

Abstract
Induced pluripotent stem (iPS) cells may be a powerful tool in regenerative medicine, but their potential tumorigenicity is a significant challenge for the clinical use of iPS cells. Previously, we succeeded in converting miPS cells into cancer stem cells (CSCs) under the conditions of tumor microenvironment. Both stem cells and tumor cells are profoundly influenced by bi-directional communication with their respective microenvironment, which dictates cell fate determination and behavior. The microenvironment derived from iPS cells has not been well studied. In this paper, we have investigated the effects of secreted factors from Nanog-mouse iPS (miPS) cells on mouse Lewis lung cancer (LLC) cells that are found in the conditioned media. The results demonstrated that miPS cells secrete factors that can convert the epithelia phenotype of LLC cells to a mesenchymal phenotype, and that can promote tumorigenisity, migration and invasion. Furthermore, LLC cells that have been exposed to miPS conditioned medium became resistant to apoptosis. These various biological effects suggest that the miPS microenvironment contain factors that can promote an epithelial-mesenchymal transition (EMT) through an active Snail-MMP axis or by suppressing differentiation in LLC cells.
Keywords: Mouse induced pluripotent stem cell, stem cell microenvironment, epithelial-mesenchymal transition, lung Lewis cancer cell
Introduction
Induced pluripotent stem (iPS) cells can be induced from fully differentiated non-pluripotent cells, and possess pluripotency similar to that of embryonic stem (ES) cells. In 2006, Takahashi and Yamanaka first achieved this landmark breakthrough by reprograming mouse embryonic fibroblasts (MEFs) into this new type of pluripotent stem cell via the ectopic expression of only four transcription factors, namely Oct4, Sox2, Klf4 and c-Myc [1]. Since the discovery of iPS cells, the field has attracted a great amount of scientific and public attention because of the undefined mechanism by which the developmental potential of these cells can be reverted and the potential for clinical applications using patient specific iPS derived cells [2-4]. Since the generation ofiPS cells might be a powerful tool in regenerative medicine, the potential tumorigenicity of iPS cells hints caution and a more defined assessment of microenvironmental factors that might contribute to this shift is needed. Previously, we succeeded in converting miPS cells into cancer stem cells (CSCs) under the conditions of the tumor microenvironment [5]. Although the mechanism of tumor development from treated miPS cells is not clear yet, secreted factors in the tumor microenvironment appear to be involved in converting miPS cells to CSCs. More recently, Fujimori et al utilized mouse embryonic stem cells as a model and demonstrated that environmental aberrancy during differentiation leads to the emergence of pluripotent cells showing cancerous characteristics [6].
In fact, both stem cells and tumor cells are profoundly influenced by bi-directional communication with their respective microenvironment, which dictates cell fate determination and behavior [7-9]. The microenvironment of human embryonic stem cells (hESCs) can suppress the tumorigenic phenotype of human metastatic melanoma and breast carcinoma cells. This effect is exclusive to hESCs and not to other stem cell types derived from amniotic fluid, cord blood, or adult bone marrow [9-11]. However, other research has demonstrated that factors derived from the hESC-derived cellular microenvironment provide a supportive setting for human tumor progression and metastasis, and mediated angiogenesis and vasculogenesis [12]. As a critical component of the tumor microenvironment, stem cell biologists and oncologists are interested in bone marrow derived mesenchymal stem cells (MSCs). Karnoub et al found that MSCs promoted breast cancer metastasis [13]. Similar observations were subsequently reported in breast cancer and other tumors [14-16]. In contrast, Sasser et al reported that only the growth of estrogen receptor-alpha (ERα) positive breast cancer cells were enhanced by MSCs paracrine factors, while ERα negative breast cancer cells were not [17]. Qiao et al also demonstrated MSCs inhibited the proliferation, colony-forming ability and oncogene expression in the malignant phenotypes of human liver cancer cell lines both in vitro and in vivo [18].
To date, iPS cells have been increasingly and widely researched in the field of regenerative medicine. But the microenvironment derived from iPS cells has not been well characterized. In this paper, we investigated the effects of conditioned media that are derived from Nanog-miPS cells on mouse Lewis lung cancer (LLC) cells. Here we demonstrate that the conditioned media of Nanog-miPS cells is able to convert LLC cells to a more aggressive phenotype in vivo and in vitro, which may be due to the ability of the Nanog-miPS cell microenvironment to initiate epithelial-mesenchymal transition (EMT) by activating a Snail-MMP axis or by suppressing differentiation in LLC cells.
Material and methods
Cell culture
Mouse Lewis lung cancer (LLC) cells (ATCC, USA) and mouse embryonic fibroblast (MEF) cells (Reprocell, Japan) were maintained in DMEM containing 10% fetal bovine serum (FBS). Mouse induced pluripotent stem cells (miPS; cell name: iPS-MEF-Ng-20D-17; Lot No. 012) were from Riken Cell Bank (Japan) and were maintained in medium (DMEM containing 15% FBS, 0.1 mM NEAA, 2 mM L-Glutamine, 0.1 mM 2-mercaptoethanol, 1000 U/ml LIF, 50 U/ml penicillin and 50 U/ml streptomycin) on feeder layers of mitomycin-C-treated MEF cells.
For preparing conditioned medium (CM) from miPS and MEF, medium was collected from confluent dishes and filtered using 0.45 µm filter (Millpore, Ireland). Then 3 ml CM were added into 3.5 cm dishes overnight to confirm that there were no surviving cells in the CM. Half of the medium of LLC cells was changed every day for 4 weeks and mixed with miPS CM or MEF CM, respectively. LLC cells maintained in DMEM containing 10% FBS were used as control. LLC cells were passaged every 3 days and cell morphology was photographed using a Olympus IX81 microscope equipped with a light fluorescence device (Olympus, Japan).
Animal experiments
Nude mice (Balb/c Slc-nu/nu, female, 6~8 weeks) were purchased from Charlesriver, Japan. For transplantation studies, 5 × 105 of LLC cells, LLC-miPS cells or LLC-MEF cells were suspended in 100 µl DMEM containing 10% FBS and injected subcutaneously into nude mice (n=5). Tumor sizes were measured every 3 days. After 3 weeks, tumors were excised and fixed in 10% neutral formalin buffer solution (Wako, Japan).
For lung metastases studies, 1 × 105 of LLC cells, LLC-miPS cells or LLC-MEF cells were suspended in 100 µl DMEM containing 10% FBS and injected into nude mouse tail vein (n=3). After 4 weeks, lungs were fixed for 24 hours and then processed using a routine wax-embedding procedure for histologic examination. Fifteen serial sections which were twenty micrometers thick were cut and stained with hematoxylin and eosin (HE).
Ethics statement
The plan of animal experiments was reviewed and approved by the ethics committee for animal experiments of Okayama University under the IDs OKU-2008211, OKU-2009144, OKU-2010179 and OKU-2011-305.
RNA extraction and quantitative real-time PCR (qRT-PCR)
Total RNA from cells was isolated by using RNeasy Mini Kit (QIAGEN, Germany) and TRIzol (Invitrogen, USA), respectively. One µg of total RNA was then reverse transcribed using SuperScript® II Reverse Transcriptase kit (Invitrogen, USA). Primer sequences were snail1 (forward primer, 5’-AGC TGG CCA GGC TCT CGG-3’, reverse primer, 5’-TAG CTG GGT CAG CGA GGG-3’), Slug (forward primer, 5’-ATG CCA TCG AAG CTG AGA AG-3’, reverse primer, 5’-CTC AGT GTG CCA CAC AGC AG-3’), MMP2 (forward primer, 5’-CAA GTT CCC CGG CGA TGT C-3’, reverse primer, 5’-TTC TGG TCA AGG TCA CCT GTC-3’), MMP3 (forward primer, 5’-ACA TGG AGA CTT TGT CCC TTT TG-3’, reverse primer, 5’-TTG GCT GAG TGG TAG AGT CCC-3’), MMP9 (forward primer, 5’-CTG GAC AGC CAG ACA CTA AAG-3’, reverse primer, 5’-CTC GCG GCA AGT CTT CAG AG-3’) and GAPDH (forward primer, 5’-CCC TTC ATT GAC CTC AAC TAC-3’, reverse primer, 5’-CCA CCT TCT TGA TGT CAT CAT-3’). qRT-PCR was performed with LightCycler 480 SYBR Green I Master (Roche, Germany) according to the manufacturer’s instructions. Signals were detected with a Light Cycler 480 II (Roche, Germany). The quantity of target gene mRNA was normalized to reference gene GAPDH mRNA.
In vitro cell migration and invasion assays
Migration of the cells was determined using a BD FalconTM HTS-24-multiwell Insert System (BD Biosciences, USA) with a transwell insert of 8-µm pore size. Cells (LLC, LLC-miPS, LLC-MEF) were cultured to approximately 80% confluence and serum starved for 12 hours. A volume of 100 µL containing 7.5 × 103, 1.5 × 104, 3 × 104 cells was plated onto each insert and 600 µL medium containing 10% FBS was added to the wells. After 24 hours of culture, cells migrating to the lower chamber were quantified by counting 3 randomly selected microscope fields at 100 × magnifications.All samples were assayed in triplicate.
As for invasion assays,the BD BioCoatTM 24-Multiwell Invasion System (BD Biosciences, USA) pre-coated with BD MatrigelTM Matrix were used according to the manufacturer’s protocol. The insert well and bottom well were prepared by rehydrating the BD MatrigelTM Matrix layer with warm (37°C) bicarbonate based culture medium for 2 hours at 37°C. The rehydration solution was then carefully removed, and 500 μl of cell suspension was added to the apical chambers (1.5 ×104 cells). Then 750 μl of DMEM medium containing 10% FBS was added to the each of the basal chambers. Assay plates were incubated for 22 hours at standard culturing conditions. Cells that did not invade through the pores of the transwell inserts were manually removed with cotton swabs and were fixed in cold methanol for 10 minutes and then stained with 0.01% crystal violet. The membranes from the insert housing were removed carefully and mounted with a microscope slide with a small drop of immersion oil. All of the invading cells in the membranes were counted after taking the photo using microscope fields at 40 × magnifications.All samples were assayed in triplicate.
Gelatin zymography
Samples of conditioned media were subjected to electrophoresis on 10% SDS-polyacrylamide gel containing 0.1% gelatin. The volume of each medium sample analyzed was normalized according to the cell number. After electrophoresis, the gel was incubated in the Zymogram Denaturing Buffer (25% Triton X-100 in water) with gentle agitation for 30 min at room temperature. The gel was then incubated in the Zymogram Developing Buffer (50 mM Tris-HCl, pH 8.0, 0.2 M NaCl2, 5 mM CaCl2, 0.02% Brij35) for 30 minutes at room temperature with gentle agitation and then replaced with fresh Zymogram Developing Buffer and incubated at 37°C for at least 4 hours. The gel was then stained with Coomassie Blue R-250 for 30 minutes followed by destaining solution (methanol: acetic acid: water, 50:10:40).
Statistical analysis
Data is expressed as mean ± SD. The data of Real-time PCR was 2-ΔΔCt-transformed and analyzed using ANOVA. The results of the animal experiments, apoptosis assay, migration and invasion assays were analyzed using independent samples t-test and ANOVA. P<0.05 was considered statistically significant.
Results
miPS conditioned media induced phenotypical changes and an increase in tumorigenicity of LLC cells
LLC cells have two types of morphology when cultured in 10% serum on plastic, an epithelial cell phenotype and the formation of loosely attached small cellular cluster (Figure 1A). LLC cells were observed daily after being treated with miPS CM or MEF CM. After two weeks, the morphology of LLC-miPS CM cells became more fibroblastic and cells formed small clonies in contrast to the phenotype of LLC-MEF CM cells. After four weeks, the mesenchymal phenotype of LLC-miPS CM cells was even more prominence and large colonies of mesenchymal-like cells which were loosely attached were observed as compared to LLC-MEF CM cells (Figure 1A).
Figure 1.
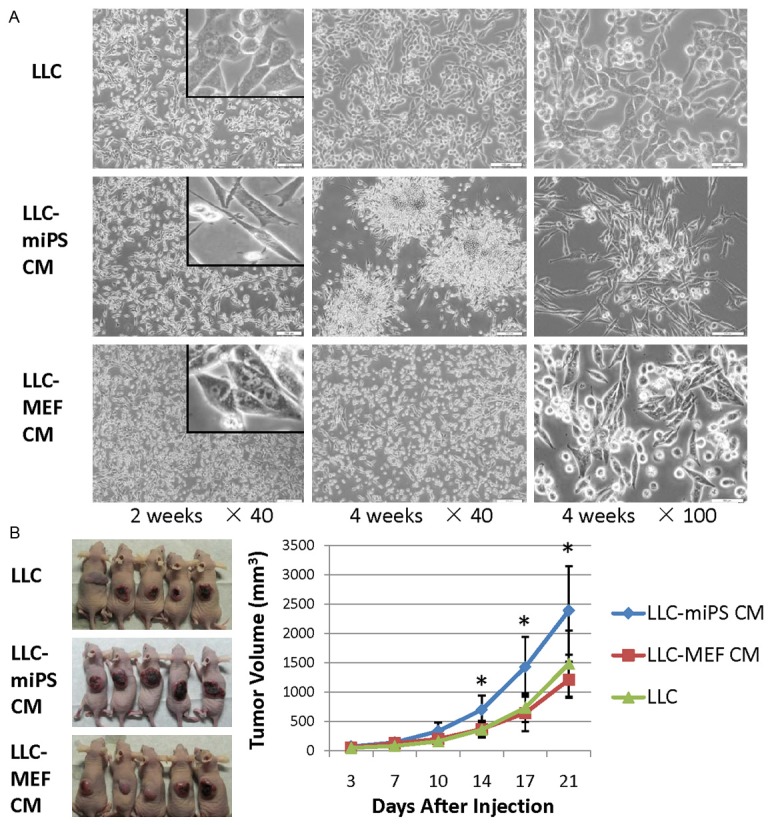
miPS microenvironment induced phonotypical changes and an increased tumorigenticity in LLC cells. A. Phase-contrast images of LLC, LLC-miPS CM and LLC-MEF CM cells treated with DMEM, miPS conditioned medium or MEF conditioned medium for 2 weeks and 4 weeks, respectively. Insert is high magnification photos. Scale bar: 200 μm, 50 μm. B. 5 × 105 of LLC cells, LLC-miPS CM cells or LLC-MEF CM cells were injected subcutaneously into nude mice (n=5). In vivo tumor formation after 21 days is shown in the left panel. Tumor volumes are significantly different at the time points indicated by an asterisk (*, P<0.05).
To investigate any potential changes in tumorigenicity, LLC-miPS CM cells were injected subcutaneously into nude mice, and compared to the tumorigenic potential of the parental LLC and LLC-MEF CM cells. Small tumor nodules appeared after 3 days in all groups and tumor size was measured every three days. After two weeks, the tumor growth curves displayed significant difference between the LLC-miPS CM group and LLC group which persisted through 21 days of tumor growth (P=0.03, P=0.02, and P=0.03 in 14 days, 17 days and 21 days, respectively). The enhanced tumor growth of the LLC-miPS CM group was also significantly different as compared to the LLC-MEF CM group (P=0.02, P=0.01, and P=0.02 in 14 days, 17 days and 21 days, respectively) (Figure 1B). The tumorigenticity of LLC-miPS CM cells was significantly increased when compared to the LLC or LLC-MEF CM cells. Using terminal deoxynucleotidyl transferase biotin-dUTP nick-end labeling (TUNEL) as a measure of apoptosis, we found that exposure to miPS conditioned medium significantly decreased apoptosis in the LLC cells as compared to LLC or LLC-MEF CM cells (Figure S1A). A significant reducing in apoptosis was also observed in tumors derived form LLC-miPS CM cells as compared to tumors arising from parental LLC or LLC-MEF CM cells (Figure S1B).
miPS conditioned media promoted LLC metastasis
We found that LLC cells treated with conditioned medium from miPS cells (LLC-miPS CM cells) had exhibited enhanced tumor growth. To investigate the metastasis ability of LLC-miPS CM cells, 1 × 105 of LLC cells, LLC-miPS CM cells or LLC-MEF CM cells were injected into nude mouse tail veins. Four weeks later, the lungs of mice were excised. Metastatic nodules could not be observed by macroscopy, but metastatic foci were observed by microscopy in the serial fifteen sections (n=3, Figure 2A). The number of metastatic foci of LLC-miPS CM group were much more than those observed from LLC or LLC-MEF CM group (P=0.005, and P=0.02, respectively), while LLC and LLC-MEF CM group (P=0.5) (Figure 2A).
Figure 2.
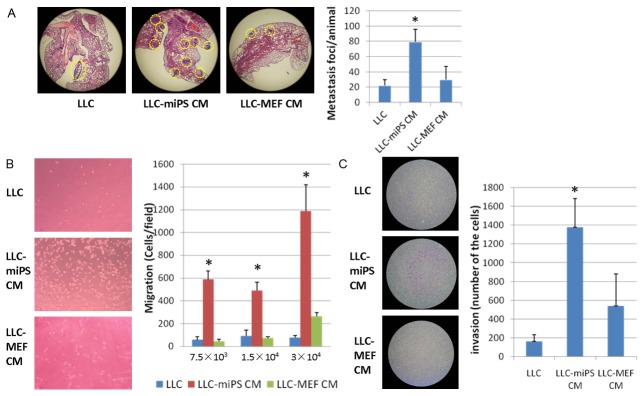
miPS microenvironment promoted LLC cells metastasis in vivo and in vitro. A. Lung metastatic foci were observed by microscopy in fifteen serial sections (n=3). Representative haematoxylin-and-eosin-stained sections of lungs of mice are shown in the left panel. Metastatic foci are delineated by a yellow dashed line. The average number of metastatic foci of LLC, LLC-miPS CM or LLC-MEF CM cells which were statistically significantly different are indicated by an asterisk (*, P<0.05). B. Transwell migration assay.7.5 × 103, 1.5 × 104 or 3 × 104 of LLC, LLC-miPS CM or LLC-MEF CM cells were added in the upper chambers, respectively. Quantification of the migration is expressed as the number of migrating cells to the lower chamber per high-power field (HPF; × 100), and significantly different indicated by an asterisk (*, P<0.05). Representative micrographs of the transwell migration assay (100 × magnification) are shown in the left panel. C. Transwell invasion assay. 1 × 104 of LLC, LLC-miPS CM or LLC-MEF CM cells were added in the upper chambers, respectively. Quantification of the invasion is expressed as the number of invading cells in the membranes and statistic significant differences are indicated by an asterisk (*, P<0.05). Representative micrographs of the transwell invasion assay (40 × magnification) are showed in the left panel.
Motility of LLC-miPS CM cells was then assessed in vitro using transwell Boyden chamber migration assays. The results demonstrated that migration capacity of LLC-miPS CM cells was enhanced as compared to the migration of LLC cells and LLC-MEF CM cells. The P value was less than 0.05 regardless of how many cells (7.5 × 103, 1.5 × 104 and 3 × 104) were seeded in the upper chambers (Figure 2B). The invasive capacity of LLC-miPS CM cells was also enhanced as compared to the invasive ability of the LLC or LLC-MEF CM cells (P<0.05, Figure 2C).
miPS conditioned media initiated EMT by activating a Snail-MMP axis
The morphological transition from an epithelia phenotype into mesenchymal phenotype together with the increasing ability of motility and invasion are properties which are generally ascribed to EMT. The EMT markers snail, slug and E-cadherin were checked by real-time PCR and confocal microscopy. mRNA and protein expression of E-cadherin could not be detected in the parental LLC cells by real-time PCR and confocal microscopy (data not shown). Expression levels of snail in LLC-miPS CM cells were significant higher than the other two control cells, while slug gene expression was down-regulated in the LLC-miPS CM cells as compared to the LLC or LLC-MEF CM cells (Figure 3A). Corresponding to the enhanced metastatic phenotype of the LLC-miPS CM cells, the expression of different MMP family members was assessed by real-time PCR. A significant up-regulation of MMP2 and MMP9 expression was found in LLC-miPS CM cells, as compared to a decrease of MMP3 expression in both of LLC-miPS and LLC-MEF CM cells when normalized against LLC cells (Figure 3B). To investigate if the secretion of MMP2 and MMP9 proteins were increased in LLC-miPS CM cells, we performed gelatin zymography. As shown in Figure 3C, LLC-miPS CM cells produced more active MMP2 (66 KDa) and MMP9 (82 KDa) than the other two cell lines. Thus, the increased secretion of active MMP2 and MMP9 may contribute to the higher metastatic capacity of the LLC-miPS CM cells in combination with increased Snail expression.
Figure 3.
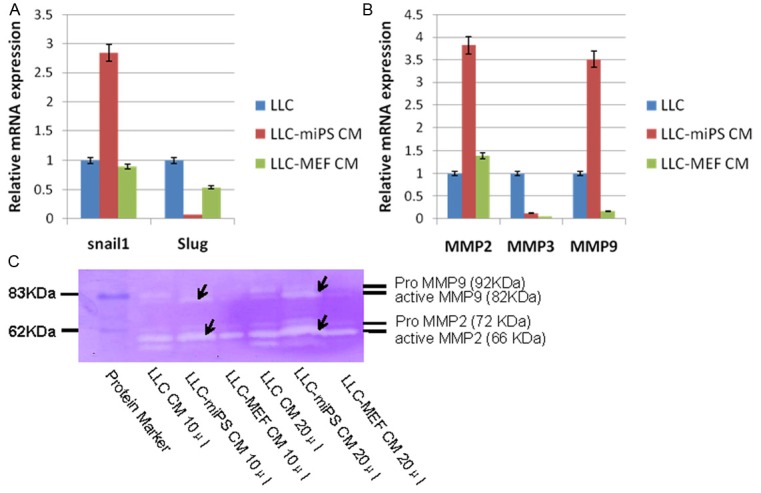
miPS microenvironment up-regulated expression of Snail1, MMP2 and MMP9. A. Real-time PCR analysis of Snail1 and Slug in LLC, LLC-miPS CM and LLC-MEF CM cells. B. Real-time PCR analysis of MMP2, MMP3 and MMP9 in LLC, LLC-miPS CM and LLC-MEF CM cells. C. 10 μl and 20 μl medium of LLC, LLC-miPS CM and LLC-MEF CM cells were subjected to zymography using gelatin as a substrate. The molecular mass standard contains pro-MMP9 (92 kDa), active MMP9 (82 kDa), pro-MMP2 (72 kDa) and active MMP2 (66 kDa). Arrow is used to indicate the zymographic bands of active MMP9 and active MMP2.
Discussion
Induced pluripotent stem (iPS) cellshave a significant potential in contributing therapies for regenerative medicine. However, following the initial excitement over the enormous prospects of this technology, several reports have raised significant concerns regarding its safety for clinical applications and reproducibility for laboratory applications [19,20]. The microenvironment is recognized as a critical component in regulating cell behavior and fate [21]. Bi-directional microenvironmental communication propagated among different types of cells can occur through several distinct mechanisms: direct cell-cell contact, autocrine and paracrine signaling driven by soluble secreted factors, and modeling (or remodeling) of the extracellular matrix [21]. Previously, we were able to convert miPS cells into cancer stem cells using the conditioned medium of cancer cell lines (including LLC, P19, B16 and MC.E12), which mimicked the tumor microenvironment [5]. However, the converse - an evaluation of the effects of an iPS microenvironment on tumor cells - has not yet been sufficiently explored.
In this study, the results demonstrated that miPS cells secrete some soluble factors that can promote the growth, migration and invasion of LLC cancer cells in vivo and in vitro, which are potentially distinct from ES microenvironment of soluble or extracellular matrix-associated factors [9-11]. In addition, the miPS cell microenvironment also altered LLC cell morphology from an epithelia phenotype to mesenchymal phenotype, increased the capacity for migration and invasion, induced resistance to apoptosis, as well as increased the production of the transcription factor of Snail1 which are all properties that are associated with EMT. EMT is a transdifferentiation program that converts epithelial cells into individual migratory cells that is critical for embryonic development and the oncogenic progression of tumor cells into cancer stem cells [22]. Converting an epithelial cell into a mesenchymal cell requires alterations in morphology, cellular architecture, adhesion, and migration. All of these properties are associated with EMT and are generated by secreted factor(s) from miPS cells [23,24]. Moreover, the downregulation of E-cadherin expression and nuclear localization of active β-catenin are considered as key markers of EMT [22]. However, in this study we failed to detect expression of E-cadherin both of the RNA level and of the protein level in LLC cells, but we did observed an increase β-catenin translocated from cytoplasm to the nucleus in a few of the LLC cells after treatment with miPS conditioned medium (Figure S2). Usually, β-catenin is part of a complex of proteins that constitute the adherens junctions [25]. Since LLC cells lack E-cadherin, the identity of the cadherin isotype that is binding β-catenin and that is translocated to the nucleus in LLC cells is unknown. Liu et al found when β-catenin is not assembled into complexes with cadherins, it can form a complex with axin [26]. While bound to axin, β-catenin can be phosphorylated by GSK-3, which creates a signal for the rapid ubiquitin-dependent degradation of β-catenin by proteosomes. Various signals such as the canonical Wnt signalling pathway can inhibit GSK-3-mediated phosphorylation of β-catenin, allowing β-catenin to translocate to the nucleus, interact with transcription factors, and regulate gene transcription [26].
In this study, we also found an increased secretion of activated MMP2 and MMP9, which may be correlated with an upregulation of Snail1 expression. This may be the case since an upregulation of MMP expression by Snail family transcription factors that can contribute to enhanced metastasis has also been reported [27,28]. EMT can also enhance the acquisition of properties that are associated with CSCs [29]. Zhang et al identified a side population of cells from LLC cells which possess well-known characteristics of CSCs, and comprised 1.1% of the total LLC population [30]. In our study, we could not detect expression of embryonic stem cell markers, such as Nanog, Oct4 or Cripto which are often associated with CSCs (data not shown). It may be because the number of CSCs in the LLC cells population is extremely low or that in this system these markers are not associated with a CSCs phenotype. Brabletz et al considered if the vast majority of cells leaving a primary tumor and disseminating to distant sites lack self-renewal capability, their ability to contribute to metastatic foci in end organs is compromised from the outset because of their limited proliferative potential [31]. Thus, EMT may a significant lead to increase in the number of self-renewing cells that can initiate the invasion-metastasis cascade [31]. Qiao et al concluded that Snail may up-regulate MMP2 or MMP9 thereby stimulating the early stages of EMT, while Slug may share a role with Snail in maintaining longer-term EMT [27]. Therefore, the miPS conditioned medium function as a stem cell niche to promote CSC renewal in LLC cells.
In conclusion, the miPS cell microenvironment can initiate EMT in LLC cells by activating a Snail-MMP axis, which results in a change in cellular morphology from an epithelial phenotype to a mesenchymal phenotype, increased capacity for migration and invasion, and an increased resistance to apoptosis. These results therefore should be taken into account before iPS is used in a clinical setting where latent and undetectable tumor cells may exist.
Acknowledgements
L. Chen was supported by Grant-in-Aid for JSPS Fellows. This work was partly supported by Grant-in-Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology (No. 21300179) in Japan.
Supporting Information
References
- 1.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Hoekstra M, Mummery CL, Wilde AA, Bezzina CR, Verkerk AO. Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front Physiol. 2012;3:346. doi: 10.3389/fphys.2012.00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sánchez-Danés A, Richaud-Patin Y, Carballo-Carbajal I, Jiménez-Delgado S, Caig C, Mora S, Di Guglielmo C, Ezquerra M, Patel B, Giralt A, Canals JM, Memo M, Alberch J, López-Barneo J, Vila M, Cuervo AM, Tolosa E, Consiglio A, Raya A. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med. 2012;4:380–395. doi: 10.1002/emmm.201200215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen L, Kasai T, Li Y, Sugii Y, Jin G, Okada M, Vaidyanath A, Mizutani A, Satoh A, Kudoh T, Hendrix MJ, Salomon DS, Fu L, Seno M. A model of cancer stem cells derived from mouse induced pluripotent stem cells. PLoS One. 2012;7:e33544. doi: 10.1371/journal.pone.0033544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujimori H, Shikanai M, Teraoka H, Masutani M, Yoshioka K. Induction of Cancerous Stem Cells during Embryonic Stem Cell Differentiation. J Biol Chem. 2012;287:36777–36791. doi: 10.1074/jbc.M112.372557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizutani A, Chen L, Kasai T, Kudoh T, Murakami H, Fu L, Seno M. A cancer stem cell model: an insight into the conversion of induced pluripotent stem cells to cancer stem-like cells. In: Rajasekhar VK, editor. Cancer Stem Cell. John Wiley & Sons; [Epub ahead of print] [Google Scholar]
- 8.Murakami H, Mizutani A, Chen L, Kasai T, Kudoh T, Fu L, Seno M. Cancer stem cell derived from mouse induced pluripotent stem cells. In: Hayat MA, editor. Stem Cells and Cancer Stem Cells. Volume 10. Springer; [Epub ahead of print] [Google Scholar]
- 9.Hendrix MJ, Seftor EA, Seftor RE, Kasemeier-Kulesa J, Kulesa PM, Postovit LM. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat Rev Cancer. 2007;7:246–255. doi: 10.1038/nrc2108. [DOI] [PubMed] [Google Scholar]
- 10.Postovit LM, Seftor EA, Seftor RE, Hendrix MJ. A three-dimensional model to study the epigenetic effects induced by the microenvironment of human embryonic stem cells. Stem Cells. 2006;24:501–505. doi: 10.1634/stemcells.2005-0459. [DOI] [PubMed] [Google Scholar]
- 11.Postovit LM, Margaryan NV, Seftor EA, Kirschmann DA, Lipavsky A, Wheaton WW, Abbott DE, Seftor RE, Hendrix MJ. Human embryonic stem cell microenvironment suppresses the tumorigenic phenotype of aggressive cancer cells. Proc Natl Acad Sci U S A. 2008;105:4329–4334. doi: 10.1073/pnas.0800467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tzukerman M, Rosenberg T, Reiter I, Ben-Eliezer S, Denkberg G, Coleman R, Reiter Y, Skorecki K. The influence of a human embryonic stem cell-derived microenvironment on targeting of human solid tumor xenografts. Cancer Res. 2006;66:3792–3801. doi: 10.1158/0008-5472.CAN-05-3467. [DOI] [PubMed] [Google Scholar]
- 13.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–563. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 14.Albarenque SM, Zwacka RM, Mohr A. Both human and mouse mesenchymal stem cells promote breast cancer metastasis. Stem Cell Res. 2011;7:163–171. doi: 10.1016/j.scr.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Molloy AP, Martin FT, Dwyer RM, Griffin TP, Murphy M, Barry FP, O’Brien T, Kerin MJ. Mesenchymal stem cell secretion of chemokines during differentiation into osteoblasts, and their potential role in mediating interactions with breast cancer cells. Int J Cancer. 2009;124:326–332. doi: 10.1002/ijc.23939. [DOI] [PubMed] [Google Scholar]
- 16.Huang WH, Chang MC, Tsai KS, Hung MC, Chen HL, Hung SC. Mesenchymal stem cells promote growth and angiogenesis of tumors in mice. Oncogene. 2013;32:4343–4354. doi: 10.1038/onc.2012.458. [DOI] [PubMed] [Google Scholar]
- 17.Sasser AK, Mundy BL, Smith KM, Studebaker AW, Axel AE, Haidet AM, Fernandez SA, Hall BM. Human bone marrow stromal cells enhance breast cancer cell growth rates in a cell line-dependent manner when evaluated in 3D tumor environments. Cancer Lett. 2007;254:255–264. doi: 10.1016/j.canlet.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 18.Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC, Ye L, Zhang X. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008;18:500–507. doi: 10.1038/cr.2008.40. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura M, Okano H. Cell transplantation therapies for spinal cord injury focusing on induced pluripotent stem cells. Cell Res. 2013;23:70–80. doi: 10.1038/cr.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong SG, Dunbar CE, Winkler T. Assessing the Risks of Genotoxicity in the Therapeutic Development of Induced Pluripotent Stem Cells. Mol Ther. 2013;21:272–281. doi: 10.1038/mt.2012.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abbott DE, Bailey CM, Postovit LM, Seftor EA, Margaryan N, Seftor RE, Hendrix MJ. The epigenetic influence of tumor and embryonic microenvironments: how different are they? Cancer Microenviron. 2008;1:13–21. doi: 10.1007/s12307-008-0004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119:1417–1419. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Müller T, Bain G, Wang X, Papkoff J. Regulation of epithelial cell migration and tumor formation by beta-catenin signaling. Exp Cell Res. 2002;280:119–133. doi: 10.1006/excr.2002.5630. [DOI] [PubMed] [Google Scholar]
- 26.Liu X, Rubin JS, Kimmel AR. Rapid, Wnt-induced changes in GSK3beta associations that regulate beta-catenin stabilization are mediated by Galpha proteins. Curr Biol. 2005;15:1989–1997. doi: 10.1016/j.cub.2005.10.050. [DOI] [PubMed] [Google Scholar]
- 27.Qiao B, Johnson NW, Gao J. Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-beta 1 is Snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int J Oncol. 2010;37:663–668. doi: 10.3892/ijo_00000715. [DOI] [PubMed] [Google Scholar]
- 28.Lin CY, Tsai PH, Kandaswami CC, Chang GD, Cheng CH, Huang CJ, Lee PP, Hwang JJ, Lee MT. Role of tissue transglutaminase 2 in the acquisition of the mesenchymal-like phenotype in highly invasive A431 tumor cells. Mol Cancer. 2011;10:87. doi: 10.1186/1476-4598-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S, Baba H, Mori M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010;101:293–299. doi: 10.1111/j.1349-7006.2009.01419.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang AM, Fan Y, Yao Q, Ma H, Lin S, Zhu CH, Wang XX, Liu J, Zhu B, Sun JG, Chen ZT. Identification of a Cancer Stem-like Population in the Lewis Lung Cancer Cell Line. Asian Pac J Cancer Prev. 2012;13:761–766. doi: 10.7314/apjcp.2012.13.3.761. [DOI] [PubMed] [Google Scholar]
- 31.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–747. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.