

Abstract
Vascular endothelial growth factor A (VEGF), a key factor in angiogenesis, plays an essential role in skeletal development and postnatal homeostasis. VEGF serves as a survival factor for chondrocytes and couples the resorption of cartilage with bone formation during endochondral ossification. Recently, it has also been found to regulate the balance between osteoblast and adipocyte differentiation in bone marrow mesenchymal stem cells. Surprisingly, this regulatory function of VEGF is not based on paracrine signaling involving cell surface receptor activation. Instead, the mechanism appears to utilize intracellular VEGF, which is functionally linked to the nuclear envelope protein lamin A. Lamin A and VEGF control osteoblast and adipocyte differentiation by regulating the levels of the osteoblast and adipocyte transcription factors Runx2 and PPARγ, respectively. These data raise the intriguing possibility that loss of bone mass during aging may be manipulated by controlling the levels and activity of intracellular VEGF in bone marrow mesenchymal stem cells.
Keywords: VEGF, bone remodeling, osteoblast, transcription, skeletal development
Introduction
Multiple cell types in the skeleton are known to express vascular endothelial growth factor-A (VEGFA or just VEGF). VEGF was initially identified as one of the key paracrine factors in both angiogenesis and vasculogenesis (Carmeliet et al. 1996; Ferrara et al. 1996). Later, it became clear that it also has other important roles, including cellular survival during bone and cartilage development. In contrast to the paracrine functions of VEGF in vascular development and angiogenesis, the survival of endothelial cells (Li and Keller 2000), hematopoietic stem cells (Gerber et al. 2002) and tumor cells (Lee et al. 2007; Samuel et al. 2011) has been linked to intracrine/autocrine functions of VEGF. The different functions of VEGF during the development and maintenance of bone are still incompletely understood.
VEGF Isoforms and Receptors
Alternative splicing of the VEGF primary transcript generates several isoforms of human VEGF mRNAs that encode proteins of 121, 145, 165, 189 and 206 amino acid residues (Ferrara and Keyt 1997; Poltorak et al. 1997). The corresponding mouse isoforms are one amino acid residue shorter than the human proteins and named accordingly (VEGF120 and so forth). The VEGF gene consists of eight exons and all isoforms include the amino acid sequence encoded by exon 3, which regulates covalent dimer formation and the binding to VEGF receptor 1 (VEGFR1), as well as exon 4, which encodes the VEGF receptor 2 (VEGFR2) binding site. Exons 6 and 7 encode heparin-binding domains; both of these domains are missing in VEGF121/120, whereas only the domain encoded by exon 6 is missing in VEGF165/164. Consequently, different VEGF isoforms have different properties. VEGF121/120 is freely diffusible, whereas VEGF189/188 avidly binds to cell surface molecules and extracellular matrix components; the predominant isoform VEGF165/164 exhibits a combination of these properties.
Paracrine VEGF signaling is mediated by the tyrosine kinase receptors VEGFR1 (Flt1) and VEGFR2 (KDR/Flk1) (Carmeliet and Collen 1999; de Vries et al. 1992; Fong et al. 1995; Shalaby et al. 1997; Shalaby et al. 1995; Terman et al. 1992). All VEGF isoforms can bind to both receptors although the affinity of VEGF for VEGFR1 is about 10-fold higher than for VEGFR2. VEGFR2 has strong tyrosine kinase activity and thus is the main receptor involved in cell signaling. Phosphorylation of specific tyrosine residues in the intracellular domain of VEGFR2 induces activation of various signaling pathways, including the activation of MAPK, PI3K/AKT, Src, and Rac signaling (Koch et al. 2011). In contrast, VEGFR1 has weak tyrosine kinase activity and mainly serves as a decoy receptor by competing with VEGFR2 for binding of ligand. The Flt1 gene encodes transcripts for both the full-length VEGFR1 tyrosine kinase receptor as well as for a secreted form that lacks the transmembrane and intracellular tyrosine kinase domains as the result of an alternative splicing event. Furthermore, VEGF binds to the co-receptors neuropilin 1 (NRP1) and neuropilin 2 (NRP2), which can stimulate VEGFR2 activation (Neufeld et al. 1999; Soker et al. 1998).
Paracrine VEGF Functions in Endochondral Ossification
The vertebrate skeleton develops by processes called intramembranous and endochondral ossifications (Karsenty 1999; Olsen et al. 2000). Intramembranous bone formation is characterized by the differentiation of condensed mesenchymal cells into osteoblasts, and this occurs in the cranial vault, jaws and in part of the clavicle. In the rest of the skeleton, condensing mesenchymal cells differentiate into chondrocytes and form the cartilage templates of future bones in a process known as endochondral bone formation. In this process, chondrocytes express high levels of VEGF as they mature and undergo hypertrophy and this is rapidly followed by invasion of blood vessels, osteoclasts (chondroclasts) and osteoprogenitor cells from the perichondrial areas into regions of hypertrophic cartilage (Fig. 1). This results in the formation of the primary ossification center, which is characterized by the resorption of the cartilage and the deposition of bone marrow and trabecular bone.
Figure 1.
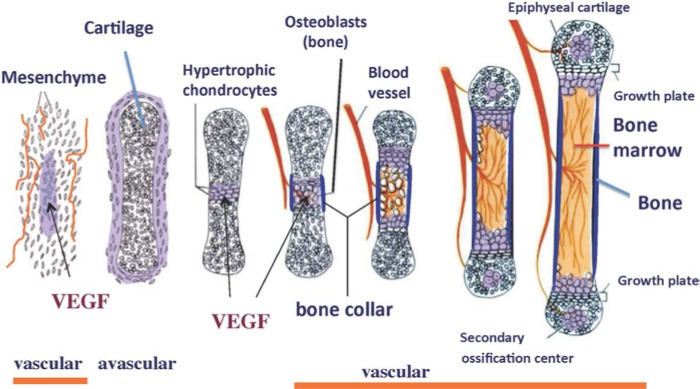
Diagram illustrating the stages of endochondral ossification and the expression of VEGF during the process. First expressed by cells in mesenchymal condensations and in perichondrial cells of avascular cartilage models of the future bone, VEGF is highly expressed by hypertrophic chondrocytes. This high level of expression stimulates the invasion of endothelial cells, osteoclasts, osteoblastic progenitor cells and hematopoietic cells into the hypertrophic cartilage during the formation of the primary ossification center.
Studies by Gerber et al. (1999) revealed that VEGF-mediated blood vessel invasion is essential for coupling resorption of cartilage with bone formation during endochondral ossification. A lack of blood vessel invasion results in reduced apoptosis of hypertrophic chondrocytes and thickening of the growth plate cartilage. VEGF is known to be a chemotactic factor for endothelial cells and monocytes/macrophages (Barleon et al. 1996; Hiratsuka et al. 1998). Mice expressing only the VEGF120 isoform showed delayed recruitment of blood vessels into the perichondium as well as delayed invasion of vessels into the metaphyseal region of the primary ossification center (Zelzer et al. 2002). In contrast, mice that only expressed the VEGF188 isoform displayed abnormalities in the capillary network in the epiphyseal region and increased hypoxia and massive cell death in the interior of the growth plate cartilage (Maes et al. 2004). This suggests that soluble VEGF isoforms that also contain a heparin-binding domain are required to efficiently attract blood vessel growth into the epiphyseal regions of long bones.
Further support for the crucial role of VEGF expression in hypertrophic cartilage was revealed by a conditional deletion of VEGF in chondrocytes (Zelzer et al. 2004). The conditional knock-out (CKO) mice showed delayed invasion of blood vessels into primary ossification centers and delayed removal of terminally differentiated hypertrophic chondrocytes. Furthermore, massive cell death occurred in joint and epiphyseal regions of VEGF CKO endochondral bones, similar to the abnormalities seen following the conditional deletion of hypoxia-inducible factor 1α (HIF1α) in chondrocytes. This suggests that HIF1α and its target gene VEGF are part of a key mechanism that supports chondrocyte survival during embryonic endochondral bone development.
During endochondral ossification, osteoblast precursor cells—which migrate into the cartilage from the perichondrium in response to VEGF produced by hypertrophic chondrocytes—also express high levels of VEGF (Maes et al. 2010) (Fig. 2). The cells are closely associated with invading blood vessels in a pericyte-like manner and later differentiate into trabecular osteoblasts, osteocytes, and mesenchymal stem/stromal cells within the developing bones. The conditional deletion of Vegf in Osterix-expressing osteoblast lineage cells causes a delay in the invasion of blood vessels and osteoclasts and results in reduced size, thickness, and mineralization of craniofacial and long bones in newborn mice (unpublished results). As the mice grow older, a more striking skeletal phenotype develops, consisting of a progressive loss in bone mass and a dramatic increase in marrow fat (Fig. 3).
Figure 2.

Diagram illustrating how Osterix-expressing osteoblast progenitor cells in the perichondral region of endochondral bones respond to VEGF produced by hypertrophic chondrocytes during endochondral ossification.
Figure 3.
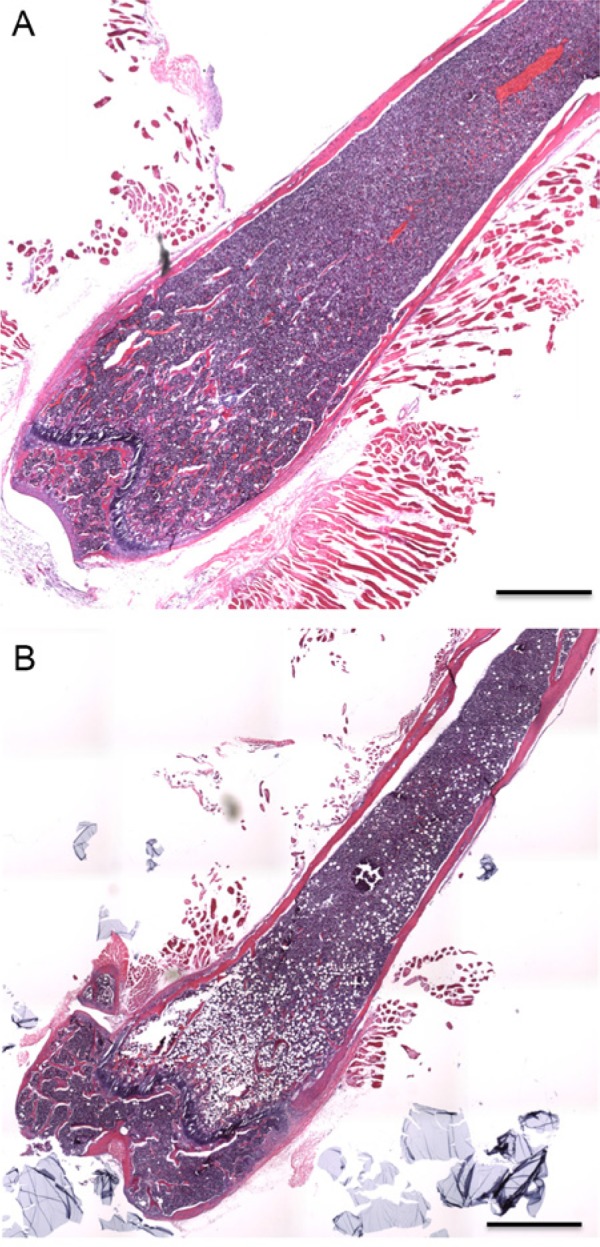
(A) Section of the femur from a 2 month-old wild type mouse. (B) Section of the femur from a mouse with a conditional loss of VEGF expression in Osterix-expressing osteoblastic cells. The loss of VEGF expression is associated with decreased trabecular and cortical bone mass and an increased number of adipocytes in the bone marrow. Scale bars = 1 mm.
Progenitor-derived VEGF Regulates Bone Homeostasis
Further analyses of the conditional VEGF mutants have led to the conclusion that VEGF controls the balance between osteoblast and adipocyte differentiation in bone marrow mesenchymal stem cells (MSCs) (Liu et al. 2012). The use of the transgene tdTomato as a fluorescent lineage marker reveals that the marrow adipocytes in the mutant mice are derived from Osterix-expressing progenitor cells (unpublished results). In vitro differentiation experiments using bone marrow MSCs demonstrate that the defects in osteoblastogenesis and adipogenesis resulting from the loss of VEGF can be rescued by retroviral-mediated expression of VEGF but not by the addition of recombinant VEGF. This strongly suggests that the VEGF-mediated control of stem cell fate is regulated by intracellular but not paracrine VEGF (Fig. 4). MSCs are known to express reduced levels of VEGF during aging, coinciding with the incidence of osteoporotic features, such as a loss in trabecular bone mass and an increase in marrow fat. This raises the possibility that age-dependent osteoporosis may be, in part, due to a progressive lossof VEGF-dependent mechanisms that stimulate osteoblast differentiation and repress the differentiation of adipocytes.
Figure 4.

Diagram illustrating how VEGF, produced by osteoblast progenitor cells, serves to stimulate osteoblast differentiation via an intracrine mechanism but stimulates osteoclastogenesis and angiogenesis as a paracrine factor. Neutralizing antibodies against VEGF can inhibit the paracrine function of VEGF but not its intracrine function.
Mechanisms of Intracrine VEGF Signaling
Experiments using a conditional deletion of the VEGF receptors VEGFR1 and VEGFR2 in osteoblastic lineage cells indicate that both receptors are required for the stimulatory effect of VEGF on osteoblastogenesis, but they are not critical for the ability of VEGF to repress adipocyte differentiation. Mice deficient in the nuclear envelope protein lamin A have been reported to have decreased bone formation and osteopenia (Li et al. 2011), whereas suppression of lamin A expression promoted adipocyte lineage commitment (Naito et al. 2012). Interestingly, there is a functional interaction between lamin A and VEGF: a knock-down of VEGF in MSCs increases lamin A protein expression whereas reduced VEGF protein levels are detected upon the loss of one Lmna allele in MSCs. Furthermore, both lamin A and VEGF appear to control osteoblast and adipocyte differentiation by regulating the levels of the osteoblast and adipocyte transcription factors Runx2 and PPARγ, respectively.
The precise mechanisms of intracrine VEGF function in bone marrow MSCs are not yet clear. Furthermore, whether intracrine VEGF requires binding and/or activation of VEGF receptors to control bone marrow stem cell fate is not known. Finally, since osteoblastic cells are known to stimulate osteoclastogenesis and angiogenesis by paracrine VEGF-dependent mechanisms (27, 30, 31) (Fig. 5), an important question is whether and/or how the cells control the balance between paracrine and intracrine VEGF functions during embryonic development and postnatal growth and aging.
Figure 5.
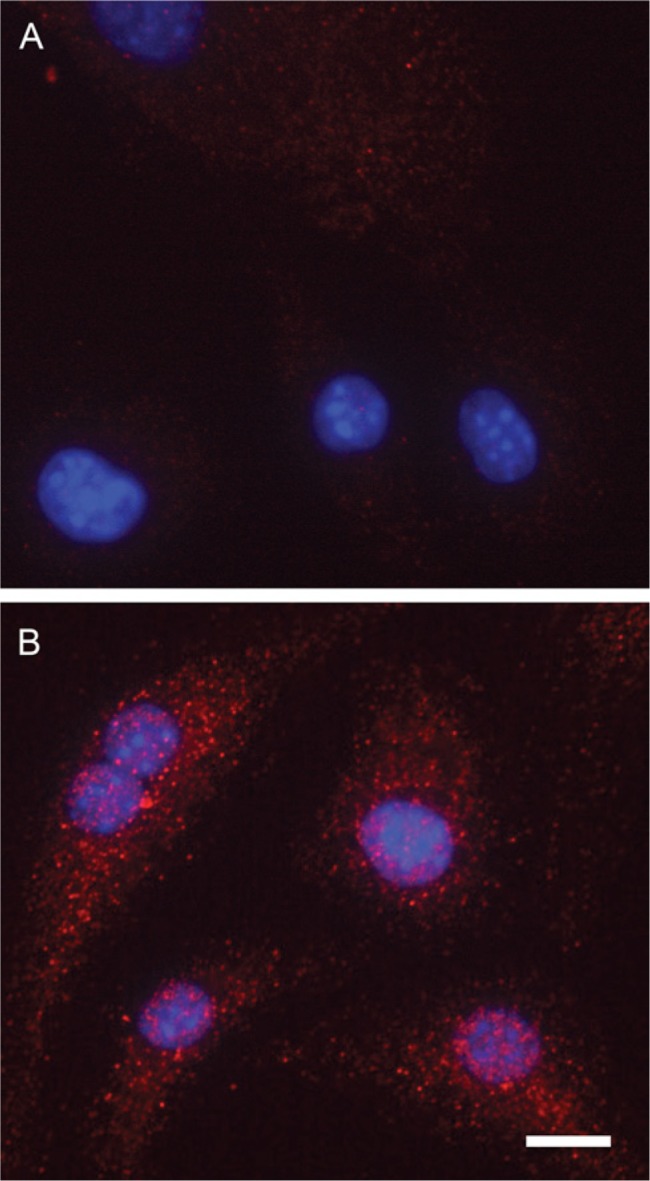
Confocal microscopy of bone marrow-derived mesenchymal stem cells stained with (A) non-immune IgG or (B) antibodies against VEGF. Staining shows reactivity in both the endoplasmic reticulum/Golgi and the nucleus. DAPI nucleic acid stain was used to stain cell nuclei. Scale bar = 10 µm.
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors received financial support (AR036819; PI: Bjorn R. Olsen) from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health for the research carried out in their laboratory and reviewed in the article.
References
- Barleon B, Sozzani S, Zhou D, Weich HA, Mantovani A, Marme D. (1996). Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 87:3336-3343 [PubMed] [Google Scholar]
- Carmeliet P, Collen D. (1999). Role of vascular endothelial growth factor and vascular endothelial growth factor receptors in vascular development. Curr Top Microbiol Immunol 237:133-158 [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. (1996). Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380:435-439 [DOI] [PubMed] [Google Scholar]
- de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT. (1992). The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 255:989-991 [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. (1996). Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380:439-442 [DOI] [PubMed] [Google Scholar]
- Ferrara N, Keyt B. (1997). Vascular endothelial growth factor: basic biology and clinical implications. EXS 79:209-232 [DOI] [PubMed] [Google Scholar]
- Fong GH, Rossant J, Gertsenstein M, Breitman ML. (1995). Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376:66-70 [DOI] [PubMed] [Google Scholar]
- Gerber HP, Malik AK, Solar GP, Sherman D, Liang XH, Meng G, Hong K, Marsters JC, Ferrara N. (2002). VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 417:954-958 [DOI] [PubMed] [Google Scholar]
- Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. (1999). VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nature Med 5:623-628 [DOI] [PubMed] [Google Scholar]
- Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. (1998). Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A 95:9349-9354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsenty G. (1999). The genetic transformation of bone biology. Genes Dev 13:3037-3051 [DOI] [PubMed] [Google Scholar]
- Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. (2011). Signal transduction by vascular endothelial growth factor receptors. Biochem J 437:169-183 [DOI] [PubMed] [Google Scholar]
- Lee TH, Seng S, Sekine M, Hinton C, Fu Y, Avraham HK, Avraham S. (2007). Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed VEGFR1/FLT1. PLoS Med 4:e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Keller G. (2000) VEGF nuclear accumulation correlates with phenotypical changes in endothelial cells. J Cell Sci 113 (Pt 9):1525-1534 [DOI] [PubMed] [Google Scholar]
- Li W, Yeo LS, Vidal C, McCorquodale T, Herrmann M, Fatkin D, Duque G. (2011). Decreased bone formation and osteopenia in lamin a/c-deficient mice. PloS One 6:e19313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Berendsen AD, Jia S, Lotinun S, Baron R, Ferrara N, Olsen BR. (2012). Intracellular VEGF regulates the balance between osteoblast and adipocyte differentiation. J Clin Invest 122:3101-3113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM. (2010). Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 19:329-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes C, Stockmans I, Moermans K, Van Looveren R, Smets N, Carmeliet P, Bouillon R, Carmeliet G. (2004) Soluble VEGF isoforms are essential for establishing epiphyseal vascularization and regulating chondrocyte development and survival. J Clin Invest 113:188-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito M, Omoteyama K, Mikami Y, Takagi M, Takahashi T. (2012). Suppression of lamin A/C by short hairpin RNAs promotes adipocyte lineage commitment in mesenchymal progenitor cell line, ROB-C26. Histochem Cell Biol 137:235-247 [DOI] [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. (1999). Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13:9-22 [PubMed] [Google Scholar]
- Olsen BR, Reginato AM, Wang W. (2000). Bone development. Annu Rev Cell Dev Biol 16:191-220 [DOI] [PubMed] [Google Scholar]
- Poltorak Z, Cohen T, Sivan R, Kandelis Y, Spira G, Vlodavsky I, Keshet E, Neufeld G. (1997). VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J Biol Chem 272:7151-7158 [DOI] [PubMed] [Google Scholar]
- Samuel S, Fan F, Dang LH, Xia L, Gaur P, Ellis LM. (2011). Intracrine vascular endothelial growth factor signaling in survival and chemoresistance of human colorectal cancer cells. Oncogene 30:1205-1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby F, Ho J, Stanford WL, Fischer KD, Schuh AC, Schwartz L, Bernstein A, Rossant J. (1997). A requirement for Flk1 in primitive and definitive hematopoiesis and vasculogenesis. Cell 89:981-990 [DOI] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. (1995). Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376:62-66 [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. (1998). Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92:735-745 [DOI] [PubMed] [Google Scholar]
- Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, Bohlen P. (1992). Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem Biophys Res Commun 187:1579-1586 [DOI] [PubMed] [Google Scholar]
- Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E, Olsen BR. (2004). VEGFA is necessary for chondrocyte survival during bone development. Development 131:2161-2171 [DOI] [PubMed] [Google Scholar]
- Zelzer E, McLean W, Ng YS, Fukai N, Reginato AM, Lovejoy S, D’Amore PA, Olsen BR. (2002). Skeletal defects in VEGF(120/120) mice reveal multiple roles for VEGF in skeletogenesis. Development 129:1893-1904 [DOI] [PubMed] [Google Scholar]