

Abstract
The retinoblastoma gene Rb is a prototype tumor suppressor, which encodes a protein that is inactivated in a broad range of human cancers through different mechanisms. Rb functions to regulate cell proliferation, differentiation, as well as cell death. Therefore, even though Rb inactivation promotes cancer development, this may also open up certain vulnerabilities of cancers that can potentially be targeted with drug intervention. Based on the assumption that cancers that have mutation, deletion, or rearrangement in the Rb locus represent strong loss of Rb function while cancers with WT Rb on average retain some Rb function, we searched Genomics of Drug Sensitivity in Cancer database to identify cancer drugs that are particularly effective to cancers with Rb genomic alterations. Three mitotic inhibitors were identified from this analysis. We further tested the effects of two mitotic inhibitors, Taxol and STLC, on prostate and breast cancer cells. We demonstrate that the Rb status affects cancer cell sensitivity to these mitotic drugs and that the sensitizing effects of Rb are mediated in part by its regulation of the cell cycle checkpoint protein Mad2. Since the mitotic inhibitors identified in our analysis inhibit mitosis through distinct targets, it is possible that the Rb functional status may serve as a general biomarker for cancer sensitivity to mitotic inhibitors. Because the Rb pathway is inactivated in a large number of human cancers, identification of agents that are particularly effective or ineffective based on the Rb status in cancers can potentially be used generally to matching patients with appropriate treatments to achieve better therapeutic outcome.
Keywords: Drug sensitivity, Rb, retinoblastoma tumor suppressor, Mad2, cell death, mitotic inhibitor, Taxol, S-Trityl-L-cysteine, STLC
Introduction
Cancer is the second leading cause of death in the US and the economic cost to the society is staggering. It is increasingly clear that cancer is not a single disease but are a collection of lots of diseases with different underlying causes. Therefore a major challenge in cancer treatment is to identify suitable biomarkers that can be used to stratify patients and inform therapeutic decisions.
The retinoblastoma tumor suppressor (Rb) is a prototype tumor suppressor inactivated in a broad range of human cancers [1,2]. Rb can be inactivated in cancers by mutation, deletion, or loss of expression of Rb, by overexpression of D type Cyclins, mutation of p16 family of Cdk inhibitors, or by expression of viral oncoproteins. The Retinoblastoma protein pRb functions by regulating the expression of its target genes. The expression of diverse cellular targets including genes involved in cell cycle regulation, DNA replication, DNA repair, cell cycle checkpoint, apoptosis as well as differentiation have been found to be regulated by Rb. Consistent with its diverse functions, Rb inactivation can either lead to increased cell proliferation or increased cell death [3]. Therefore while Rb inactivation often promotes cancer development, this may also open up certain vulnerabilities of such cancers to certain drug intervention. Furthermore, since Rb inactivation in cancers is quite common, approaches that can promote the effect of cell death in conjunction with Rb inactivation through synthetic lethality can potentially be used to treat a large fraction of cancers [4]. Interestingly, a recent study showed that the Rb pathway inactivation is associated with improved response to neoadjuvant chemotherapy in breast cancers [5], suggesting that Rb status can potentially be used to stratify patients and inform treatment.
The rapid advances in cancer genome projects have resulted in a large number of drug sensitivity data that are available from the public domain. We have taken advantage of the available data from the Genomics of Drug Sensitivity in Cancer database [6] to identify cancer drugs that are particularly effective to cancers with Rb genomic alterations. Since the Rb pathway is inactivated in a large number of human cancers, identification of agents that are particularly effective or ineffective based on the Rb status in cancers can potentially be used generally to matching patients with appropriate treatments to achieve better therapeutic outcome. In this study, we analyzed public genomic drug sensitivity database and identified several drugs that target mitosis to be particularly effective toward cancers with Rb mutation.
Materials and methods
Statistical analysis of drug-Rb interaction
We obtained “Multivariate ANOVA for all compounds” dataset from the Genomics of Drug Sensitivity in Cancer database at www.cancerrxgene.org. This dataset contains output of a multivariate analysis of variance (MANOVA) that correlates drug responses with genetic alterations in cancer cell lines. Among output that contains drug interaction with Rb gene, We ordered and chose the 15 drugs that have calculated Benjamini-Hochberg adjusted p-values significant at 20% False Discovery Rate. With estimated “Anova_effect” from MANOVA analysis, we calculated IC50_effect as 10^ (Anova_effect divided by LN (10)), which indicates the folds change of IC50 values in respect to loss-of-Rb function. And we generated an indicator called “Drug Sensitivity Change with Rb Mutation” as the inverse of IC50_effect. In addition, as statistically significant but small difference in drug sensitivity may not be useful in clinical practice, we set up an artificial threshold of 20% difference, and focused only on drugs that are 20% more or less sensitive in Rb mutant cells.
Cell culture
PC3, Du145 and MDA-MB-231 cells were obtained from the American Type Culture Collection. Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine and 50 IU penicillin/streptomycin in a humidified atmosphere with 5% CO2 at 37°C. Paclitaxel and STLC treatment were carried out in medium with 1% FBS.
Lentiviral preparation and infection
The pLKO.1 lentiviral RNAi expression system was used to generate shRb, shMad2 and shGFP constructs, which shGFP was used to prepare control virus. The sequences of shRb used in this study were described in [7], the sequences of shMad2 and shGFP controls are: 5’-CCGGTAATACGGACTCACCTTGCTTGCTCGAGCAAGCAAGGTGAGTCCGTATTTTTTTT-3’ (shMad2); 5’-CCGGTACGTCTATATCATGGCCGACAACTAGTTGTCGGCCATGATATAGACGTTTTTTG-3’ (shGFP). Viral packaging was done according to the previously described protocol [7]. Briefly, RNAi expression plasmids, pCMV-dR8.91 and pCMV-VSV-G were cotransfected into 293 T cells using the calcium phosphate method and incubated for 6 h. The transfection medium was then replaced with fresh medium. Virus was collected 48 h after transfection and concentrated using 20% sucrose buffer at 20,000 g for 4 h. The virus was re-dissolved in the complete growth medium and stocked at -80°C. Cells were infected with the viruses in the presence of Polybrene (10 μg/ml) for 48 h, selected with puropmycin (2 μg/ml) for 48 h, and then treated as desired.
Cell proliferation analysis
Cell proliferation was assessed by MTT assay. Briefly, cells (3 × 103 cells/well) were seeded into 96-well plates. After treatments described in result section, culture medium was replaced with fresh medium containing 0.5 mg/ml MTT and incubated for 2 h at 37°C. After removing the medium, 100 μl of DMSO was added to each well to solubilize the formazan present in viable cells. The plates were analyzed by measuring the optical density at 540 nm. Experiments were repeated three times.
Luciferase reporter assay
For the luciferase activity assay, 104 cells were seeded into 48-well plates for 24 h and were co-transfected with 0.5 μg of the E2F luciferase reporter construct and 10 ng of the phRL-TK plasmid using Lipofectamine 2000 Transfection Reagent (Invitrogen). At 48 h post-transfection, the luciferase activity was measured with Dual-Luciferase Reporter Assay System (Promega) on a Monolight luminometer (Becton Dickinson).
Clonogenic assay
Cells (1 × 103) were seeded in triplicate in 6-well plates one day before treatment. Cells were then treated with STLC or DMSO as indicated in result section for 10 days. Survived colonies were fixed and stained with the fixing/staining solution (0.5% w/v crystal violet and 10% formalin in PBS) for 30 min. After removing the staining solution, washing with water to remove residual crystal violet, and air drying, the remaining crystal violet was extracted with 500 μl of 10% acetic acid. Clonogenic growth was determined by measuring the optical density at 630 nm. The value for treated cells was normalized to those corresponding control cells. Data was representative of three independent experiments.
FACS analysis of cell cycle and cell death
For the cell cycle assay, 105 cells were seeded in 6-well plate. After treatment for 48 h, cells were harvested and fixed gently by adding 75% ethanol and placing at -20°C for 4-16 h. The cells were washed twice with PBS, resuspended in 300 μl PBS containing 100 μg/ml propidium iodide and 0.1 mg/ml RNase, incubated for 30 min at room temperature in dark, and analyzed using a FACScanto flow cytometer (BD Biosciences).
For the cell death assay, 105 cells were seeded into 6-well plates. After treatment for 48 h described in the result section, the adherent and detached cells were collected and washed twice with Annexin V binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), and resuspended at 1 × 106 cells/mL in 100 μl of the binding buffer. 5 μl of Annexin V-FITC and 5 μl of propidium iodide (stock concentration, 100 μg/ml) were added to the cell suspension. The cells were incubated for 15 min at room temperature in dark, and then 400 μl of binding buffer was added to each sample. Quantification of cell death was performed with a FACScanto flow cytometer after staining, and PI- and Annexin V-positive cells were considered as dead cell.
FlowJo 7.1.0 software (Tree Star, Ashland, OR) were used for data analysis and at least 10,000 cells were counted for each measurement. All data was representative of 3 independent assays.
Western blotting
After treatments, cells were washed with PBS and lysed in homogenization buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride) for 30 min on ice with brief sonication. Equal amounts of protein were loaded. A Li-Cor Odyssey image reader was used for western detection. The following antibodies were used: sc 69879 against β-actin (Santa Cruz Biotechnology) and Rb 4.1 against Rb (DSHB). The goat anti-mouse immunoglobulin G (IgG) and goat anti-rabbit IgG secondary antibodies were obtained from Li-Cor.
RNA isolation and RT-PCR
Total RNA was extracted with the RNeasy Mini Kit according to the manufacturer’s instructions (Qiagen, Valencia, CA, USA). cDNA was synthesized using M-MLV reverse transcriptase from Promega (Madison, WI, USA). Real time RT-PCR was determined using the Thermo Scientific™ DyNAmo™ SYBR™ Green qPCR Kit on an Opticon 2 Real-time PCR detector (Bio-Rad). GAPDH mRNA level was used as normalization control. Primer pairs used for RT-PCR are: human Mad2, 5’-GGAAGCGCGTGCTTTTGTTT-3’ (Forward), and 5’-TCTTTCAGTTGTTCCACCACA-3’ (Reverse); human GAPDH, 5’-CTCTGACTTCAACAGCGACAC-3’ (Forward), and 5’-CATACCAGGAAATGAGCTTGACAA-3’ (Reverse).
Results
Analysis of genomics of drug sensitivity in cancer database to identify drugs that show increased or decreased efficacy toward cancer cells with genomic alterations in the Rb locus
To identify drugs that show increased efficacy toward cancers with Rb mutations (genomic alterations), we took advantages of the Genomics of Drug Sensitivity in Cancer database [6], a publicly available IC50 dataset of 147 anticancer agents on over 1000 tumor cell lines (Yang, Soares et al. 2013). We explored results from multivariate analysis of variance that correlate drug responses to 71 common cancer-related genomic alterations, including point mutations, amplifications and deletions of common cancer genes, cancer gene rearrangements and microsatellite instability, and applied the following two criteria: 1) the drug response to Rb genomic alterations gives a Benjamini-Hochberg adjusted p-value that is statistically significant at 20% False Discovery Rate; 2) the estimated difference in drug sensitivity between WT and mutant Rb cancer cells is greater than 20%. Upon analysis, seven anticancer agents, VX-702, BI-D1870, GW843682X, Metformin, PLX4720, Paclitaxel and S-Trityl-L-cysteine, are more effective toward Rb mutant cells; while three, PD-0332991, PD-0325901 and Bexarotene, are less effective (Figure 1).
Figure 1.
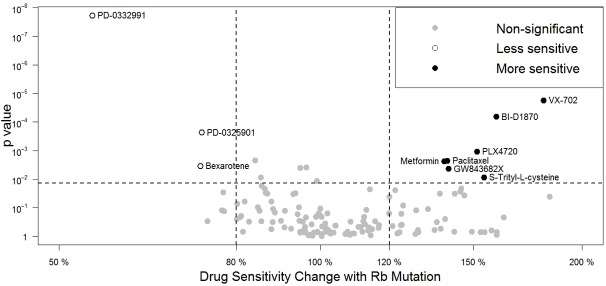
Influence of Rb mutation on drug sensitivity. An Rb-drug interaction plot shows the relative change of drug sensitivity with the existence Rb mutation (x-axis, in percentage), and significance (y-axis, in log-scaled p-value). Each circle represents a single Rb-drug interaction calculated from the MANOVA analysis. In this plot, drugs on the left are more sensitive in cells with WT Rb, while those on the right are more sensitive in Rb mutant cells. The Rb-drug interaction is statistically significant when it is above the 20% horizontal False Discovery Rate (FDR) line. The interaction is practically significant when Rb mutant leads to more than 20% change in drug sensitivity from that of cells with WT Rb. In particular, empty circles indicate drugs that are less effective to Rb mutant, while circles filled with black indicate drugs that are more effective to Rb mutant. All other Rb-drug interaction that does not satisfy both statistical and practical significance were filled with grey and without labeled names.
PD-0332991 is a highly selective inhibitor of the Cyclin D/Cdk4 and Cdk6 inhibitor that functions by blocking Rb phosphorylation [8,9]. Therefore Rb mutant cells are expected to be resistant to PD-0332991. Among all the drugs that show selectivity based on Rb genomic alteration status, PD-0332991 exhibits the most significant interaction: a more than 45% decrease in drug sensitivity with a p-value of less than 2 × 10-8 (Figure 1). The identification of PD-0332991, which is a targeted drug that requires functional Rb, validates the accuracy of our analysis and suggests that our analysis can identify useful drug sensitivity information.
Among the drugs that display Rb mutant sensitivity include three drugs that inhibit mitosis: S-Trityl-L-cysteine (STLC) is a selective inhibitor of mitotic kinesin Eg5; GW843682X is a selective inhibitor of polo-like kinase 1 and 3; while paclitaxel (Taxol) is an inhibitor of mitotic spindle formation [10-13]. Compared to those with WT Rb, cancer cells with Rb mutations are on average 40% time more sensitive to paclitaxel or GW843682X and 54% times more sensitive to STLC. These observations suggest that Rb inactivation sensitizes cancer cells to mitotic inhibitors. It should be noted that our analysis are comparing the drug sensitivity of cancer cells that have mutant Rb with those that have WT Rb. Since cancer cells often have inactivated Rb through different mechanisms, which inactivate pRb function to different extent and/or duration, it is likely that cancer cells will have different extent of pRb inactivation. Therefore comparing the drug sensitivity of the Rb mutant cells with cancer cells that have partially inactivated WT Rb impacted the statistical significance observed in our analysis.
Rb modulates cancer cell sensitivity to paclitaxel and STLC treatment
To directly test if Rb status affects the anticancer effects of mitotic inhibitors, we first examined the growth inhibitory effects of Taxol and STLC in a pair of prostatic cancer cell lines, PC3 and DU145, as these two drugs are often used to treat or have promising effect on prostate cancers [14,15]. Both PC3 and DU145 are derived from type-I ADI prostate cancer [16], with a major difference that DU145 has mutated Rb while PC3 retains significant level of Rb function [17,18]. After treating both cancer cells with increasing concentrations, both STLC and Taxol displayed stronger growth inhibition on DU145 cells compared with that on PC3 (Figure 2A, 2B).
Figure 2.
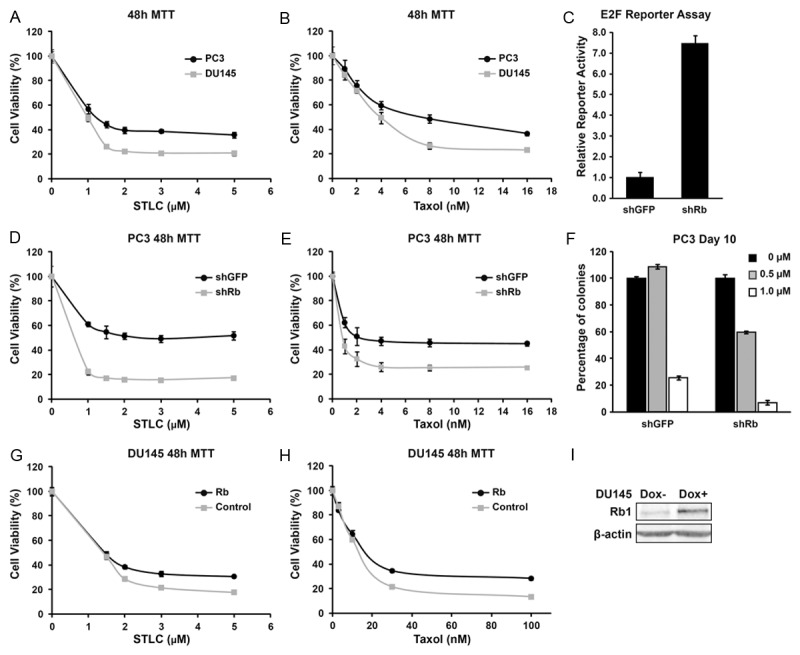
Loss of Rb increases cell sensitivity to Paclitaxel and STLC treatment. (A, B, D, E, G, H) The effect of increasing or decreasing Rb levels on cell growth inhibition by mitotic inhibitors. MTT assays were used to measure the amount of live cells after 48 hours drug treatment. (C) Knockdown efficiency of shRb construct in PC3 cells. (F) Formation of PC3 colonies 10 days after STLC treatment. (I) Western blot of Rb and β-actin expression in DU145 cells upon Dox induction. Error bars represent the standard error of the mean (SEM) in (A-H).
To further examine directly whether Rb affects the sensitivity of cancer cells to these two mitotic inhibitors, we tested the effect of Rb knockdown using shRb lentiviral constructs [7]. The efficacy of Rb knockdown in PC3 cells was confirmed by more than seven folds of increased activity in E2F1-Luciferase Report Assay compared with shGFP, a control shRNA construct against GFP gene (Figure 2C). These results also confirm that PC3 cells retain significant level of Rb function. As expected, knockdown of Rb significantly sensitized PC3 cells to the growth inhibitory effect of either STLC (Figure 2D) or Taxol (Figure 2E) in all the concentrations we have tested. Furthermore, clonogenic assays showed that STLC, cause a much faster and more significant decrease in the surviving PC3shRb colonies compared to that of PC3shGFP (Figure 2F). Taken together, these results demonstrate that inactivation of Rb can induce increased sensitivity of Rb WT PC3 cancer cells to mitotic inhibitors.
We further tested the effect of restoring WT Rb expression in DU145 cells by constructing an inducible Rb expression lentiviral system in which WT Rb protein can be induced by addition of doxycycline in the medium [19] (Figure 2I). Compared to DU145Rb- cells in which Rb was not expressed, WT Rb expression in DU145 cells (DU145Rb+) significantly increased cell viability when treated with STLC or Taxol (Figure 2G, 2H). Taken together, these results demonstrate that growth inhibitory effects of Taxol and STLC are influenced by the pRb function.
Loss of Rb increased mitotic inhibitor paclitaxel and STLC induced cell death
As inactivation of Rb can increase cell proliferation as well as increase cell death, the observation of decreased growth rate in paclitaxel and STLC caused by loss of Rb function prompted us to determine the effect of Rb inactivation on cell cycle progression and cell death in the presence or absence of these mitotic inhibitors. As expected, knockdown of Rb promotes G1/S progression and moderately decreased fractions of cells in G1 in both the PC3 and the MDA-MB-231 cells (Figure 3A, 3C, 3E and 3G), and addition of mitotic inhibitor induces G2/M arrest in both cell type with or without the Rb knockdown (Figure 3A-H). However, in the presence of STLC, significantly increased sub G1 population of cells were observed in both the PC3 and the MDA-MB-231 Rb knockdown cells, indicating Rb knockdown increased level of cell death (Figure 3A-H).
Figure 3.

Loss of Rb increases cell death to Paclitaxel and STLC treatment. A-H: Cell cycle profiles of PC3 and MDA-MB-231 cells with or without Rb knockdown at 48 hours after drug treatment as indicated. The fractions of sub-G1 and aneuploidy (>4n) cells were also labeled. I-L: Effects of Rb knockdown on PC3 and MDA-MB-231 cell death at 48 hours after drug treatment. Error bars represent the standard error of the mean.
Annexin V and propidium iodide staining were used to further characterize the effect of Rb knockdown on STLC-induced cell death in PC3. Although shRb only has a small effect on cell death without drug treatment, a significantly elevated cell death were observed in PC3shRb treated with either 1.5 or 3 μM STLC (Figure 3I). A similar effect of Rb knockdown was also observed in STLC treated MDA-MB-231, a frequently studied human metastatic breast carcinoma cell line [20] (Figure 3J). Similarly, knockdown of Rb also increased paclitaxel-induced cell death assays in both PC3 and MDA-MB-231 cells (Figure 3K-L). Therefore knockdown of Rb in PC3 or MDA-MB-231 cells significantly increased the sensitivity of these cancer cells to mitotic inhibitor induced cell death.
Increased Mad2 expression partially mediates the effect of Rb on mitotic inhibitor-induced cell death
Mitotic inhibitors such as Taxol and STLC activate spindle assembly checkpoint and blocks cells in mitosis. Prolonged arrest in mitosis has been linked with increased likelihood of death in mitosis by apoptosis [21]. Mad2 is a central regulator of spindle assembly checkpoint and is a target gene regulated by E2F and Rb [22]. Inactivation of the Rb family proteins leads to upregulated Mad2 expression, which is sufficient to promote aneuploidy and tumorigenesis in mice [22-24]. Interestingly, down-regulation of a mitotic spindle check point gene, Mad2, has been shown to induce paclitaxel resistance through cellular senescence in both breast cancer and ovarian cancer cells [25,26]. These observations raised possibility that increased Mad2 expression in the absence of Rb prolongs mitotic arrest in the presence of mitotic inhibitor and increased the level of cell death.
To test this hypothesis, we first determined if Rb regulates Mad2 expression in MDA-MB-231 and PC3 cells by comparing the level of Mad2 with or without Rb knockdown. Real-time PCR showed that the expression level of Mad2 was increased by four folds in MDA-MB-231shRb and 1.5 folds in PC3shRb compared to their corresponding shGFP control cells (Figure 4A, 4B). To further determine if increased Mad2 expression contribute to the effect of Rb on mitotic inhibitor-induced cell death, we generated lentiviral shMad2 construct [27]. shMad2 caused more than 70% reduction in Mad2 expression (Figure 4C). By a two-step transduction procedure in which MDA-MB-231 or PC3 were first transduced with shRb or shGFP control, followed by a second transduction of shMad2 or shGFP control, we tested the effect of knockdown Mad2 on shRb-induced sensitivity to mitotic inhibitors. Consistent with the previous finding, knockdown of Rb expression induced elevated cell death in both PC3 and MDA-MD-231 cancer cells response to STLC (Figure 4D, 4E) or Taxol (Figure 4F, 4G). Interestingly, knockdown of Mad2 significantly reduced shRb-induced sensitivity to STLC induced cell death in MDA-MD-231 cells (Figure 4D, compare shRb+shGFP and shRb+shMad2, p<0.01) as well as in PC3 cells (Figure 4E, compare shRb+shGFP and shRb+shMad2, p<0.01). Similarly, knockdown of Mad2 also significantly decreased shRb induced sensitivity to paclitaxel-induced cell death (Figure 4F, 4G). The observation that knockdown of Mad2 partially rescues shRb induced sensitivity to mitotic inhibitors in conjunction with the finding that Mad2 expression is up-regulated in Rb knockdown cells suggest that Rb modulates sensitivity to mitotic inhibitors, at least in part, through increased Mad2 expression.
Figure 4.
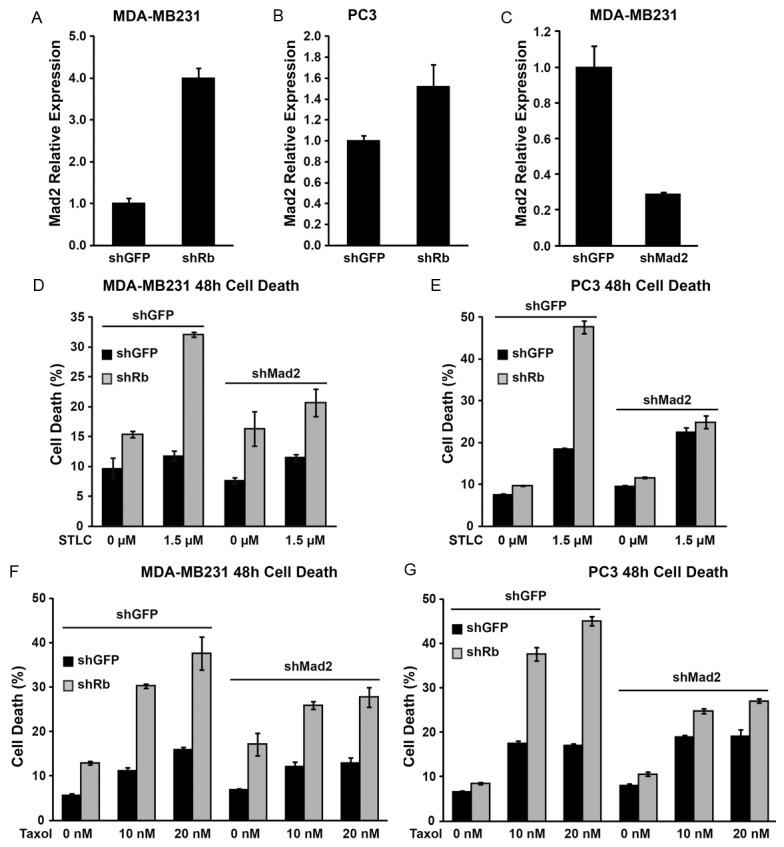
Rb modulates mitotic inhibitor-induced cell death partially through increased Mad2 expression. (A, B)Mad2 expression upon Rb knockdown in PC3 and MDA-MB-231 cells measured by real time PCR. (C) Knockdown efficiency of shMad2 construct in MDA-MB-231 cells measured by real time PCR. (D-G) Effects of Mad2 knockdown on cell death in PC3 (D, F) or MDA-MB-231 (E, G) cells at 48 hours after treatment with STLC (D, E) or Paclitaxel (F, G). In each figure above, error bars represent the standard error of the mean.
Discussion
Our analysis of the Genomics of Drug Sensitivity in Cancer database have revealed that the Rb mutant cancer cells are associated with increased sensitivity toward three mitotic inhibitors: STLC, GW843682X, and paclitaxel. By knocking down Rb in PC3 cells and by expressing WT Rb in DU145 cells, we confirmed that Rb functional status affects the sensitivity of cancer cells to mitotic inhibitors. We further showed that the effects of Rb are mediated in part by the upregulation of mitotic regulator Mad2. Since these three mitotic inhibitors function through distinct mechanisms: STLC inhibits mitotic kinesin Eg5, GW843682X inhibits polo-like kinase 1 and 3, while Taxol binds to polymerized tubulin and inhibits mitotic spindle formation, we speculate that it is a general relationship between Rb inactivation and the process of mitotic progression rather than a specific mechanism during mitosis that increased efficacy of mitotic inhibitors. Thus Rb functional status may serve as a general biomarker in drug sensitivity for mitotic inhibitors (Figure 5).
Figure 5.
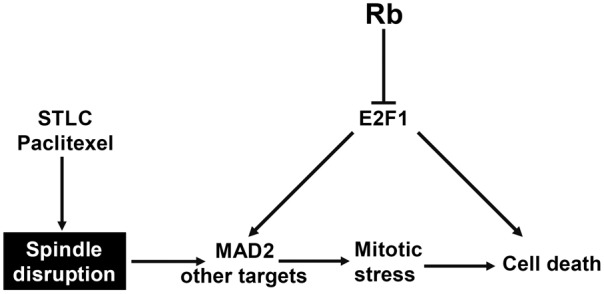
A working model for loss of Rb promote cell death upon mitotic inhibitor treatment. Mitotic inhibitor such as STLC or Paclitaxel disrupts spindle formation or dynamics during mitosis, which leads to mitotic stress and finally cell death. Loss of Rb function induces increased E2F1 activity, which in turn induces not only cell death, but also elevated expression of Mad2. This increased Mad2 expression prolongs mitotic arrest in the presence of mitotic inhibitor and further increased the level of cell death.
It should be noted that although the Rb pathway is often inactivated in cancers, multiple mechanisms contribute to Rb inactivation in cancers. While Rb inactivation by mutation, deletion, or loss of expression will remove Rb function all the time, it is possible that functional Rb inactivation by mutation of p16 or overexpression of D type cyclins may not completely inactivate Rb at all the times. Therefore it will be interesting to compare how inactivation of Rb by different mechanisms such as overexpression of Cyclin D, loss of p16, or expression of viral oncogenes that inactivate Rb, affect the sensitivity to mitotic inhibitors.
Since the Rb pathway is often inactivated in cancers, understanding how mutation or inactivation of Rb affects drug sensitivity is of great interest because this will allow most cancers to be stratified and matched with appropriated treatment. Furthermore, such studies can potentially lead to the identification of drugs that are particularly effective to cancers that lost Rb. Although the loss of functional Rb pathway results in certain advantages for a cancer cell in terms of unregulated entry into S-phase and proliferation, other apoptotic programs and vulnerabilities can be unmasked by the deregulated Rb pathway [7,18], which can potentially be exploited in developing novel cancer therapies.
In addition to the mitotic inhibitors, our statistical analysis also showed that several inhibitors of the MAP kinase pathway show some differential effects depending on the Rb status. VX-702 is a p38 MAPK inhibitor [28,29], PLX4720 is a selective BRAF inhibitor [30], and BI-D1870 is a p90 RSK inhibitor [31]. It is possible that the Rb mutant cancer cells are more sensitive to inhibitors of the MAPK pathway. However, Rb cells also showed less sensitivity to the MEK inhibitor PD-0325901 [32-34]. It is possible that additional targets of these kinase inhibitors affected the sensitivity or resistance of the Rb mutant cancer cells. Further studies on the targets of VX-702 and PD-0325901 will be needed to elucidate the mechanisms that mediate the observed sensitivity or resistance.
Acknowledgements
We would like to thank Drs. R. Hiipakka and members of the Du lab for many discussions and supply of reagents. The Rb monoclonal antibody developed by Dr. Sage was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology. This work was supported in part by the following grants: NIH CA149275, NIH/NCCAM AT004418, NIH GM074197.
Disclosure of conflict of interest
The authors declare no conflict of interest.
References
- 1.Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714–24. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gordon GM, Du W. Conserved RB functions in development and tumor suppression. Protein Cell. 2011;2:864–878. doi: 10.1007/s13238-011-1117-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon GM, Du W. Targeting Rb inactivation in cancers by synthetic lethality. Am J Cancer Res. 2011;1:773–786. [PMC free article] [PubMed] [Google Scholar]
- 5.Witkiewicz AK, Ertel A, McFalls J, Valsecchi ME, Schwartz G, Knudsen ES. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res. 2012;18:5110–5122. doi: 10.1158/1078-0432.CCR-12-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, Ramaswamy S, Futreal PA, Haber DA, Stratton MR, Benes C, McDermott U, Garnett MJ. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li B, Gordon GM, Du CH, Xu J, Du W. Specific killing of Rb mutant cancer cells by inactivating TSC2. Cancer Cell. 2010;17:469–480. doi: 10.1016/j.ccr.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, Los G, Slamon DJ. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK, Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–1438. [PubMed] [Google Scholar]
- 10.Spaniol K, Boos J, Lanvers-Kaminsky C. An in-vitro evaluation of the polo-like kinase inhibitor GW843682X against paediatric malignancies. Anticancer Drugs. 2011;22:531–542. doi: 10.1097/CAD.0b013e3283454526. [DOI] [PubMed] [Google Scholar]
- 11.Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, Noro N, Furuta M, Emmitte KA, Gilmer TM, Mook RA Jr, Cheung M. In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol Cancer Ther. 2007;6:450–459. doi: 10.1158/1535-7163.MCT-06-0543. [DOI] [PubMed] [Google Scholar]
- 12.Long BH, Fairchild CR. Paclitaxel inhibits progression of mitotic cells to G1 phase by interference with spindle formation without affecting other microtubule functions during anaphase and telephase. Cancer Res. 1994;54:4355–4361. [PubMed] [Google Scholar]
- 13.Skoufias DA, DeBonis S, Saoudi Y, Lebeau L, Crevel I, Cross R, Wade RH, Hackney D, Kozielski F. S-trityl-L-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J Biol Chem. 2006;281:17559–17569. doi: 10.1074/jbc.M511735200. [DOI] [PubMed] [Google Scholar]
- 14.Xing ND, Ding ST, Saito R, Nishizawa K, Kobayashi T, Inoue T, Oishi S, Fujii N, Lv JJ, Ogawa O, Nishiyama H. A potent chemotherapeutic strategy in prostate cancer: S-(methoxytrityl)-L-cysteine, a novel Eg5 inhibitor. Asian J Androl. 2011;13:236–241. doi: 10.1038/aja.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gan L, Chen S, Wang Y, Watahiki A, Bohrer L, Sun Z, Huang H. Inhibition of the androgen receptor as a novel mechanism of taxol chemotherapy in prostate cancer. Cancer Res. 2009;69:8386–8394. doi: 10.1158/0008-5472.CAN-09-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Litvinov IV, Antony L, Dalrymple SL, Becker R, Cheng L, Isaacs JT. PC3, but not DU145, human prostate cancer cells retain the coregulators required for tumor suppressor ability of androgen receptor. Prostate. 2006;66:1329–1338. doi: 10.1002/pros.20483. [DOI] [PubMed] [Google Scholar]
- 17.Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O’Meara S, Santarius T, Avis T, Barthorpe S, Brackenbury L, Buck G, Butler A, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Hunter C, Jenkinson A, Jones D, Kosmidou V, Lugg R, Menzies A, Mironenko T, Parker A, Perry J, Raine K, Richardson D, Shepherd R, Small A, Smith R, Solomon H, Stephens P, Teague J, Tofts C, Varian J, Webb T, West S, Widaa S, Yates A, Reinhold W, Weinstein JN, Stratton MR, Futreal PA, Wooster R. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther. 2006;5:2606–2612. doi: 10.1158/1535-7163.MCT-06-0433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danos AM, Liao Y, Li X, Du W. Functional inactivation of Rb sensitizes cancer cells to TSC2 inactivation induced cell death. Cancer Lett. 2013;328:36–43. doi: 10.1016/j.canlet.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin JY, Zhang L, Clift KL, Hulur I, Xiang AP, Ren BZ, Lahn BT. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One. 2010;5:e10611. doi: 10.1371/journal.pone.0010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker-Nasir E, Codington JF, Jahnke MR, Fuller TC, Jeanloz RW. Isolation and partial characterization of surface components of cell line MDA-MB-231 derived from a human metastatic breast carcinoma. J Natl Cancer Inst. 1982;69:371–80. [PubMed] [Google Scholar]
- 21.Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell. 2004;7:637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, Lowe SW, Cordon-Cardo C. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- 23.Sotillo R, Hernando E, Díaz-Rodríguez E, Teruya-Feldstein J, Cordón-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuyler SC, Wu YF, Kuan VJ. The Mad1-Mad2 balancing act - a damaged spindle checkpoint in chromosome instability and cancer. J Cell Sci. 2012;125:4197–206. doi: 10.1242/jcs.107037. [DOI] [PubMed] [Google Scholar]
- 25.Hao X, Zhou Z, Ye S, Zhou T, Lu Y, Ma D, Wang S. Effect of Mad2 on paclitaxel-induced cell death in ovarian cancer cells. J Huazhong Univ Sci Technolog Med Sci. 2010;30:620–625. doi: 10.1007/s11596-010-0553-y. [DOI] [PubMed] [Google Scholar]
- 26.Prencipe M, Fitzpatrick P, Gorman S, Tosetto M, Klinger R, Furlong F, Harrison M, O’Connor D, Roninson IB, O’Sullivan J, McCann A. Cellular senescence induced by aberrant MAD2 levels impacts on paclitaxel responsiveness in vitro. Br J Cancer. 2009;101:1900–8. doi: 10.1038/sj.bjc.6605419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci U S A. 2004;101:4459–64. doi: 10.1073/pnas.0306069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Damjanov N, Kauffman RS, Spencer-Green GT. Efficacy, pharmacodynamics, and safety of VX-702, a novel p38 MAPK inhibitor, in rheumatoid arthritis: results of two randomized, double-blind, placebo-controlled clinical studies. Arthritis Rheum. 2009;60:1232–41. doi: 10.1002/art.24485. [DOI] [PubMed] [Google Scholar]
- 29.Ding C. Drug evaluation: VX-702, a MAP kinase inhibitor for rheumatoid arthritis and acute coronary syndrome. Curr Opin Investig Drugs. 2006;7:1020–5. [PubMed] [Google Scholar]
- 30.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KS, Fong D, Zhu YL, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim SH, Schlessinger J, Zhang KY, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, Bollag G. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P, Alessi DR. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem J. 2007;401:29–38. doi: 10.1042/BJ20061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haura EB, Ricart AD, Larson TG, Stella PJ, Bazhenova L, Miller VA, Cohen RB, Eisenberg PD, Selaru P, Wilner KD, Gadgeel SM. A phase II study of PD-0325901, an oral MEK inhibitor, in previously treated patients with advanced non-small cell lung cancer. Clin Cancer Res. 2010;16:2450–7. doi: 10.1158/1078-0432.CCR-09-1920. [DOI] [PubMed] [Google Scholar]
- 33.LoRusso PM, Krishnamurthi SS, Rinehart JJ, Nabell LM, Malburg L, Chapman PB, DePrimo SE, Bentivegna S, Wilner KD, Tan W, Ricart AD. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16:1924–37. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- 34.Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR, Przybranowski SA, Sebolt-Leopold J, Van Becelaere K, Doherty AM, Kennedy RM, Marston D, Howard WA Jr, Smith Y, Warmus JS, Tecle H. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett. 2008;18:6501–6504. doi: 10.1016/j.bmcl.2008.10.054. [DOI] [PubMed] [Google Scholar]