

Abstract
Neuroserpin, the major inhibitor of tissue plasminogen activator (tPA) in brain, has been shown to be up-regulated in Alzheimer’s disease (AD). Inhibition of tPA activity leads to reduced brain levels of plasmin, one of the main enzymes responsible for the degradation and clearance of amyloid-beta and its plaques from the brain. Thyroid hormone is one of the few factors known to enhance expression of neuroserpin in neurons. Thyroid hormone acts on neurons by binding to its receptors THR1α and THR1β, which then function in the nucleus to up-regulate the expression of numerous genes including the RNA-binding protein HuD. HuD acts post-transcriptionally to enhance expression of numerous proteins including neuroserpin by stabilizing their mRNAs. A series of Alzheimer’s disease brain tissues were compared to age-matched control brains for their expression of neuroserpin, THRβ1 and HuD by western blotting. Alzheimer’s disease brain tissues with elevated neuroserpin protein also showed increased expression of THRβ1 and HuD. Pair-wise analyses showed significant correlation p-values between neuroserpin, THRβ1 and HuD levels; suggesting that the up-regulation of neuroserpin in Alzheimer’s disease brain may result from an activation of the thyroid hormone response system in these individuals. These findings provide evidence for a potential relationship between thyroid hormone disorders and Alzheimer’s disease.
Keywords: Alzheimer’s disease, neuroserpin, thyroid hormone receptor-β1, tissue plasminogen activator, hyperthyroidism, human disease brain
Introduction
Alzheimer’s disease (AD), the leading cause of dementia and cognitive decline in aged individuals (Selkoe, 2001), is characterized by the accumulation of the amyloid-beta (Aβ) protein and its extracellular plaques in the AD brain. Inefficient clearance of the Aβ peptide in aged-adults is a potential mechanism for pathogenic Aβ plaque formation leading to neuronal death in AD. Plasmin (EC#3.4.21.7), which can cleave both fibrillar and oligomeric Aβ, is one of several proteases thought to regulate Aβ levels in the brain (Tucker et al. 2000). Both the pro-enzyme plasminogen (Tsirka et al. 1997; Basham and Seeds 2001) and its activator, tissue plasminogen activator (tPA) (EC#3.4.21.68) are produced and secreted by neurons (Krystosek and Seeds 1981; Moonen et al. 1982; Pittman 1985). Furthermore, neuronal tPA is directly implicated in synaptic activity associated with hippocampal long-term potentiation, cerebellar motor learning, and amygdala-mediated fear and anxiety (Qian et al. 1993; Seeds et al. 1995, 1999, 2003; Pawlak et al. 2003). The localization of both Aβ and plasminogen/plasmin in neuronal plasma membrane rafts lends further support to a plasmin–Aβ interaction in vivo (Ledesma et al., 2000). Thus, tPA-dependent activation of plasminogen to plasmin may be a critical step in synaptic plasma membrane amyloid precursor protein turnover/degradation and clearance of Aβ, thus maintaining brain homeostasis.
Earlier studies showed that the activity of plasmin (Ledesma et al. 2000) and tPA (Fabbro and Seeds 2009) are both dramatically reduced in Alzheimer’s disease brain compared to age-matched control brain. This reduced tPA activity correlated with a major increase of the tPA-specific inhibitor neuroserpin in the AD brain tissue, and tPA-neuroserpin complexes (Fabbro & Seeds, 2009). Immunohistochemistry showed that both tPA (Kinghorn et al., 2006) and neuroserpin (Fabbro & Seeds, 2009) co-localized with Aβ plaques in the AD brain tissue. However, recent studies by Barker et al. (2010) using a much broader age group (54–98yr.) of Alzheimer’s disease brain with a mean age of 79yr. as compared to the younger population (mean age 68yr.) used by Fabbro & Seeds (2009) also found lower plasmin protein and activity in the frontal cortex of the AD brains, but not statistically significant differences as compared to age-matched controls. Interestingly, in this same older group of AD brain tissues Barker et al. (2012) found reduced levels of neuroserpin, probably reflecting the more pronounced neuron cell death seen in this older AD population. Alternatively, the neuroserpin antibody used by Barker et al. in their ELISA assay may not have recognized neuroserpin/tPA complexes, which readily dissociate on SDS-PAGE and are quantified as part of the neuroserpin/cleaved neuroserpin duplex at 45–50kDa in the western blot assay for neuroserpin protein used by Fabbro & Seeds.
An attempt to use the plasmin system in cerebral spinal fluid as an indicator of Alzheimer’s disease (Martorana et al., 2012) failed to find any difference in tPA zymographic activity between the AD and control population. However, this difference in tPA activity from frontal cortex may reflect significant tPA expression in the ventricular region and choroid plexus (Friedman & Seeds, 1994) that can influence tPA levels in the CSF where neuroserpin is low.
In addition, studies with a mouse model of AD, expressing a mutated-human precursor protein transgene leading to Aβ plaques and spatial learning deficits, showed that knocking-out the neuroserpin gene dramatically reduced brain levels of Aβ, plaque formation and restored near-normal spatial learning to these mice (Fabbro et al., 2011). These findings support the premise that neuroserpin inhibition of tissue plasminogen activator plays an important role both in the accumulation of brain amyloid plaques and the loss of cognitive abilities in AD. Similar studies with PAI-1 inhibitors (Jacobsen et al., 2008) and PAI-1 knockout mice (Liu et al., 2011) support the proposal that increased tPA activity and plasmin lead to lower brain Aβ levels and the reversal of cognitive deficit in these mice.
We have explored what factors may lead to the dramatic increase in neuroserpin protein in the AD brain. Increased synaptic activity and thyroid hormone are both known to up-regulate, expression of neuroserpin (Navarro-Yubero et al., 2004). Thyroid hormone acting on its receptors THRα & THRβ influences expression of numerous genes including the receptors themselves (Lebel et al., 1993; Vallortigara et al., 2009), as well as the neuron-specific RNA-binding protein HuD (Cuadrado et al., 2003; Navarro-Yubero et al., 2004). HuD acts to stabilize mRNAs including neuroserpin mRNA (Bolognani and Perrone-Bizzozero, 2008); thus, leading to enhanced translation and elevated tissue levels of neuroserpin. Therefore to assess this relationship, we have compared neuroserpin levels with THRβ1 and HuD levels in the Alzheimer’s disease brain to those levels in age-matched control brains.
Experimental Procedures
Homogenate preparation
Snap-frozen pieces of frontal cortex brain tissue from patients with AD and age-matched controls were obtained from University of Colorado Department of Pathology, Harvard Brain Tissue Resource Center, and New York Brain Bank at Columbia University, and their Braak staging of AD severity was noted. Tissues were homogenized in a Tris-HCl buffer (0.1 M Tris, pH 8.1, with 1% Triton X-100) and the suspensions centrifuged at 14,000×g for 45 min at 4°C. The supernatants were collected and stored at −80°C. Total protein concentration in each homogenate was determined by BCA assay kit (Pierce Biotechnology).
Western blot and protein quantification
Fifty micrograms of total protein from frontal cortex homogenates of AD and age-matched controls, as well as increasing amounts (1ng to 1ug) of purified THRβ1 (Protein One, Rockville, MD), Neuroserpin (Abcam, #ab63224), and HuD (gift of Dr. Nora Perrone-Bizzozero, University of New Mexico) protein standards were resolved using 10% SDS-polyacrylamide gel electrophoresis. The proteins from the gels were transferred to PVDF (polyvinylidene difluoride) membrane (Millipore, Bedford, MA, USA) using a semi-dry transfer system (BioRad Laboratories, Hercules, CA, USA). The PVDF membranes were then treated with blocking buffer (Odyssey; LI-COR, Lincoln, NE) and incubated with primary antibodies against THRβ1 (Pierce Biotechnology, mouse monoclonal, 1:1000), Neuroserpin (ProScience Inc, rabbit polyclonal, 1:1000 dilution) and HuD (Santa Cruz Biotechnology, mouse monoclonal E-1, 1:1000 dilution) overnight at 4°C. Actin antibody reactivity was used to confirm equal protein loading of the samples. After washing three times with PBST (phosphate buffered saline containing 0.05% Tween 20), the membranes were incubated with appropriate secondary antibodies, IRDye800 conjugated anti-rabbit and anti-mouse secondary antibodies, (1:10000, Odyssey; LI-COR Biosciences, Lincoln, NE)) at 25°C for 45 min. After three washes with PBST, the blot was transferred to PBS. Protein bands on the membrane were visualized and analyzed with an infrared imaging system (Odyssey; LI-COR Biosciences), and the signal intensity was determined with imaging software (Image Studio, LI-COR Biosciences) and exported for graphic representation as the mean ± SEM (Prism; GraphPad, San Diego, CA). Quantification of the proteins was assessed from western blots using the same concentrations of primary and secondary antibodies but with increasing known concentrations (1, 3,10,30,100,300, 1000ng) of each standard protein (THRβ1, NS and HuD) to obtain a linear equation from which each protein of interest in the brain homogenates could be quantified, and its densitometric linearity confirmed using two different concentrations of each brain homogenate, and densitometric exposure for two different time periods.
Statistical Analysis
Quantitative data of neuroserpin, THRβ1, and HuD concentration are presented as the mean ± SEM (standard error of means) in figures. A students t-test was used to determine statistical differences of neuroserpin, THRβ1 and HuD expression between AD vs Control (GraphPad Software). A p value of < 0.05 was considered statistically significant. Pair-wise correlation of NS Vs THRβ1, NS Vs HuD, and THRβ1 Vs HuD were calculated for both AD and Control samples separately. Further, to delineate the overall correlation of Disease state (disease=1, no disease=0) Vs NS, Disease state Vs THRβ1, Disease state Vs HuD, NS Vs THRβ1, NS Vs HuD, and THRβ1 Vs HuD, pair-wise correlation coefficients were calculated from all the observations of these variables from both AD and control samples. In order to fit a regression model to assess overall association of expression of NS, THRβ1 and HuD, a binary logistic regression with the disease status as dependent variable (Disease or No Disease) and the expression levels of NS, THRβ1 and HuD as independent variables were tried (STATA). However, no valid model could be estimated using NS, THRβ1 and HuD mainly because of the high co-linearity among the variables and the three variables in limited number of observations may be perfectly overfitting the data.
Results
The possibility that thyroid hormone plays a role in the up-regulation of neuroserpin in Alzheimer’s disease was explored. However, quantifying active thyroid hormone T3 in post-mortem brain tissue would be difficult and is further complicated by the necessity of separating free thyroid hormone from receptor-bound and metabolized forms, as well as cellular thyroid hormone from vascular and CSF forms. Therefore, we have chosen to assess THRβ1 which is found primarily in cortical neurons and whose expression is regulated by T3 levels in a variety of tissues (Zandieh-Doulabi et al., 2004; Rabier et al., 2006; Liu et al., 2007; Vallortigara et al., 2009). Since neuroserpin up-regulation in cultured cells by thyroid hormone has been shown to act through the RNA-binding protein HuD (Navarro-Yubero et al., 2004), HuD has also been assessed in these Alzheimer’s disease brain tissues and their age-matched controls.
An initial comparison of neuroserpin, THRβ1 and HuD in five Alzheimer’s disease brain and five control age-matched brain homogenates is seen in Figure 1 western blots. It is readily apparent that all three proteins are expressed at higher levels in the AD brain. This observation was confirmed by quantifying neuroserpin, THRβ1 and HuD western blots of twelve Alzheimer’s disease (mean age 67.7yr.) frontal cortex homogenates as compared to twelve age-matched control (mean age 68.8yr.) frontal cortex homogenates (Table 1A & 1B). All the AD cortical tissue contained numerous amyloid plaques, and the brain banks’ Braak staging (Braak & Braak, 1991) of the neurofibrillary tangle involvement of each Alzheimer’s disease tissue is indicated (Table 1). The mean average of each of the three proteins in AD brain homogenates is significantly (* p<0.05) greater than those mean values for the age-matched control brain samples (Table 2).
Figure 1. Neuroserpin, THR-β1, and HuD up-regulated in the Alzheimer’s disease brain.

A) Neuroserpin, THR-β1 and HuD concentration of Alzheimer’s disease brain samples
B) Neuroserpin, THR-β1 and HuD concentration of age-matched control brain samples. Sample numbers (1–5) correspond to those in Table 1A & 1B. All three protein amounts are lower in the age-matched controls in these 50ug total protein loads.
Table 1A.
Neuroserpin, THRβ1 and HuD concentration of Alzheimer’s disease brain samples
| Sample #s (Age/Gender/Braak stage) | Neuroserpin (ng/50μg protein) | THRβ1 (ng/50μg protein) | HuD (ng/50μg protein) |
|---|---|---|---|
|
| |||
| 1) 59yr. Female V | 84.2 | 357.2 | 154.3 |
| 2) 62yr. Female VI | 255.6 | 559.6 | 100.8 |
| 3) 66yr. Female VI | 246.7 | 113.9 | 28.9 |
| 4) 68yr. Male V | 526.3 | 374.1 | 99.6 |
| 5) 62yr. Female VI | 514.2 | 238.0 | 80.7 |
| 6) 72yr. Male I | 319.9 | 740.4 | 108.7 |
| 7) 70yr. Male V | 412.3 | 754.8 | 83.2 |
| 8) 73yr. Female VI | 169.0 | 263.3 | 40.4 |
| 9) 75yr. Male VI | 274.1 | 752.4 | 52.2 |
| 10) 64yr. Male VI | 139.0 | 545.2 | 33.4 |
| 11) 74yr. Male VI | 397.0 | 834.3 | 54.3 |
| 12) 67yr. Male VI | 415.5 | 759.6 | 31.2 |
Table 1B.
Neuroserpin, THRβ1 and HuD concentration of age-matched control brain samples
| Sample #s (Age/Gender) | Neuroserpin (ng/50μg protein) | THRβ1 (ng/50μg protein) | HuD (ng/50μg protein) |
|---|---|---|---|
|
| |||
| 1) 62yr. Male | 119.9 | 148.8 | 26.6 |
| 2) 63yr. Female | 58.8 | 11.4 | 12.3 |
| 3) 69yr. Male | 163.9 | 81.3 | 18.9 |
| 4) 70yr. Male | 221.8 | 101.8 | 20.2 |
| 5) 63yr. Female | 107.2 | 17.5 | 10.4 |
| 6) 72yr. Male | 160.7 | 107.8 | 71.4 |
| 7) 70yr. Male | 100.8 | 209.0 | 82.8 |
| 8) 74yr. Female | 40.9 | 187.3 | 22.8 |
| 9) 74yr. Male | 11.2 | 98.2 | 22.7 |
| 10) 65yr. Male | 126.3 | 172.9 | 87.6 |
| 11) 74yr. Male | 172.8 | 211.4 | 15.1 |
| 12) 70yr. Male | 100.2 | 178.9 | 22.1 |
Table 2.
Results of student t-test of expression levels of NS, THRβ1, and HuD from AD versus age-matched control brain homogenatesa
| Variable | t value | Significance (two tailed) | 95% Confidence interval
|
|
|---|---|---|---|---|
| Lower | Higher | |||
| NS | 4.4 | p = .0002 | −290.4 | −104.5 |
| THRβ1 | 5.4 | p = <.0001 | −555.1 | −243.4 |
| HuD | 2.7 | p = .0124 | −66.7 | −9.1 |
Degrees of freedom = 22
Pair-wise correlation coefficient of NS, THR-β1 and HuD was calculated separately for Control and AD without a statistically significant positive relationship in NS Vs THRβ1, NS Vs HuD, and THRβ1 Vs HuD (Table 3). Interestingly, when neuroserpin amounts were plotted versus the THRβ1 amounts for each sample, the correlative scatter-plot shows that all the AD brain samples fall outside of those values for the control brain samples (Figure 2). Furthermore, the relationship of neuroserpin levels to the levels of THRβ1 in each of these brain samples compared using Pearson’s correlation coefficient shows a significant positive association between neuroserpin levels and THRβ1 levels in these brain samples (Figure 2 and Table 4). Similarly, correlative plots of THRβ1 and HuD, as well as neuroserpin and HuD for each of the tissue samples suggest a moderately positive relationship (Figures 3 & 4) with these proteins. Significant associations between THRβ1 and HuD, as well as between neuroserpin and HuD in Alzheimer’s disease was confirmed by these correlations. The box plot (Figure 5) clearly shows the significant differences in the mean values for NS, THRβ1 and HuD proteins between Alzheimer’s disease brain and control brain tissues.
Table 3.
Pair-wise correlation coefficient of NS, THR-β1 and HuD was calculated separately for Control and AD. The inferential statistic is given as r2 value; p valuea
| Con-NS | AD-NS | Con- THRβ1 | AD- THRβ1 | Con- HuD | AD- HuD | |
|---|---|---|---|---|---|---|
| Con-NS | 1 | 0.372; 0.234 | ||||
| AD-NS | 0.372; 0.234 | 1 | ||||
|
|
||||||
| Con-THRβ1 | 1 | 0.416; 0.178 | ||||
| AD-THRβ1 | 0.416; 0.178 | 1 | ||||
|
|
||||||
| Con- HuD | 1 | 0.006; 0.984 | ||||
| AD-HuD | 0.006; 0.984 | 1 | ||||
Degrees of freedom = 12
Figure 2. THR-β1 versus neuroserpin levels.

Correlative scatter plot of all AD brain samples (▲) comparing neuroserpin (densitometric units) versus THR-β1 (densitometric units) to those of age-matched control brain samples (●). Pair-wise correlation coefficient of NS Vs THRβ1 of all observations from AD and age-matched control brain homogenates were positively correlated with a Pearson’s r(24)=0.599; p=0.002
Table 4.
Pair-wise correlation coefficient of independent variables (disease state, NS, THRβ1 and HuD) of all observations from AD and age-matched control brain homogenates. The inferential statistic is given as r2 value; p valuea
| Disease AD | NS | THRβ1 | HuD | |
|---|---|---|---|---|
| Disease | ||||
| AD | 1 | |||
| NS | 0.685; .0002 | 1 | ||
| THRβ1 | 0.752; .00002 | 0.599; .002 | 1 | |
| HuD | 0.502; 0.12 | 0.341; 0.103 | 0.403;.051 | 1 |
Degrees of freedom = 24
Figure 3. THR-β1 levels versus HuD.

Correlative plot of THR-β1 amounts versus HuD amounts in each of the AD brain samples (▲) or age-matched control brain samples (●). Pair-wise correlation coefficient of THRβ1 Vs HuD of all observations from AD and age-matched control brain homogenates were moderately correlated with a Pearson’s r(24)=0.403; p=0.051.
Figure 4. Neuroserpin versus HuD levels.

Correlative plot of neuroserpin amounts versus HuD amounts in AD brain samples (▲) and age-matched control brain samples (●). Pair-wise correlation coefficient of NS Vs HuD of all observations from AD and age-matched control brain homogenates were moderately correlated with a Pearson’s r(24)=0.502; p= 0.12.
Figure 5. NS (A), THRβ1 (B), and HuD (C) protein concentrations from AD and age-matched control brains.
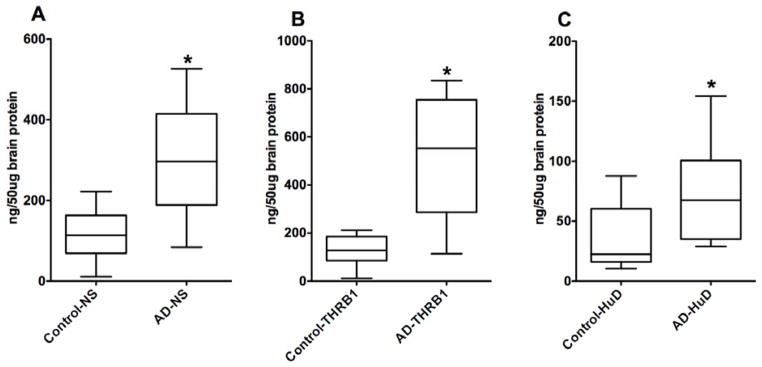
Values are means ± S.E.M. of twelve samples per group. Student t test was used to determine differences among brain tissue means (AD Vs Control) on each protein. The AD brain samples were significantly different from the control brain samples for each of the three brain proteins. An asterisk indicates that the mean is different (P<0.05) from that of the control.
Discussion
These studies show that the brain tissue of Alzheimer’s disease individuals expresses major increases in the tPA inhibitor neuroserpin, THRβ1 and HuD proteins. Pair-wise analyses indicate that there is a significant association between these three proteins and Alzheimer’s disease brain as compared to age-matched control brain. The strong correlation between these three proteins and Alzheimer’s disease is likely due to the action of brain thyroid hormone. Direct measurement of T3 levels in these post-mortem brain tissues would have been subject to a multitude of variables as indicated above and was not undertaken. However, a number of studies have shown a relationship between tissue levels of thyroid hormone and its receptor THRβ1 (Cheng et al., 2010). Hyperthyroidism leads to an up-regulation of THRβ1 in rat liver (Zandieh-Doulabi et al., 2004), whereas THRβ1 expression is down-regulated in the muscle of hypothyroid rats (Liu et al, 2007) and the brains of hypothyroid mice (Vallortigara et al., 2009). More importantly, T3 administration increases THRβ1 expression in cultured cerebral neurons (Lebel et al., 1993) and increases THRβ1 expression in mouse brain (Vallortigara et al., 2009). These findings indicate that when brain THRβ1 is elevated so is brain thyroid hormone.
T3 and THRβ1 complexes have several targets within the nucleus; one of the more interesting is the neuronal RNA-binding protein HuD. HuD acts to stabilize a number of mRNAs including neuroserpin, where it binds to the 3′-UTR. A 70-fold increase in neuroserpin protein (Bolognani et al., 2009) and a 57-fold increase in transthyretin, the transporter of thyroid hormone T4 into brain (Perrone-Bizzozero et al., 2011) are seen in mice over-expressing HuD; these mice also have major learning deficits (Bolognani et al., 2007). In addition, neuroserpin RNA and protein levels are down-regulated in many brain areas including the hippocampus, cortical layers II/III and VIa, retrospinal cortex, and the medial habenular nucleus of the hypothyroid rat brain (Navarro-Yubero et al., 2004). Furthermore, neuroserpin mRNA and protein levels are increased upon T3 administration to PC12 cells stably expressing a THR-transgene (Navarro-Yubero et al., 2004). However, HuD expression in these same PC12 cells was reduced by T3 treatment. This suggests that some of the regulatory effects of T3 on neuroserpin expression in cultured cells and different brain areas may depend on factors other than HuD to control gene expression and neuroserpin RNA stability. While these studies provide further support for an association between thyroid hormone and neuroserpin in cognitive deficits, we cannot rule out that the increases seen in neuroserpin, THRβ1 and/or HuD in the Alzheimer’s disease brain occur by mechanisms other than thyroid hormone. For example we cannot rule out that ischemia in the Alzheimer’s disease brain may lead to an increase in neuroserpin expression, since mouse studies with middle cerebral artery occlusion induced ischemia (Wu et al., 2010) showed increased neuroserpin in the hippocampus and cerebral cortex.
An earlier report (Amadio, et al., 2009) using immunohistochemistry with a nELAV family (HuB, HuC & HuD) antibody showed a decrease in the nELAV family of proteins in the hippocampus of Alzheimer’s disease brain tissue. In contrast, our studies used a HuD-specific antibody and focused on brain frontal cortex. These studies with Alzheimer’s disease brain tissue and those animal studies described above further demonstrate that the various Hu/ELAV proteins have a complex pattern of expression in the brain and indicate that the relative HuD expression among ELAV proteins varies in different brain regions, as well as the total HuD protein expression.
Both hyper- and hypothyroidism have been implicated in increasing the risk of developing Alzheimer’s disease (Tan & Vasan, 2009). Although thyroid hormone levels in serum showed no change in Alzheimer’s disease (McKhann et al., 1984), circulating thyroid hormone levels do not accurately reflect thyroid hormone metabolism in the CNS where T3 levels are tightly regulated within a narrow range (Dratman et al., 1983); thus suggesting that small deviations in normal cerebral T3 levels may result in cognitive dysfunction (Loosen, 1992). The Honolulu-Asia aging study showed that higher total and free thyroxine levels were associated with increased risk of dementia and Alzheimer’s disease (de Jong et al., 2009). Similarly, the Sao Paulo aging study found an association of subclinical hyperthyroidism with dementia and Alzheimer’s disease (Bensenor et al., 2010). In another study high T3 levels were seen in patients with mild cognitive impairment and a neuropsychological profile typical of Alzheimer’s disease (Quinlan et al., 2010). Although our studies did not measure thyroid hormone levels directly in the brain tissues, the increased levels of THRβ1, HuD and neuroserpin in the frontal cortex of Alzheimer’s disease patients as compared to those of age-matched control patients suggest they had elevated brain thyroid hormone levels. However, the tissues used for these studies are from brain banks so we do not know the patient’s thyroid history or whether they may have been receiving hormone therapy. Interestingly, the increased expression of THRβ1 in the Alzheimer’s disease brain may suggest an adaptive response to elevated amyloid precursor protein (APP) mRNA, since THRs have been shown (Belakavadi et al., 2011) to repress APP gene expression by directly binding to the APP gene and actively recruiting histone modifying enzymes that repress APP transcription. Yet, the statistically significant correlation between these three proteins and Alzheimer’s disease indicates a definitive association of thyroid hormone system action and Alzheimer’s disease.
Acknowledgments
The authors would like to express our gratitude to Dr. Paul McGuire for providing the space and facilities to complete these studies. We sincerely appreciate the helpful advice and insight provided by Dr. Nora Perrone-Bizzozero and Dr. Andrea Allan with the HuD studies and statistical analyses. The brain tissues were kindly provided by the Harvard Brain Tissue Resource Center, which is supported in part by PHS grant R24MH068855, and the New York Brain Bank at Columbia University. Our studies were supported by the Cure Alzheimer’s Fund and in part by NIH grant NS044129.
Abbreviations
- AD
Alzheimer’s disease
- Aβ
amyloid-beta protein
- NS
neuroserpin
- THRβ1
thyroid hormone receptor-beta 1
- HuD
Hu protein D
- APP
amyloid precursor protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amadio M, Pascale A, Wang J, Ho L, Quattrone A, Gandy S, Haroutunian V, Racchi M, Pasinetti GM. nELAV proteins alteration in Alzheimer’s disease brain: a novel putative target for amyloid-β reverberating on AβPP processing. J Alz Dis. 2009;16:409–419. doi: 10.3233/JAD-2009-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker R, Love S, Kehoe P. Plasminogen and plasmin in Alzheimer’s disease. Brain Res. 2010;1355:7–15. doi: 10.1016/j.brainres.2010.08.025. [DOI] [PubMed] [Google Scholar]
- Barker R, Kehoe PG, Love S. Activators and inhibitors of the plasminogen system in Alzheimer’s disease. J Cell Mol Med. 2012;16:865–876. doi: 10.1111/j.1582-4934.2011.01394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basham ME, Seeds NW. Plasminogen expression in the neonatal and adult mouse brain. J Neurochem. 2001;77:318–325. doi: 10.1046/j.1471-4159.2001.t01-1-00239.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathologica. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Belakavadi M, Dell J, Grover GJ, Fondell JD. Thyroid hormone suppression of b-amyloid precursor protein gene expression in the brain involves multiple epigenetic regulatory events. Mol Cell Endorinol. 2011;339:72–80. doi: 10.1016/j.mce.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Bensenor IM, Lotufo P, Menezes P, Scazufca M. Subclinical hyperthyroidism and dementia: the San Paulo Ageing and Health Study. BMC Public Health. 2010;10:298–303. doi: 10.1186/1471-2458-10-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognani F, Qui S, Tanner D, Paik J, Perrone-Bizzozero NI, Weber E. Associative and spatial learning and memory deficits in transgenic mice over-expressing the RNA-binding protein HuD. Neurobiol Learn & Mem. 2007;87:635–643. doi: 10.1016/j.nlm.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Bolognani F, Perrone-Bizzozero NI. RNA-protein interactions and control of mRNA stability in neurons. J Neurosci Res. 2008;86:481–489. doi: 10.1002/jnr.21473. [DOI] [PubMed] [Google Scholar]
- Bolognani F, Contente-Cuomo T, Perrone-Bizzozero NI. Novel recognition motifs and biological functions of the RNA-binding protein HuD revealed by genome-wide identification of its targets. Nucleic Acids Res. 2009;38:117–130. doi: 10.1093/nar/gkp863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SY, Leonard J, Davis P. Molecular aspects of thyroid hormone actions. Endocr Rev. 2010;31:139–70. doi: 10.1210/er.2009-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong FJ, Masaki K, Chen H, Remaley A, Breteler M, Petrovich H, White L, Launer LJ. Thyroid function, the risk of dementia and neuropathologic changes: the Honolulu-Asia aging study. Neurobiol Aging. 2009;30:600–606. doi: 10.1016/j.neurobiolaging.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dratman MB, Crutchfield F, Gordon J, Jennings A. Iodothyronine homeostasis in rat brain during hypo and hyperthyroidism. Am J Physiol. 1983;245:E185–193. doi: 10.1152/ajpendo.1983.245.2.E185. [DOI] [PubMed] [Google Scholar]
- Fabbro S, Seeds NW. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J Neurochem. 2009;109:303–315. doi: 10.1111/j.1471-4159.2009.05894.x. [DOI] [PubMed] [Google Scholar]
- Fabbro S, Schaller K, Seeds NW. Amyloid-beta levels are significantly reduced and spatial memory defects are rescued in a novel neuroserpin-deficient Alzheimer’s disease transgenic mouse model. J Neurochem. 2011;118:928–938. doi: 10.1111/j.1471-4159.2011.07359.x. [DOI] [PubMed] [Google Scholar]
- Friedman GC, Seeds NW. Tissue plasminogen activator expression in the embryonic nervous system. Dev Brain Res. 1994;81:41–49. doi: 10.1016/0165-3806(94)90066-3. [DOI] [PubMed] [Google Scholar]
- Kinghorn KJ, Crowther DC, Sharp LK, Nerelius C, Davis RL, Chang HT, Green C, Gubb DC, Johansson J, Lomas DA. Neuroserpin binds A-beta and is a neuroprotective component of amyloid plaques in Alzheimer disease. J Biol Chem. 2006;281:29268–29277. doi: 10.1074/jbc.M600690200. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Comery T, Martone R, Elokdah H, Crandall D, Oganesian A, Aschmies S, Kirksey Y, Gonzales C, Xu J, Zhou H, Atchison K, Wagner E, Zaleska M, Das I, Arias R, Bard J, Riddell D, Gardell S, Abou-Gharbia M, Robichaud A, Magolda R, Vlasuk G, Bjornsson T, Reinhart P, Pangalos M. Enhanced clearance of Aβ in brain by sustaining the plasmin proteolysis cascade. Proc Nat Acad Sci USA. 2008;105:8754–8759. doi: 10.1073/pnas.0710823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystosek A, Seeds NW. Plasminogen activator release at the neuronal growth cone. Science. 1981;213:1532–1534. doi: 10.1126/science.7197054. [DOI] [PubMed] [Google Scholar]
- Lebel JM, L’Hérault S, Dussault J, Puymirat J. Thyroid hormone up-regulates thyroid hormone receptor beta gene expression in rat cerebral hemisphere astrocyte cultures. Glia. 1993;9:105–112. doi: 10.1002/glia.440090203. [DOI] [PubMed] [Google Scholar]
- Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP alpha-cleavage and A-beta degradation and is reduced in Alzheimer’s disease brains. EMBO Rep. 2000;1:530–535. doi: 10.1093/embo-reports/kvd107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CR, Li LY, Zang X, Liu YM, Sun Y, Kan B. Effects of hyper- and hypothyroid on expression of thyroid hormone receptor mRNA in rat myocardium. J Endocrinol. 2007;195:429–438. doi: 10.1677/JOE-07-0253. [DOI] [PubMed] [Google Scholar]
- Liu RM, van Groen T, Katre A, Cao D, Kadisha I, Ballinger C, Wang L, Carroll S, Li L. Knockout of plasminogen activator inhibitor 1 gene reduces amyloid beta peptide burden in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2011;32:1079–1089. doi: 10.1016/j.neurobiolaging.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loosen T. Effects of thyroid hormones on central nervous system in aging. Psychoneuroendocrinology. 1992;17:355–374. doi: 10.1016/0306-4530(92)90041-5. [DOI] [PubMed] [Google Scholar]
- Martorana A, Sancesario G, Esposito Z, Nuccetelli M, Sorge R, Formosa A, Dinallo V, Bernardi G, Bernardini S, Sancesario G. Plasmin system of Alzheimer’s disease patients: CSF analysis. J Neural Transm. 2012;119:763–769. doi: 10.1007/s00702-012-0778-y. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan E. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS ADRDA Work Group. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Moonen G, Grau-Wagemans M, Selak I. Plasminogen activator-plasmin system and neuronal migration. Nature. 1982;298:753–755. doi: 10.1038/298753a0. [DOI] [PubMed] [Google Scholar]
- Navarro-Yubero C, Cuadrado A, Sonderegger P, Munoz A. Neuroserpin is post-transcriptionally regulated by thyroid hormone. Brain Res Mol Brain Res. 2004;123:56–65. doi: 10.1016/j.molbrainres.2003.12.018. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. 2003;6:168–174. doi: 10.1038/nn998. [DOI] [PubMed] [Google Scholar]
- Perrone-Bizzozero NI, Tanner D, Mounce J, Bolognani F. Increased expression of axogenesis-related genes and mossy fiber length in dentate granule cells from adult HuD over-expressor mice. ASN Neuro. 2011;3:259–270. doi: 10.1042/AN20110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittman RN. Release of plasminogen activator and a calcium-dependent metalloprotease from cultured sympathetic and sensory neurons. Dev Biol. 1985;110:91–101. doi: 10.1016/0012-1606(85)90067-3. [DOI] [PubMed] [Google Scholar]
- Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- Quinlan P, Nordlund A, Lind K, Gustafson D, Edman A, Wallin A. Thyroid hormones are associated with poorer cognition in mild cognitive impairment. Dement Geriatr Cogn Disord. 2010;30:205–211. doi: 10.1159/000319746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabier B, Williams AJ, Mallein-Gerin F, Williams GR, Chassande O. Thyroid hormone-stimulated differentiation of primary rib chondrocytes in vitro requires thyroid hormone receptor beta. J Endocrinol. 2006;191:221–228. doi: 10.1677/joe.1.06838. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Williams BL, Bickford PC. Tissue plasminogen activator induction in Purkinje neurons after cerebellar motor learning. Science. 1995;270:1992–1994. doi: 10.1126/science.270.5244.1992. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Haffke SP. Neuronal migration is retarded in mice lacking the tissue plasminogen activator gene. Proc Natl Acad Sci U S A. 1999;96:14118–14123. doi: 10.1073/pnas.96.24.14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Ferguson JE. Absence of tissue plasminogen activator gene or activity impairs mouse cerebellar motor learning. J Neurosci. 2003;23:7368–7375. doi: 10.1523/JNEUROSCI.23-19-07368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Clearing the brain’s amyloid cobwebs. Neuron. 2001;32:177–180. doi: 10.1016/s0896-6273(01)00475-5. [DOI] [PubMed] [Google Scholar]
- Tan ZS, Vasan RS. Thyroid function and Alzheimer’s disease. J Alz Dis. 2009;16:503–507. doi: 10.3233/JAD-2009-0991. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashi T, Price D, Walker D, Scheff S, McGillis JP, Rydel RE, Estus S. The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci. 2000;20:3937–3946. doi: 10.1523/JNEUROSCI.20-11-03937.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallortigara J, Chassande O, Higueret P, Enderlin V. Thyroid hormone receptor alpha plays an essential role in the normalization of adult-onset hypothyroidism-related hypoexpression of synaptic plasticity target genes in striatum. J Neuroendocrinol. 2009;21:49–56. doi: 10.1111/j.1365-2826.2008.01802.x. [DOI] [PubMed] [Google Scholar]
- Wu J, Echeverry R, Guzman J, Yepes M. Neuroserpin protects neurons from ischemia-induced plasmin-mediated cell death independently of tissue-type plasminogen activator inhibition. Am J Pathol. 2010;177:2576–2584. doi: 10.2353/ajpath.2010.100466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandieh-Doulabi B, Platvoet-Ter Schiphorst M, Kalsbeek A, Fliers E, Bakker O, Wiersinga WM. Biurnal variation in rat liver thyroid hormone receptor mRNA is dependent on the biological clock in the suprachiasmatic nucleus, whereas diurnal variation of THRβ1 mRNA is modified by food intake. Endocrinology. 2004;145:1284–1289. doi: 10.1210/en.2003-0791. [DOI] [PubMed] [Google Scholar]