoform-specific regions of the full-length autoantigens studied (Table I), 24 (92%) contained isoform specific regions that encode 2642 HLA-A2. 1–restricted CD8+ T-cell antigen epitopes. Similarly, 23 (88%) of these autoantigens contained isoform-specific regions of autoantigens, which encode potential HLA-DR4–restricted CD4+ T-cell antigen epitopes (not shown). These results suggest that upregulation of these novel HLA-restricted T-cell antigen epitopes, under autoimmune-prone conditions, may break immune tolerance and elicit T-cell autoimmunity to the cells and tissues expressing these isoforms, as shown previously in MHC class I transgenic mice.36
DISCUSSION
This is the first comprehensive bioinformatic analysis of the mechanistic role of alternative splicing on the expression of immunogenic autoantigens. The 45 randomly selected autoantigens analyzed are associated with a broad spectrum of autoimmune diseases. Among these autoantigens, the majority (62%) have not been previously documented to have alternatively spliced isoforms.10–21,37,38 Our in silico studies demonstrate that alternative splicing occurred in 100% of the transcripts for this broad spectrum of autoantigens. This rate for alternative splicing for autoantigens is much higher than the 38% to 42% rate of alternative splicing observed in more than 9500 randomly selected human genes (P < .001).3,4 Importantly, our studies also revealed that 70% of the autoantigen isoform-specific regions encoded antibody binding domains, 92% encoded MHC class I–restricted T-cell antigen epitopes, and 88% encoded MHC class II–restricted T-cell antigen epitopes. These results are consistent with our hypothesis that alternative splicing potentially modulates the immunogenicity of autoantigens by the generation of novel tolerance-breaking epitopes within the autoantigens.
It is not surprising that alternative splicing of a transcript that affects the ORF and protein structure would result in changes to the proteins’ immunogenic properties. In particular, changes in protein structure that affect surface accessibility, hydrophilicity, proximity to a site recognized by helper T cells, and protein epitope mobility will likely alter the immunogenicity of the protein antigens. Two autoantigens, cardiac-specific α-myosin and PM/Scl-100, have also been previously demonstrated to have alternative splicing of their transcripts, resulting in production of isoforms that are more immunogenic than their other isoforms. In addition, 2 other previous studies showed that differential or aberrant expression of autologous proteins, in which alternative splicing resulted in a protein structure against which the host had previously not been tolerized, could lead to the breaking of immune tolerance and elicitation of an immune response to the self-antigen in experimental autoimmune encephalomyelitis (EAE)22 and in autoimmune diabetes.21 Our panoramic analysis of the human genome databases supports these studies and indicates that alternative splicing is likely to be a major mechanism that expands the autoantigen epitope repertoire and regulates the immunogenicity of autologous proteins (Fig 2).
FIG 2.
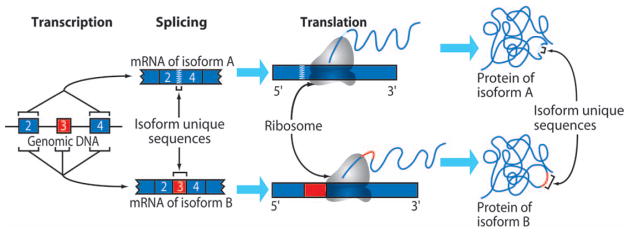
Diagrammatic representation of an autoantigen undergoing alternative splicing, resulting in at least 2 possible mRNA isoforms, which are then translated into 2 distinct protein isoforms. Isoform specific regions can result from inclusion of extra exons (red) or at the junctional region where an exon is spliced out (zigzag). These potentially encode the untolerized epitopes that may lead to autoimmune response (see Fig E1 in the Journal’s Online Repository at www.mosby.com/jaci for a more detailed diagrammatic representation of this figure).
Other studies that have contributed significantly to insights into potential pathogenic mechanisms contributing to autoimmunity include the following: (1) upregulation of MHC class I by skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies36; (2) unique peptide fragments generated by granzyme B may be an exclusive property of autoantigens39; and (3) posttranslational modifications of autologous proteins may also contribute to the immunogenicity of autoantigens.34 Our analyses indicate that alternative splicing is yet another potentially important molecular mechanism underlying the pathogenesis of autoimmunity.
On the basis of our results, we propose a “permissive splicing” model of autoimmunity (Fig 3), in which (1) autoantigens transcripts should have a high degree of alternative splicing, but alternative splicing per se does not make autologous proteins become autoantigens; (2) in healthy conditions, there is predominant expression of the tolerized isoforms of these autologous proteins; (3) the expression of untolerized isoforms of these autoantigen transcripts should be upregulated in autoimmune-prone conditions; and (4) genes with no or a low degree of alternative splicing are unlikely to encode autoantigens. This model provides a hypothesis-generating tool for further analyses of autoimmune targets by narrowing down the possible candidate genes with these proposed criteria.
FIG 3.

Permissive splicing in autoimmunity working model. Autoimmunity depends on host susceptibility (eg, autoimmune diseases–associated HLA), and environmental insults (eg, viruses) as well as the frequency of splicing. The model predicts that for a protein to be a pathogenic autoantigen, it must have a high frequency of alternative splicing, which can be modulated by inflammatory cytokines. Of the 6 possible scenarios, model B fulfills these criteria.
There are several published studies that support our model. First, approximately 75% of alternative spliced isoforms of proteins are involved in signaling and regulation, suggesting that alternative splicing is vital where information must be processed and regulated differently over time and in response to a variety of stimuli.6 In fact, several known autoantigens are closely involved with posttranscriptional modifications. One such example is RA33 (Table I), a member of the heterogeneous nuclear ribonucleoproteins, a protein complex involved in RNA splicing. Second, there is evidence that tumor antigens undergo a significantly higher rate of noncanonical alternative splicing compared with randomly selected gene transcripts. Because we also found a significantly higher rate of noncanonical splicing in autoantigens compared with randomly selected genes (80% vs <1%), there may be similar posttranscriptional processing of tumor and autoantigen transcripts by non-canonical spliceosomes.33 Third, there are 5 previous studies that demonstrated the predominance of one autoantigen isoform over another isoform in autoimmune diseases.10,14,16,21,22 These studies suggested that the dominant expression of one alternatively spliced isoform over the other may be contributing to their differences in immunogenicity, which is similar to our report on self-tumor antigen CML66-L. Fourth, overexpression of self-antigens is one of the major proposed mechanisms of autoimmune diseases. Zinkernagel et al8 suggested that the overexpression of self-antigens, or novel antigenic structure of the autoantigens, overcomes the threshold of antigen concentration at which an immune response is initiated.40 This threshold may be lower for the untolerized regions of certain autoantigen isoforms. Such an untolerized region may be encoded by the extra exons introduced by alternative splicing, resulting in formation of chimeric autoantigens. Further evidence to support this concept comes from a study that showed a fine specificity of the autoimmune response to SSA and SSB ribonucleo-proteins.7 These chimeric autoantigens may impose the endogenous danger signals for the host immune system to react, similar to that of exogenous danger signals elaborated by pathogens. Fifth, differential expression of autoantigen isoforms has been shown to occur in experimentally induced autoimmune myocarditis. A quantitative RT-PCR analysis showed a 32-fold overexpression of the isoform specific domain of CD44, which is encoded by exon 15 (a unique extra exon in this splice variant of CD44). This overexpression is temporally associated with the autoimmune process of the affected myocardial tissue, which is not observed in normal myocardial tissue. Two other studies showed that the expression of autoantigens can be induced by proinflammatory cytokines: (1) Ku70 expression by IL-13 and IL-4,41 and (2) proteinase 3 by IFN-α.42 These results clearly demonstrate that aberrant overexpression of alternatively spliced isoforms, with untolerized protein sequence encoded by newly spliced exons, can contribute to the autoimmune process. However, the generation of new alternatively spliced isoforms is likely an ongoing process of autoimmunity, and therefore, it is difficult to distinguish whether the increased prevalence of alternative splicing is a cause or a consequence of autoimmunity.
Several groups have reported that peptide analogues of disease-inducing epitopes of an antigen can effectively reverse the disease process in animal models of EAE and adjuvant arthritis. It has also been shown that diabetes can be prevented in nonobese diabetic mice by early administration of a variety of β-cell autoantigens. Therefore, identification of novel antigenic epitopes of autoantigens may enhance our ability to develop novel, antigen-specific DNA tolerizing vaccines, peptide tolerizing vaccines, or splice-modulating antisense oligonucleotides. In diseases with unclear autoimmune etiology, the “permissive splicing” model, described herein, can be applied to predict potential autoantigen targets. An example is chronic urticaria, where we found that a possible autoimmune target, the alpha chain of the high-affinity Fc receptor for IgE, can indeed undergo alternative splicing to produce distinct transcripts. Furthermore, this approach could also lead to the future development of high-throughput tools, such as gene arrays or protein arrays, for diagnosis and prognosis of autoimmune diseases.
Acknowledgments
Partially supported by National Institutes of Health grants AI054514, AI36936, P30 DK56238 (the Texas Gulf Coast Digestive Diseases Center), and P20 CA103698 (Baylor Cancer Center); the Kostas Family Foundation; the Caroline Weiss Law Foundation for Molecular Medicine; and the Myositis Association of America. Dr Yang is a Chao Family Scholar of Medicine.
We are grateful to Dr T. Cooper, Department of Pathology, Baylor College of Medicine, for insightful discussions, and to Dr J. Brendese, Ms K. Franks, K. Jolivette, M. Rogers, and A. Wirt for their assistance in preparation of this manuscript.
Abbreviations used
- DM
Diabetes mellitus
- EAE
Experimental autoimmune encephalomyelitis
- EST
Expression sequence tag
- IA-2
Insulinoma-associated tyrosine phosphatase-like protein
- NCBI
National Center for Biotechnological Information
- ORF
Open reading frame
- PLP
Proteolipid protein
References
- 1.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 2.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 3.Modrek B, Resch A, Grasso C, Lee C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001;29:2850–9. doi: 10.1093/nar/29.13.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brett D, Hanke J, Lehmann G, Haase S, Delbruck S, Krueger S, et al. EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett. 2000;474:83–6. doi: 10.1016/s0014-5793(00)01581-7. [DOI] [PubMed] [Google Scholar]
- 5.Stamm S. Signals and their transduction pathways regulating alternative splicing: a new dimension of the human genome. Hum Mol Genet. 2002;11:2409–16. doi: 10.1093/hmg/11.20.2409. [DOI] [PubMed] [Google Scholar]
- 6.Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30:13–9. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- 7.Scofield RH, Farris AD, Horsfall AC, Harley JB. Fine specificity of the autoimmune response to the Ro/SSA and La/SSB ribonucleoproteins. Arthritis Rheum. 1999;42:199–209. doi: 10.1002/1529-0131(199902)42:2<199::AID-ANR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 8.Zinkernagel RM, Hengartner H. Regulation of the immune response by antigen. Science. 2001;293:251–3. doi: 10.1126/science.1063005. [DOI] [PubMed] [Google Scholar]
- 9.Ladd AN, Cooper TA. Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-11-reviews0008. reviews0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaufman KM, Kirby MY, McClain MT, Harley JB, James JA. Lupus autoantibodies recognize the product of an alternative open reading frame of SmB/B′. Biochem Biophys Res Commun. 2001;285:1206–12. doi: 10.1006/bbrc.2001.5302. [DOI] [PubMed] [Google Scholar]
- 11.Chan EK, Tan EM, Ward DC, Matera AG. Human 60-kDa SS-A/Ro ribonucleoprotein autoantigen gene (SSA2) localized to 1q31 by fluorescence in situ hybridization. Genomics. 1994;23:298–300. doi: 10.1006/geno.1994.1502. [DOI] [PubMed] [Google Scholar]
- 12.Matera AG, Wu W, Imai H, O’Keefe CL, Chan EK. Molecular cloning of the RNA polymerase I transcription factor hUBF/NOR-90 (UBTF) gene and localization to 17q21. 3 by fluorescence in situ hybridization and radiation hybrid mapping. Genomics. 1997;41:135–8. doi: 10.1006/geno.1997.4647. [DOI] [PubMed] [Google Scholar]
- 13.Machiels BM, Zorenc AH, Endert JM, Kuijpers HJ, van Eys GJ, Ramaekers FC, et al. An alternative splicing product of the lamin A/C gene lacks exon 10. J Biol Chem. 1996;271:9249–53. doi: 10.1074/jbc.271.16.9249. [DOI] [PubMed] [Google Scholar]
- 14.Niccoli P, Fayadat L, Panneels V, Lanet J, Franc JL. Human thyroperoxidase in its alternatively spliced form (TPO2) is enzymatically inactive and exhibits changes in intracellular processing and trafficking. J Biol Chem. 1997;272:29487–92. doi: 10.1074/jbc.272.47.29487. [DOI] [PubMed] [Google Scholar]
- 15.Okumura M, Yamakawa H, Ohara O, Owaribe K. Novel alternative splicings of BPAG1 (bullous pemphigoid antigen 1) including the domain structure closely related to MACF (microtubule actin cross-linking factor) J Biol Chem. 2002;277:6682–7. doi: 10.1074/jbc.M109209200. [DOI] [PubMed] [Google Scholar]
- 16.Richardson RT, Batova IN, Widgren EE, Zheng LX, Whitfield M, Marzluff WF, et al. Characterization of the histone H1-binding protein, NASP, as a cell cycle-regulated somatic protein. J Biol Chem. 2000;275:30378–86. doi: 10.1074/jbc.M003781200. [DOI] [PubMed] [Google Scholar]
- 17.Roth HJ, Kronquist KE, Kerlero de Rosbo N, Crandall BF, Campagnoni AT. Evidence for the expression of four myelin basic protein variants in the developing human spinal cord through cDNA cloning. J Neurosci Res. 1987;17:321–8. doi: 10.1002/jnr.490170402. [DOI] [PubMed] [Google Scholar]
- 18.Tang TK, Tang CJ, Chen YL, Wu CW. Nuclear proteins of the bovine esophageal epithelium, II: the NuMA gene gives rise to multiple mRNAs and gene products reactive with monoclonal antibody W1. J Cell Sci. 1993;104:249–60. doi: 10.1242/jcs.104.2.249. [DOI] [PubMed] [Google Scholar]
- 19.Yu Q, Mirski SE, Sparks KE, Cole SP. Two COOH-terminal truncated cytoplasmic forms of topoisomerase II alpha in a VP-16-selected lung cancer cell line result from partial gene deletion and alternative splicing. Biochemistry. 1997;36:5868–77. doi: 10.1021/bi962400y. [DOI] [PubMed] [Google Scholar]
- 20.Jakymiw A, Raharjo E, Rattner JB, Eystathioy T, Chan EK, Fujita DJ. Identification and characterization of a novel Golgi protein, golgin-67. J Biol Chem. 2000;275:4137–44. doi: 10.1074/jbc.275.6.4137. [DOI] [PubMed] [Google Scholar]
- 21.Park YS, Kawasaki E, Kelemen K, Yu L, Schiller MR, Rewers M, et al. Humoral autoreactivity to an alternatively spliced variant of ICA512/IA-2 in Type I diabetes. Diabetologia. 2000;43:1293–301. doi: 10.1007/s001250051525. [DOI] [PubMed] [Google Scholar]
- 22.Klein L, Klugmann M, Nave KA, Tuohy VK, Kyewski B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat Med. 2000;6:56–61. doi: 10.1038/71540. [DOI] [PubMed] [Google Scholar]
- 23.Cui L, Yu WP, DeAizpurua HJ, Schmidli RS, Pallen CJ. Cloning and characterization of islet cell antigen-related protein-tyrosine phosphatase (PTP), a novel receptor-like PTP and autoantigen in insulin-dependent diabetes. J Biol Chem. 1996;271:24817–23. [PubMed] [Google Scholar]
- 24.Cianci CD, Zhang Z, Pradhan D, Morrow JS. Brain and muscle express a unique alternative transcript of alphaII spectrin. Biochemistry. 1999;38:15721–30. doi: 10.1021/bi991458k. [DOI] [PubMed] [Google Scholar]
- 25.Hutchison S, LeBel C, Blanchette M, Chabot B. Distinct sets of adjacent heterogeneous nuclear ribonucleoprotein (hnRNP) A1/A2 binding sites control 5′ splice site selection in the hnRNP A1 mRNA precursor. J Biol Chem. 2002;277:29745–52. doi: 10.1074/jbc.M203633200. [DOI] [PubMed] [Google Scholar]
- 26.Diez J, Park Y, Zeller M, Brown D, Garza D, Ricordi C, et al. Differential splicing of the IA-2 mRNA in pancreas and lymphoid organs as a permissive genetic mechanism for autoimmunity against the IA-2 type 1 diabetes autoantigen. Diabetes. 2001;50:895–900. doi: 10.2337/diabetes.50.4.895. [DOI] [PubMed] [Google Scholar]
- 27.Klein L, Kyewski B. “Promiscuous” expression of tissue antigens in the thymus: a key to T-cell tolerance and autoimmunity? J Mol Med. 2000;78:483–94. doi: 10.1007/s001090000146. [DOI] [PubMed] [Google Scholar]
- 28.Yan Y, Phan L, Yang F, Talpaz M, Yang Y, Xiong ZY, et al. A novel mechanism of alternative promoter and splicing regulates the epitope generation of tumor antigen CML66-L. J Immunol. 2004;172:651–60. doi: 10.4049/jimmunol.172.1.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan EM. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv Immunol. 1989;44:93–151. doi: 10.1016/s0065-2776(08)60641-0. [DOI] [PubMed] [Google Scholar]
- 30.Mahler M, Bluthner M, Pollard KM. Advances in B-cell epitope analysis of autoantigens in connective tissue diseases. Clin Immunol. 2003;107:65–79. doi: 10.1016/s1521-6616(03)00037-8. [DOI] [PubMed] [Google Scholar]
- 31.Jameson BA, Wolf H. The antigenic index: a novel algorithm for predicting antigenic determinants. Comput Appl Biosci. 1988;4:181–6. doi: 10.1093/bioinformatics/4.1.181. [DOI] [PubMed] [Google Scholar]
- 32.Wolfsberg TG, Landsman D. A comparison of expressed sequence tags (ESTs) to human genomic sequences. Nucleic Acids Res. 1997;25:1626–32. doi: 10.1093/nar/25.8.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Will CL, Schneider C, Reed R, Luhrmann R. Identification of both shared and distinct proteins in the major and minor spliceosomes. Science. 1999;284:2003–5. doi: 10.1126/science.284.5422.2003. [DOI] [PubMed] [Google Scholar]
- 34.Utz PJ, Gensler TJ, Anderson P. Death, autoantigen modifications, and tolerance. Arthritis Res. 2000;2:101–14. doi: 10.1186/ar75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Renkvist N, Castelli C, Robbins PF, Parmiani G. A listing of human tumor antigens recognized by T cells. Cancer Immunol Immunother. 2001;50:3–15. doi: 10.1007/s002620000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, et al. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci U S A. 2000;97:9209–14. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan EK, Di Donato F, Hamel JC, Tseng CE, Buyon JP. 52-kD SS-A/Ro: genomic structure and identification of an alternatively spliced transcript encoding a novel leucine zipper-minus autoantigen expressed in fetal and adult heart. J Exp Med. 1995;182:983–92. doi: 10.1084/jem.182.4.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Troster H, Metzger TE, Semsei I, Schwemmle M, Winterpacht A, Zabel B, et al. One gene, two transcripts: isolation of an alternative transcript encoding for the autoantigen La/SS-B from a cDNA library of a patient with primary Sjogrens’ syndrome. J Exp Med. 1994;180:2059–67. doi: 10.1084/jem.180.6.2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casciola-Rosen L, Andrade F, Ulanet D, Wong WB, Rosen A. Cleavage by granzyme B is strongly predictive of autoantigen status: implications for initiation of autoimmunity. J Exp Med. 1999;190:815–26. doi: 10.1084/jem.190.6.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001;1:147–53. doi: 10.1038/35100573. [DOI] [PubMed] [Google Scholar]
- 41.Kelavkar U, Wang S, Badr K. KU 70/80 lupus autoantigen is the transcription factor induced by interleukins (IL)-13 and -4 leading to induction of 15-lipoxygenase (15-LO) in human cells. Adv Exp Med Biol. 2002;507:469–81. doi: 10.1007/978-1-4615-0193-0_73. [DOI] [PubMed] [Google Scholar]
- 42.Burchert A, Wolfl S, Schmidt M, Brendel C, Denecke B, Cai D, et al. Interferon-alpha, but not the ABL-kinase inhibitor imatinib (STI571), induces expression of myeloblastin and a specific T-cell response in chronic myeloid leukemia. Blood. 2003;101:259–64. doi: 10.1182/blood-2002-02-0659. [DOI] [PubMed] [Google Scholar]