

Abstract
Insulin-like growth factor binding protein 3 (IGFBP3), a hypoxia-inducible gene, regulates a variety of cellular processes including cell proliferation, senescence, apoptosis and epithelial-mesenchymal transition (EMT). IGFBP3 has been linked to the pathogenesis of cancers. Most previous studies focus upon proapoptotic tumor suppressor activities of IGFBP3. Nevertheless, IGFBP3 is overexpressed in certain cancers including esophageal squamous cell carcinoma (ESCC), one of the most aggressive forms of squamous cell carcinomas (SCCs). The tumor-promoting activities of IGFBP3 remain poorly understood in part due to a lack of understanding as to how the tumor microenvironment may influence IGFBP3 expression and how IGFBP3 may in turn influence heterogeneous intratumoral cell populations. Here, we show that IGFBP3 overexpression is associated with poor postsurgical prognosis in ESCC patients. In xenograft transplantation models with genetically engineered ESCC cells, IGFBP3 contributes to tumor progression with a concurrent induction of a subset of tumor cells showing high expression of CD44 (CD44H), a major cell surface receptor for hyaluronic acid, implicated in invasion, metastasis and drug resistance. Our gain-of-function and loss-of-function experiments reveal that IGFBP3 mediates the induction of intratumoral CD44H cells. IGFBP3 cooperates with hypoxia to mediate the induction of CD44H cells by suppressing reactive oxygen species (ROS) in an insulin-like growth factor-independent fashion. Thus, our study sheds light on the growth stimulatory functions of IGFPB3 in cancer, gaining a novel mechanistic insight into the functional interplay between the tumor microenvironment and IGFBP3.
Keywords: CD44, esophageal, squamous cell carcinoma, hypoxia, IGFBP3 and reactive oxygen species
Introduction
Esophageal squamous cell carcinoma (ESCC) is one of the deadliest forms of all human squamous cell carcinomas (SCCs), characterized by poor prognosis and limited therapeutic intervention. Treatment failure can be linked to tumor-cell heterogeneity due to diverse cell types (cancer cells, fibroblasts, immune cells, blood vessels) in tumors. ESCC cells themselves contribute to intratumoral heterogeneity. Amongst the cell surface markers defining distinct subsets of cancer cells is CD44. As a major receptor for hyaluronic acid, CD44 has a role in cancer cell invasion, metastasis and drug resistance by mediating crosstalk between cancer cells and the surrounding microenvironment [1-4]. High CD44 (CD44H) expression has been attributed to tumorigenic cells or those referred to as tumor initiating cells or cancer stem cells (CSCs). Tumor initiating capability of CD44H cells has been documented in many tumor types including head and neck SCC (HNSCC) and ESCC [4-7]. CD44 is upregulated in cancer cells adjacent to tumor stroma [6]. Cancer cells display increased generation of reactive oxygen species (ROS) and adaptation to oxidative stresses [8]. CD44 has been implicated in regulation of antioxidant capacity of CSCs [9-11]. In the tumor microenvironment, hypoxia regulates CD44 expression via hypoxia inducible factor (HIF)-1α [12]; however, it remains elusive as to how ESCC cells cope with oxidative stress under hypoxic conditions.
Insulin-like growth factor binding protein 3 (IGFBP3) regulates cell proliferation and apoptosis in insulin-like growth factor (IGF)-dependent and independent manners. IGFBP3 has been linked to the pathogenesis of cancers. Most previous studies focus upon proapoptotic tumor suppressor activities of IGFBP3, inhibiting the prosurvival IGF-mediated signaling [13]. Nevertheless, IGFBP3 is overexpressed in ESCC and other tumor types where its biological significance remains unclear [14-16]. Hypoxia activates IGFBP3 transcription through HIF-1α [17]. IGFBP3 is often localized to the intratumoral hypoxic area [17]. Under experimental conditions, ectopically expressed IGFBP3 neutralizes IGFs to trigger apoptosis in ESCC cells upon xenograft transplantation [18]; however, endogenous IGFBP3 is induced in tumors and permissive for tumor formation. Moreover, IGFBP3I56G/L80G/L81G (GGG-IGFBP3) lacking IGF-binding capability not only negates apoptosis, but facilitates invasive tumor growth in vitro and in vivo [16,17,19-21]. Thus, IGFBP3 may have context-dependent tumor-promoting activities.
The tumor-promoting activities of IGFBP3 remain poorly understood in part due to limited understanding of the roles of IGFBP3 influencing tumor cell heterogeneity in the hypoxic tumor microenvironment. We hypothesized that IGFBP3 has a unique biological activity in established tumors. Herein, we demonstrate for the first time that IGFBP3 may have a novel IGF-independent antioxidant activity, suppressing ROS-mediated cytotoxicity, thereby increasing the intratumoral CD44H cells in the hypoxic tumor microenvironment.
Materials and methods
Tissue microarrays and immunohistochemistry
ESCC tissues and adjacent non-cancerous mucosa were procured via surgery at the Kagoshima University Hospital (YK and SN) and used to generate tissue microarray as described previously [22]. All of the clinical materials were obtained from informed-consent patients in accordance with Institutional Review Board standards and guidelines. Immunohistochemistry was performed and scored as described previously [23]. In brief, sections were incubated with anti-human IGFBP-3 mouse monoclonal antibody (Clone 84728.111) (R&D Systems, Minneapolis, MN) at 1:250 dilution, followed by incubation with biotinylated secondary IgG and signal development using the DAB Peroxidase Substrate Kit (Vector). The staining was assessed independently by two of the authors (SN and AJK). The intensity was scored as negative (0), weakly positive (0.5), definitively positive (1) or strongly positive (2).
Cells and treatment
ESCC cell lines (TE11, TE12 and T.T), T-TeRas and derivatives including those expressing wild-type (WT) or GGG-mutant IGFBP3 were established and exposed to either hypoxia (0.5% O2) or normoxia (21% O2); or treated with or without hydrogen peroxide (H2O2) (Sigma-Aldrich, St. Louis, MO) or recombinant human IGFBP-3 (rhIGFBP3) as described previously [17,18,21,24].
Lentivirus or retrovirus-mediated gene transfer and transient transfection for RNA interference (RNAi)
Stable transduction of cells with tetracycline-inducible (Tet-On) or constitutively-expressed short hairpin RNA (shRNA) directed against IGFBP3 (V2LHS_111628 and V2LHS_225584; BP3-1 and BP3-2) or a non-silencing scramble control sequence (RHS4743) (Open Biosystems, Huntsville, AL) were done as described previously [21]. Small interfering RNA (siRNA) sequences directed against IGFBP3 (Stealth siRNA, HSS105267 and HSS179812; BP3-1 and BP3-2) or a non-silencing control sequence (Silencer Select Negative Control #1) (10 nM) (Invitrogen, Carlsbad, CA) were transfected transiently with LipofectamineTM RNAi Max reagent (Invitrogen), following the manufacturer’s instructions. Sixteen hours after the transfection, cells were exposed to hypoxia or normoxia, or treated with or without H2O2 for 48 h.
Flow cytometry and Fluorescence Activated Cell Sorting (FACS)
FACSCalibur (BD Biosciences, San Jose, CA) and FlowJo (Tree Star, Ashland, OR) were used for flow cytometry. Cells were suspended in Hank’s balanced salt solution (Invitrogen) containing 1% BSA (Sigma-Aldrich) and stained with PE/Cy7-anti-CD24 at 1:10 (BioLegend, San Diego, CA) and APC-anti-CD44 at 1:20 (BD Biosciences) on ice for 30 min. To purify CD44L and CD44H cells, xenograft tumors were minced into 1 mm3 pieces and incubated in Dulbecco’s modified Eagle medium (DMEM) (Invitrogen) containing 1 mg/ml collagenase I (C9263-1G, Sigma-Aldrich) at 37°C for 90 min. Following centrifugation, residual tissue pieces were digested in 0.05% trypsin-EDTA (Invitrogen) at 37°C for 10 min and then with 1 U/ml Dispase (#354235, BD Biosciences) and 100 μg/ml DNase I (#10104159001, Roche) at 37°C for 10 min. Dissociated tumor cells were filtrated with a 40 μm cell strainer (BD Biosciences), washed and incubated with the above anti-CD24 (1:10) and anti-CD44 (1:20) antibodies along with FITC-anti-mouse histocompatibility complex class I antigen H-2K[d] (1:10) (BD Biosciences, San Jose, CA) and propidium iodide (1:250) (Sigma-Aldrich). FACS Vantage SE (BD Biosciences) was used to isolate CD44high-CD24low/- cells (CD44H) and CD44low/--CD24low/- cells (CD44L) from TE11 and T-TeRas xenograft tumors. Flow cytometry was repeated for each genotype and condition at least three times.
DCF assay
ROS were determined by flow cytometry with 2’, 7’-dichlorodihydrofluorescein diacetate (DCF) dye (Invitrogen) according to the manufacturer’s instruction. In brief, cells were incubated with 10 μM DCF at 37°C for 30 min and further cultured for up to 3 hours prior to flow cytometry.
Real-time reverse-transcription polymerase chain reaction (RT-PCR)
Real-time RT-PCR for IGFBP3 was done as described previously using β-actin as an internal control [17,21].
Western blotting
Western blotting was done as described previously [17,21] using the following primary antibodies at the indicated titers; anti-IGFBP3 (DSL-R00536; Diagnostic Systems Laboratories, Webster, TX) (1:1000), anti-HIF-1α (610958; BD Biosciences) (1:1000) and anti-β-actin (AC74; Sigma-Aldrich, St. Louis, MO) (1:5000).
Statistical analyses
Data from experiments are presented as mean ± standard error (n=3) or mean ± standard deviation (n=8) and were analyzed by two-tailed Student’s t test. P<0.05 was considered significant. Overall survival curves were plotted by the Kaplan-Meier method and subjected to the log rank test. Variables with a value of P<0.05 by univariate analysis were used for subsequent multivariate analyses based on Cox’s proportional hazards model.
Results
IGFBP3 contributes to invasive tumor growth and poor prognosis in ESCC patients
To determine whether IGFBP3 expression may influence disease progression in ESCC patients, we first performed immunohistochemistry (IHC) for IGFBP3 using tissue microarrays representing paired primary tumors and adjacent normal mucosa from patients who underwent esophagectomy and lymph node dissection without prior neoadjuvant chemotherapy or radiation therapy. IGFBP3 was overexpressed in tumors in 32 out of 91 informative cases (35%) compared to adjacent normal mucosa (Figure 1A). IGFBP3 expression was weak in normal or non-cancerous esophageal mucosa; and no tumor samples showed apparent downregulation of IGFBP3 in ESCC cells compared to adjacent normal mucosa (data not shown) as shown previously [24]. ESCC cells tended to express IGFBP3 focally. Six cases (6.6%) with the most intense IGFBP3 expression showed a significantly reduced postsurgical survival rate (Figure 1B) associated significantly with tumor depth, lymph node metastasis, lymphatic invasion and venous invasion (Table 1), implying IGFBP3 overexpression in tumor progression.
Figure 1.

High IGFBP3 expression is associated with poor prognosis in ESCC patients. A: Immunohistochemistry reveals focal upregulation of IGFBP3 in primary ESCC tissues on tissue microarrays. Two representative cases scored as strongly positive IGFBP3 expression in tumors are shown. Note an intense cytoplasmic staining of IGFBP3 (arrows). IGFBP3 was expressed in spindle-shaped tumor cells reminiscent of EMT in case 1. Scale bars, 50 μm. B: A high level of IGFBP3 expression predicts a poor 5 year survival rate. Overall survival curves were plotted according to the Kaplan-Meier method, and p value was calculated using log rank test.
Table 1.
Univariate and multivariate analysis of factors affecting overall survival rate
| Variables | Number of patients | 5-year survival rate (%) | Univariate analysis | Multivariate analysis | |
|---|---|---|---|---|---|
|
| |||||
| p-value | Relative risk (confident interval) | p-value | |||
| Sex | |||||
| male | 82 | 29.3 | 0.037 | ||
| female | 9 | 37.8 | |||
| Tumor depth | |||||
| pT1, 2 | 38 | 56.3 | <0.001 | 1.591 | 0.002 |
| pT3, 4 | 53 | 15.9 | (1.188-2.180) | ||
| Lymph node metastasis | |||||
| negative | 44 | 47.0 | 0.003 | 1.200 | 0.211 |
| positive | 47 | 18.8 | (0.902-1.610) | ||
| Lymphatic invasion | |||||
| negative | 31 | 42.1 | 0.032 | ||
| positive | 60 | 28.3 | |||
| Venous invasion | |||||
| negative | 63 | 39.6 | 0.005 | 1.241 | 0.137 |
| positive | 28 | 17.9 | (0.932-1.638) | ||
| IGFBP3 expression | |||||
| Low (score 0-1) | 85 | 35.0 | 0.045 | 1.268 | 0.31 |
| High (score 2) | 6 | 0.0 | (0.780-1.880) | ||
IGFBP3 may regulate tumor growth influencing CD44H cell content in xenograft tumors
We hypothesized that IGFBP3 may influence tumor growth and CD44 expression in ESCC cells in the hypoxic tumor microenvironment. We used TE11 and T-TeRas cells since both form xenograft tumors in immunodeficient mice and that hypoxia has been documented in resulting tumors [17,25]. TE11 forms well-differentiated SCC [17] while T-TeRas gives rise to poorly-differentiated tumors [21]. TE11 cells show relatively high basal IGFBP3 expression [16]. T-TeRas cells show low IGFBP3 expression in culture and a moderate induction in xenograft tumors [18].
To perform RNAi experiments in vivo, TE11 cells were stably transduced by lentivirus expressing tetracycline inducible shRNA directed against either IGFBP3 or a non-silencing scramble control sequence. When shRNA was induced in culture with 0.5 μg/ml doxycycline (DOX), IGFBP3 expression was sharply suppressed under normoxic and hypoxic conditions as documented by Western blotting (Figure 2A). IGFBP3 knockdown did not affect cell proliferation or viability under normoxic conditions in culture (data not shown). To delineate the functional consequences of IGFBP3 knockdown in established tumors, we first allowed tumors to grow to approximately 80 mm3 without expressing shRNA where tumor growth was found to be comparable regardless the genotype (Figure 2B). Interestingly, tumor growth was significantly suppressed once DOX was administrated to induce shRNA directed against IGFBP3, but not a non-silencing control sequence (Figure 2B). Histology revealed reduced tumor cell viability upon IGFBP3 knockdown while viable tumor cells showed less proliferative and more differentiated characteristics with a lack of invasion, compared to control tumors (Figure 2C and data not shown). Interestingly, IGFBP3 knockdown decreased intratumoral CD44H cell content by 30% (Figure 2D). A non-specific inhibitory effect upon IGFBP3 expression was observed in culture and attributable to either DOX or scramble shRNA (Figure 2A); however, scramble shRNA did not affect tumor growth with or without DOX treatment (Figure 2B). Moreover, DOX alone did not affect tumor growth by cells without the shRNA transgene constructs (data not shown). These findings suggest that IGFBP3 may be required in regulation of ESCC cell proliferation, differentiation and invasion as well as CD44H cells in established tumors with hypoxic tumor microenvironment.
Figure 2.

IGFBP3 knockdown inhibits TE11 xenograft tumor growth and reduces intratumoral CD44H cells. (A) TE11 cells with Tet-On shRNA directed against IGFBP3 or a non-silencing sequence (scramble) were exposed to either hypoxia or normoxia in the presence or absence of 0.5 μg/ml DOX for 48 h for documentation of the RNAi effect upon IGFBP3 expression. Western blotting was done to evaluate indicated molecules with β-actin as a loading control. In (B-D), TE11 derivatives with indicated genotypes were xenografted. Mice were administered drinking water with or without DOX starting at day 28 for 3 weeks to be sacrificed at day 49. Tumor growth (B), histology (hematoxylin and eosin staining) (C) and intratumoral CD44H cells (D) were evaluated. Note that in (C), the tumor without IGFBP3 knockdown [top panels, DOX (-)] shows an invasive growth pattern as indicated by tumor cell nests extending into the adjacent muscle layers (arrows, top left panel) and poorly differentiated atypical cells exhibiting mitotic figures (top right panel). By contrast, the tumor with IGFBP3 knockdown [bottom panels, DOX (+)] is well-circumscribed as demarcated by arrow heads (bottom left panel) and contains more differentiated cells with fewer mitotic figures (bottom right panel). In (D), enzymatically dissociated TE11 cells were isolated from xenograft tumors for FACS analysis. *, P<0.05 vs. DOX (-); n=8 in (B) and n=3 in (D). Scale bar, 100 μm in (C).
We next tested how IGFBP3 overexpression may affect CD44H cell content in T-TeRas tumors. As we have described previously [18,21], T-TeRas tumors with GGG-mutant IGFBP3 grew faster than those with a control empty vector (Bla) only (Figure 3A). Moreover, T-TeRas cells expressing WT-IGFBP3 failed to form tumors undergoing apoptosis as a consequence of IGF neutralization by overexpressed IGFBP3 (Figure 3A; and data not shown). The intratumoral CD44H cells were significantly increased in GGG-mutant IGFBP3 expressing tumors compared to the Bla-control tumors (Figure 3B). These results suggest that IGFBP3 may have an IGF-independent function to facilitate tumor growth with a concurrent increase in CD44H cells.
Figure 3.
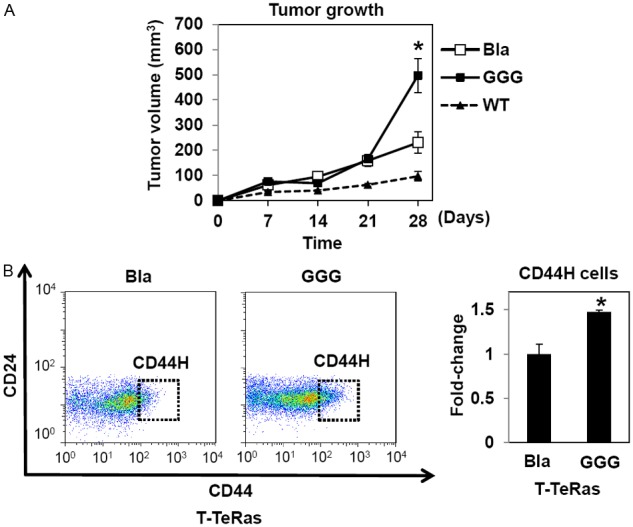
Ectopically expressed WT- and GGG-mutant IGFBP3 induces CD44H cells in T-TeRas in culture and xenograft tumors. T-TeRas derivatives with indicated genotypes [18,21] were xenografted. Cells were xenografted to evaluate tumor growth (A) and intratumoral CD44H cells (B). The relative CD44H cell level (gated subpopulation within the representative dot plots) was shown in the histogram where the number of CD44H cells from T-TeRas-Bla was set as 1. Note that FACS analysis in (B) for T-TeRas tumors with WT-IGFBP3 were precluded due to growth failure [18]. Bla, empty vector control; WT, wild-type IGFBP3; GGG, GGG-mutant IGFBP3. *, P<0.05 vs. Bla and WT; n=8 in (A) and n=3 in (B).
IGFBP3 may facilitate hypoxic induction of CD44H ESCC cells
We addressed further how IGFBP3 may influence CD44H cells under hypoxic conditions in cultured cells. Flow cytometry revealed upregulation of CD44 in both TE11 and T-TeRas cells upon hypoxic exposure (Figure 4A). In TE11, IGFBP3 knockdown suppressed CD44H cells by 50% under hypoxic, but not normoxic conditions (Figure 4B). A similar response was observed when siRNA was used instead of shRNA without having non-specific IGFBP3 inhibition by a non-silencing control sequence (data not shown). In T-TeRas cells, the hypoxic induction of CD44H cells was augmented by either WT- or GGG-mutant IGFBP3 (Figure 4C). Unlike xenograft tumors, ectopically expressed WT-IGFBP3 did not affect T-TeRas cell proliferation or viability in culture as cell culture medium contained a physiologically exceeding level of insulin as a substitute of IGFs that stimulates IGF1 receptor while negating the IGF-neutralizing effect of IGFBP3 [18,26]. These data suggest that IGFBP3 may have a role in the hypoxic induction of CD44H cells via an IGF-independent mechanism.
Figure 4.

IGFBP3 mediates hypoxic induction of CD44H cells in culture. TE11, T-Te-Ras and indicated derivatives were exposed to hypoxia (0.5% O2) or normoxia for 48 h and analyzed by FACS for CD44H cells. In (A), a segmented line indicates the CD44H cell fraction. In (B), TE11 cells with Tet-On shRNA directed against IGFBP3 or a non-silencing sequence (scramble) were treated in the presence or absence of 0.5 μg/ml DOX. In (C), Bla, empty vector control; WT, wild-type IGFBP3; GGG, GGG-mutant IGFBP3. *, P<0.05 vs. Normoxia (n=3) in (A); P<0.05 vs. Scramble and DOX (+) (n=3) in (B); and P<0.05 vs. Bla and Normoxia (n=3) in (C). #, P<0.05 vs. Bla and Hypoxia (n=3) in (C).
IGFBP3 may suppress ROS to facilitate ESCC cell adaptation and survival in the hypoxic conditions
Since both IGFBP3 and CD44 are HIF-1α target genes, we initially suspected that IGFBP3 may be upregulated in CD44H cells purified from xenograft tumors. Despite our prediction, IGFBP3 mRNA was found to be sharply suppressed in CD44H cells in both TE11 and T-TeRas cells (Figure 5), suggesting the possibility that CD44H cells per se may not necessarily depend upon IGFBP3 expression. We hypothesized that IGFBP3 may facilitate de novo generation of CD44H cells by negatively regulating ROS in ESCC cells in the hypoxic tumor microenvironment. When IGFBP3 was suppressed by siRNA, ROS production was greatly enhanced upon exposure to either hypoxia or H2O2 in TE11 cells with increased cell death (Figure 6A and 6B). In T-TeRas cells, ectopically expressed WT- or GGG-mutant IGFBP3 inhibited ROS generated in response to hypoxia or H2O2 (Figure 6C), preventing hypoxia or H2O2 from inducing cell death (Figure 6D), albeit to a modest extent.
Figure 5.
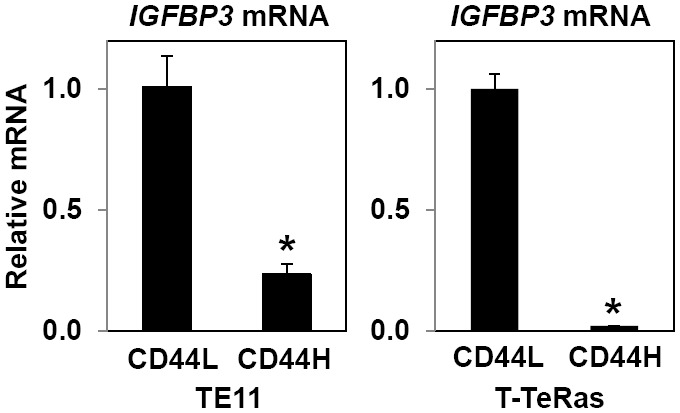
IGFBP3 mRNA is downregulated in CD44H cells from xenograft tumors. IGFBP3 mRNA was determined by qRT-PCR in CD44L and CD44H cells purified from TE11 and T-TeRas xenograft tumors. *, P<0.05 vs. CD44L; ns, not significant vs. CD44L (n=3).
Figure 6.

IGFBP3 influences ROS and cell viability in response to hypoxia or H2O2. TE11 cells were transiently transfected with siRNA directed against IGFBP3 (BP3-1 and BP3-2) or a non-silencing sequence (scr, scramble) in (A) and (B). T-TeRas derivatives with indicated genotypes were used in (C) and (D). Cells were exposed for 48 h to either hypoxia or normoxia; or H2O2 at 400 μM. In (A) and (C), ROS levels were determined by DCF assays. In (B) and (D), cell viability was determined. Bla, empty vector control; WT, wild-type IGFBP3; GGG, GGG-mutant IGFBP3. *, P<0.05 vs. scramble and either normoxia or H2O2 (-); #, P<0.05 vs. scramble and either hypoxia or H2O2 (+) (n=3) in (A) and (B). *, P<0.05 vs. Bla and either normoxia or H2O2 (-); #, P<0.05 vs. Bla and either hypoxia or H2O2 (+) (n=3) in (C) and (D).
To further validate the IGFBP3-mediated ROS regulation, we employed additional ESCC cell lines TE12 and T.T which display intermediate and modest IGFBP3 expression levels, respectively [16]. In TE12 cells, IGFBP3 knockdown increased basal ROS and increased ROS in response to hypoxia, albeit to a lesser extent than was observed in TE11 cells (data not shown). Interestingly, T.T cells were more prone to H2O2-induced cell death than TE11 or TE12 cells (data not shown). In T.T cells, ectopically expressed IGFBP3 or recombinant human (rh) IGFBP3 added into cell culture medium reduced ROS production upon hypoxic exposure (Figure 7). In aggregate, these data suggest that IGFBP3 may suppress ROS under hypoxic conditions via a unique IGF-independent antioxidant function in ESCC cells.
Figure 7.
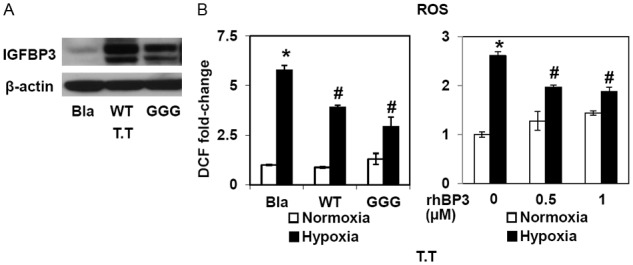
IGFBP3 regulates ROS in T.T cells. T.T cells were stably transduced with WT-IGFBP3, GGG-IGFBP3 or empty vector (Bla) to create derivatives with indicated genotypes. A: Cells were subjected to Western blotting with β-actin as a loading control. B: T.T derivatives with indicated genotypes or parental T.T cells incubated with or without rhIGFBP3 (rhBP3) were exposed to either hypoxia or normoxia for 48 h. ROS levels were determined by DCF assays.*, P<0.05 vs. either Bla or 0 nM rhBP3 and normoxia; #, P<0.05 vs. either Bla or 0 nM rhBP3 and hypoxia (n=3).
Discussion
IGFBP3 may promote disease progression in patients with ESCC featuring a hypoxic tumor microenvironment
Using well-annotated tumor tissue microarrays and genetically engineered cell lines, we show for first time that IGFBP3 overexpression may contribute to ESCC disease progression (Figure 1), influencing intratumoral CD44H cell content in the hypoxic tumor microenvironment (Figures 2, 3 and 4). CD44H cells per se show low IGFBP3 expression (Figure 5); however, IGFBP3 facilitates ESCC cell adaptation to hypoxia and increase CD44H cells by suppressing ROS in an IGF-independent manner (Figures 6 and 7).
Given context-dependent dichotomous tumor promoting and suppressive functions of IGFBP3, the prognostic value of IGFBP3 expression remains unclear in many tumor types. Earlier studies link IGFBP3 overexpression to poor prognosis in breast cancer [27,28]. IGFBP3 positive test results by IHC predicted poor prognosis in HNSCC patients showing concurrent IGF-1 receptor expression [29]. IGFBP3 expression was also found to be downregulated in 32% of tongue SCC cases (n=34) and 48% of ESCC cases (n=110), contributing to poor prognosis [30,31]. Unlike these studies, none of our primary tumor samples showed IGFBP3 downregulation in ESCC cells compared to non-cancerous epithelial cells in the adjacent normal mucosa. This is in agreement with our own previous study documenting IGFBP3 overexpression at the mRNA and protein levels in a distinct ESCC patient cohort [24]. As a hypoxia target gene, IGFBP3 was often found in the primary ESCC tumor cells surrounding a necrotic lesion [17]. Discrepancies between our data and others may be accounted for in part by intratumoral heterogeneous and focal expression of IGFBP3 (Figure 1A). They may also attributable to different scoring methods used for IHC results. In addition, some of the commercially available IGFBP3 antibodies fail to detect full length IGFBP3 by Western blot analysis (Natsuizaka et al. unpublished data). Since moderate IGFBP3 upregulation had no prognostic impact in this study, a further study is warranted with an increased sample size.
IGFBP3 may influence intratumoral CD44H ESCC cell content
How does IGFBP3 facilitate tumor growth? TE11 cells with IGFBP3 knockdown formed less invasive tumors (Figure 2). By contrast, IGFBP3 overexpression in T-TeRas cells promoted invasive tumor growth (Figure 3) associated with increased epithelial-mesenchymal transition (EMT) [21]. Interestingly, IGFBP3 promotes vascular survival and regrowth in a rodent model of retinopathy where IGFBP3 has an IGF-independent role to recruit endothelial progenitor cells [32]. IGFBP3 also facilitates migration, differentiation, and capillary formation of adult bone marrow-derived endothelial precursor cells [33]. Thus, IGFBP3 may stimulate angiogenesis, promoting tumor growth in established tumors.
Although we did not directly address the role of IGFBP3 in CSCs, influence of IGFBP3 upon intratumoral CD44H cells (Figures 2 and 3) is a novel finding. As a secretory protein, IGFBP3 interacts with numerous extracellular matrix proteins including Fibronectin [34]. The transforming growth factor (TGF)-β and EGFR signaling pathways are also influenced by IGFBP3 [24,35-39]. Since CD44 interacts also with these extracellular matrix proteins and signaling pathways, IGFBP3 and CD44 may have a mutual functional interplay. IGFBP3 was occasionally found in spindle-shaped ESCC cells reminiscent of EMT (Figure 1A). As a key constituent of the tumor microenvironment, hypoxia regulates CSCs [40] and EMT [41]. EMT has been implicated in CD44H CSC generation [42]. TGF-β mediates hypoxic induction of EMT [43]. A subset of ESCC cells undergoes EMT in the invasive tumor front [44]. In human esophageal cells, EGFR facilitates enrichment of EMT-competent cells [45] where IGFBP3 promotes TGF-β-mediated EMT [21]. Cancer cells with low CD44 (CD44L) expression may spontaneously give rise to CD44H cells [46]. Therefore, IGFBP3 may promote an EMT-mediated conversion of non-CSC to CSC.
IGFBP3 may regulate ROS via multiple mechanisms
This study reveals for the time an IGF-independent antioxidant function of IGFBP3 (Figures 6 and 7). IGFBP3 is induced upon replicative or stress-induced senescence [47-49]. Thus, IGFBP3 may have broader roles for coping with stress-inducing stimuli. How may IGFBP3 suppress ROS? Superoxide dismutase 2 (SOD2) is upregulated in T-TeRas with ectopically expressed IGFBP3 although it is unclear whether IGFBP3 may directly regulate SOD2 (Natsuizaka et al. unpublished observations). Investigation is currently underway as to how IGFBP3 may influence the cellular redox regulatory mechanisms. Although our data imply an IGF-independent role of IGFBP3, suppression of IGF-mediated signaling has been implicated in oxidative stress resistance and longevity in multiple organisms [50]. Therefore, IGFBP3 may suppress ROS in both IGF-dependent and independent manners.
In conclusion, our study sheds light on the growth stimulatory role of IGFPB3 in cancer via an IGF-independent antioxidant activity that may influence CD44H cells including putative CSCs, gaining a novel mechanistic insight into the functional interplay between the tumor microenvironment and IGFBP3.
Acknowledgements
This study was supported in part by NIH Grants R01DK077005 (to HN), K26 RR032714 (HN), P01-CA-098101 (Mechanisms of Esophageal Carcinogenesis to MN, HK, SK, KAW, SN, NLR, DB, PAG, AJK, JAD and HN), F32-CA174176 (KAW), K08 DE022842 (to DB), P30-DK050306 (to HS, SC and KJN), Pennsylvania CURE Program Grant (HN), University of Pennsylvania University Research Foundation Award (HN), University of Pennsylvania, Abramson Cancer Center Pilot Project Grant (HN), American Gastroenterological Association Foundation Student Research Fellowship Award (to SC and KJN), and the NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases (P30-DK050306). We thank Dr. Charles H. Pletcher (Flow Cytometry & Cell Sorting Facility), Mr. Adam Bedenbaugh and Ms. Daniela Budo (Molecular Pathology & Imaging Core), Dr. Gary D. Wu and Dr. Sue Keilbaugh (Molecular Biology/Gene Expression Core) for assistance in data collection and analysis. We are appreciative of discussions with the lab of Dr. Anil K Rustgi.
Disclosure of conflict of interest
The authors disclose no potential conflicts of interest.
References
- 1.Toole BP, Slomiany MG. Hyaluronan, CD44 and Emmprin: partners in cancer cell chemoresistance. Drug Resist Updat. 2008;11:110–121. doi: 10.1016/j.drup.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Twarock S, Tammi MI, Savani RC, Fischer JW. Hyaluronan stabilizes focal adhesions, filopodia, and the proliferative phenotype in esophageal squamous carcinoma cells. J Biol Chem. 2010;285:23276–23284. doi: 10.1074/jbc.M109.093146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takayama N, Arima S, Haraoka S, Kotho T, Futami K, Iwashita A. Relationship between the expression of adhesion molecules in primary esophageal squamous cell carcinoma and metastatic lymph nodes. Anticancer Res. 2003;23:4435–4442. [PubMed] [Google Scholar]
- 4.Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer. 2011;11:254–267. doi: 10.1038/nrc3023. [DOI] [PubMed] [Google Scholar]
- 5.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao JS, Li WJ, Ge D, Zhang PJ, Li JJ, Lu CL, Ji XD, Guan DX, Gao H, Xu LY, Li EM, Soukiasian H, Koeffler HP, Wang XF, Xie D. Tumor Initiating Cells in Esophageal Squamous Cell Carcinomas Express High Levels of CD44. PLoS One. 2011;6:e21419. doi: 10.1371/journal.pone.0021419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 9.Nagano O, Okazaki S, Saya H. Redox regulation in stem-like cancer cells by CD44 variant isoforms. Oncogene. 2013;32:5191–8. doi: 10.1038/onc.2012.638. [DOI] [PubMed] [Google Scholar]
- 10.Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, Masuko T, Shimizu T, Ishikawa T, Kai K, Takahashi E, Imamura Y, Baba Y, Ohmura M, Suematsu M, Baba H, Saya H. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400. doi: 10.1016/j.ccr.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 11.Yoshikawa M, Tsuchihashi K, Ishimoto T, Yae T, Motohara T, Sugihara E, Onishi N, Masuko T, Yoshizawa K, Kawashiri S, Mukai M, Asoda S, Kawana H, Nakagawa T, Saya H, Nagano O. xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013;73:1855–1866. doi: 10.1158/0008-5472.CAN-12-3609-T. [DOI] [PubMed] [Google Scholar]
- 12.Krishnamachary B, Penet MF, Nimmagadda S, Mironchik Y, Raman V, Solaiyappan M, Semenza GL, Pomper MG, Bhujwalla ZM. Hypoxia regulates CD44 and its variant isoforms through HIF-1alpha in triple negative breast cancer. PLoS One. 2012;7:e44078. doi: 10.1371/journal.pone.0044078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jogie-Brahim S, Feldman D, Oh Y. Unraveling insulin-like growth factor binding protein-3 actions in human disease. Endocr Rev. 2009;30:417–437. doi: 10.1210/er.2008-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hintz RL, Bock S, Thorsson AV, Bovens J, Powell DR, Jakse G, Petrides PE. Expression of the insulin like growth factor-binding protein 3 (IGFBP-3) gene is increased in human renal carcinomas. J Urol. 1991;146:1160–1163. doi: 10.1016/s0022-5347(17)38031-x. [DOI] [PubMed] [Google Scholar]
- 15.Chang YS, Kong G, Sun S, Liu D, El-Naggar AK, Khuri FR, Hong WK, Lee HY. Clinical significance of insulin-like growth factor-binding protein-3 expression in stage I non-small cell lung cancer. Clin Cancer Res. 2002;8:3796–3802. [PubMed] [Google Scholar]
- 16.Takaoka M, Harada H, Andl CD, Oyama K, Naomoto Y, Dempsey KL, Klein-Szanto AJ, El-Deiry WS, Grimberg A, Nakagawa H. Epidermal growth factor receptor regulates aberrant expression of insulin-like growth factor-binding protein 3. Cancer Res. 2004;64:7711–7723. doi: 10.1158/0008-5472.CAN-04-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Natsuizaka M, Naganuma S, Kagawa S, Ohashi S, Ahmadi A, Subramanian H, Chang S, Nakagawa KJ, Ji X, Liebhaber SA, Klein-Szanto AJ, Nakagawa H. Hypoxia induces IGFBP3 in esophageal squamous cancer cells through HIF-1alpha-mediated mRNA transcription and continuous protein synthesis. FASEB J. 2012;26:2620–2630. doi: 10.1096/fj.11-198598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takaoka M, Kim SH, Okawa T, Michaylira CZ, Stairs DB, Johnstone CN, Andl CD, Rhoades B, Lee JJ, Klein-Szanto AJ, El-Deiry WS, Nakagawa H. IGFBP-3 regulates esophageal tumor growth through IGF-dependent and independent mechanisms. Cancer Biol Ther. 2007;6:534–540. doi: 10.4161/cbt.6.4.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SH, Nakagawa H, Navaraj A, Naomoto Y, Klein-Szanto AJ, Rustgi AK, El-Deiry WS. Tumorigenic conversion of primary human esophageal epithelial cells using oncogene combinations in the absence of exogenous Ras. Cancer Res. 2006;66:10415–10424. doi: 10.1158/0008-5472.CAN-06-2104. [DOI] [PubMed] [Google Scholar]
- 20.Takaoka M, Kim SH, Okawa T, Michaylira CZ, Stairs DB, Johnstone CN, Andl CD, Rhoades B, Lee JJ, Klein-Szanto AJ, El-Deiry WS, Nakagawa H. IGFBP-3 Regulates Esophageal Tumor Growth Through IGF-Dependent and Independent Mechanisms. Cancer Biol Ther. 2007;6:534–40. doi: 10.4161/cbt.6.4.3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Natsuizaka M, Ohashi S, Wong GS, Ahmadi A, Kalman RA, Budo D, Klein-Szanto AJ, Herlyn M, Diehl JA, Nakagawa H. Insulin-like growth factor-binding protein-3 promotes transforming growth factor-{beta}1-mediated epithelial-to-mesenchymal transition and motility in transformed human esophageal cells. Carcinogenesis. 2010;31:1344–1353. doi: 10.1093/carcin/bgq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu K, Jiang M, Lu Y, Chen H, Sun J, Wu S, Ku WY, Nakagawa H, Kita Y, Natsugoe S, Peters JH, Rustgi A, Onaitis MW, Kiernan A, Chen X, Que J. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell. 2013;12:304–315. doi: 10.1016/j.stem.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JJ, Natsuizaka M, Ohashi S, Wong GS, Takaoka M, Michaylira CZ, Budo D, Tobias JW, Kanai M, Shirakawa Y, Naomoto Y, Klein-Szanto AJ, Haase VH, Nakagawa H. Hypoxia activates the cyclooxygenase-2-prostaglandin E synthase axis. Carcinogenesis. 2010;31:427–434. doi: 10.1093/carcin/bgp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takaoka M, Harada H, Andl CD, Oyama K, Naomoto Y, Dempsey KL, Klein-Szanto AJ, El-Deiry WS, Grimberg A, Nakagawa H. Epidermal growth factor receptor regulates aberrant expression of insulin-like growth factor-binding protein 3. Cancer Res. 2004;64:7711–7723. doi: 10.1158/0008-5472.CAN-04-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim K, Nakagawa H, Fei P, Rustgi AK, El-Deiry WS. Targeting Bcl-xL in esophageal squamous cancer to sensitize to chemotherapy plus TRAIL-induced apoptosis while normal epithelial cells are protected by blockade of caspase 9. Cell Death Differ. 2004;11:583–587. doi: 10.1038/sj.cdd.4401388. [DOI] [PubMed] [Google Scholar]
- 26.Takaoka M, Smith CE, Mashiba MK, Okawa T, Andl CD, El-Deiry WS, Nakagawa H. EGF-mediated regulation of IGFBP-3 determines esophageal epithelial cellular response to IGF-I. Am J Physiol Gastrointest Liver Physiol. 2006;290:G404–416. doi: 10.1152/ajpgi.00344.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rocha RL, Hilsenbeck SG, Jackson JG, Lee AV, Figueroa JA, Yee D. Correlation of insulin-like growth factor-binding protein-3 messenger RNA with protein expression in primary breast cancer tissues: detection of higher levels in tumors with poor prognostic features. J Natl Cancer Inst. 1996;88:601–606. doi: 10.1093/jnci/88.9.601. [DOI] [PubMed] [Google Scholar]
- 28.Rocha RL, Hilsenbeck SG, Jackson JG, VanDenBerg CL, Weng C, Lee AV, Yee D. Insulin-like growth factor binding protein-3 and insulin receptor substrate-1 in breast cancer: correlation with clinical parameters and disease-free survival. Clin Cancer Res. 1997;3:103–109. [PubMed] [Google Scholar]
- 29.Sun JM, Jun HJ, Ko YH, Park YH, Ahn YC, Son YI, Baek JH, Park K, Ahn MJ. Insulin-like growth factor binding protein-3, in association with IGF-1 receptor, can predict prognosis in squamous cell carcinoma of the head and neck. Oral Oncol. 2011;47:714–719. doi: 10.1016/j.oraloncology.2011.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Papadimitrakopoulou VA, Brown EN, Liu DD, El-Naggar AK, Jack Lee J, Hong WK, Lee HY. The prognostic role of loss of insulin-like growth factor-binding protein-3 expression in head and neck carcinogenesis. Cancer Lett. 2006;239:136–143. doi: 10.1016/j.canlet.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 31.Zhao L, He LR, Zhang R, Cai MY, Liao YJ, Qian D, Xi M, Zeng YX, Xie D, Liu MZ. Low expression of IGFBP-3 predicts poor prognosis in patients with esophageal squamous cell carcinoma. Med Oncol. 2012;29:2669–2676. doi: 10.1007/s12032-011-0133-4. [DOI] [PubMed] [Google Scholar]
- 32.Lofqvist C, Chen J, Connor KM, Smith AC, Aderman CM, Liu N, Pintar JE, Ludwig T, Hellstrom A, Smith LE. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc Natl Acad Sci U S A. 2007;104:10589–94. doi: 10.1073/pnas.0702031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang KH, Chan-Ling T, McFarland EL, Afzal A, Pan H, Baxter LC, Shaw LC, Caballero S, Sengupta N, Li Calzi S, Sullivan SM, Grant MB. IGF binding protein-3 regulates hematopoietic stem cell and endothelial precursor cell function during vascular development. Proc Natl Acad Sci U S A. 2007;104:10595–10600. doi: 10.1073/pnas.0702072104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gui Y, Murphy LJ. Insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) binds to fibronectin (FN): demonstration of IGF-I/IGFBP-3/fn ternary complexes in human plasma. J Clin Endocrinol Metab. 2001;86:2104–2110. doi: 10.1210/jcem.86.5.7472. [DOI] [PubMed] [Google Scholar]
- 35.Martin JL, Baxter RC. Transforming growth factor-beta stimulates production of insulin-like growth factor-binding protein-3 by human skin fibroblasts. Endocrinology. 1991;128:1425–1433. doi: 10.1210/endo-128-3-1425. [DOI] [PubMed] [Google Scholar]
- 36.Oh Y, Muller HL, Ng L, Rosenfeld RG. Transforming growth factor-beta-induced cell growth inhibition in human breast cancer cells is mediated through insulin-like growth factor-binding protein-3 action. J Biol Chem. 1995;270:13589–13592. doi: 10.1074/jbc.270.23.13589. [DOI] [PubMed] [Google Scholar]
- 37.Fanayan S, Firth SM, Baxter RC. Signaling through the Smad pathway by insulin-like growth factor-binding protein-3 in breast cancer cells. Relationship to transforming growth factor-beta 1 signaling. J Biol Chem. 2002;277:7255–61. doi: 10.1074/jbc.M108038200. [DOI] [PubMed] [Google Scholar]
- 38.Butt AJ, Martin JL, Dickson KA, McDougall F, Firth SM, Baxter RC. Insulin-like growth factor binding protein-3 expression is associated with growth stimulation of T47D human breast cancer cells: the role of altered epidermal growth factor signaling. J Clin Endocrinol Metab. 2004;89:1950–1956. doi: 10.1210/jc.2003-030914. [DOI] [PubMed] [Google Scholar]
- 39.Takaoka M, Smith CE, Mashiba MK, Okawa T, Andl CD, El-Deiry WS, Nakagawa H. EGF-mediated regulation of IGFBP-3 determines esophageal epithelial cellular response to IGF-I. Am J Physiol Gastrointest Liver Physiol. 2006;290:G404–16. doi: 10.1152/ajpgi.00344.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102:789–95. doi: 10.1038/sj.bjc.6605551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bao B, Azmi AS, Ali S, Ahmad A, Li Y, Banerjee S, Kong D, Sarkar FH. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim Biophys Acta. 2012;1826:272–96. doi: 10.1016/j.bbcan.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1120–30. doi: 10.1152/ajplung.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ohashi S, Natsuizaka M, Naganuma S, Kagawa S, Kimura S, Itoh H, Kalman RA, Nakagawa M, Darling DS, Basu D, Gimotty PA, Klein-Szanto AJ, Diehl JA, Herlyn M, Nakagawa H. A NOTCH3-mediated squamous cell differentiation program limits expansion of EMT-competent cells that express the ZEB transcription factors. Cancer Res. 2011;71:6836–47. doi: 10.1158/0008-5472.CAN-11-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohashi S, Natsuizaka M, Wong GS, Michaylira CZ, Grugan KD, Stairs DB, Kalabis J, Vega ME, Kalman RA, Nakagawa M, Klein-Szanto AJ, Herlyn M, Diehl JA, Rustgi AK, Nakagawa H. Epidermal growth factor receptor and mutant p53 expand an esophageal cellular subpopulation capable of epithelial-to-mesenchymal transition through ZEB transcription factors. Cancer Res. 2010;70:4174–4184. doi: 10.1158/0008-5472.CAN-09-4614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, Arendt LM, Kuperwasser C, Bierie B, Weinberg RA. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950–5. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldstein S, Moerman EJ, Baxter RC. Accumulation of insulin-like growth factor binding protein-3 in conditioned medium of human fibroblasts increases with chronologic age of donor and senescence in vitro. J Cell Physiol. 1993;156:294–302. doi: 10.1002/jcp.1041560211. [DOI] [PubMed] [Google Scholar]
- 48.Debacq-Chainiaux F, Pascal T, Boilan E, Bastin C, Bauwens E, Toussaint O. Screening of senescence-associated genes with specific DNA array reveals the role of IGFBP-3 in premature senescence of human diploid fibroblasts. Free Radic Biol Med. 2008;44:1817–1832. doi: 10.1016/j.freeradbiomed.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 49.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63:2705–2715. [PubMed] [Google Scholar]
- 50.Katic M, Kahn CR. The role of insulin and IGF-1 signaling in longevity. Cell Mol Life Sci. 2005;62:320–43. doi: 10.1007/s00018-004-4297-y. [DOI] [PubMed] [Google Scholar]