

Abstract
A widespread use of ultrasound (US) examination is contributing to an increase in the diagnosis of renal and hepatic cysts. However, the vast majority of these lesions are benign with an indolent course during the patient's lifespan. Adult polycystic kidney disease (APKD) is one of the most common diagnosed entities. APKD is a genetic disease defined by the presence of multiple kidney cysts, occasionally accompanied by hepatic cysts. The presence of hepatic cysts sparing kidneys is very rare and thereby must be assumed as a different clinical entity.
This article describes a case of an exuberant hepatomegaly due to the presence of isolated multiple hepatic cysts without renal involvement.
Background
Polycystic liver disease (PCLD) is a rare disease, and is generally found in routine abdominal echographies. Though considered benign, PCLD can greatly affect the quality of life, and in some cases can be lethal. We should not neglect these patients and must follow them closely in order to act at the right time due to the absence of an effective medical therapy that can contribute to cure. There are several surgical approaches available (as partial hepatectomy) but the only curative therapy is liver transplantation. It is important to develop different approaches (prevention, surveillance or even therapeutics) in order to decrease the volume of cysts and prevent their growth and consequently the hepatic failure.
Case presentation
We report the case of a female patient aged with 55 years, without previous known morbidities other than appendectomy in adolescence and cholecystectomy in 2006. Hepatic cysts—polycystic hepatic disease—were diagnosed by ultrasound (US) in 2006, while diagnosing gallbladder lithiasis. She had no familiar relatives with hepatic cysts. After diagnosis, the patient was irregularly followed by her family physician.
The patient presented herself to our consultation on December 2012 with dyspepsia, epigastric pain and an increased abdominal volume. She was evaluated by an abdominal US, which showed a multiple cystic formation with the biggest one measuring 16 cm.
A CT was performed to characterise hepatic cysts and to evaluate other possible and different organ localisation. The CT confirmed the presence of multiple cysts in the liver. The patient was referred to the hepatobiliary surgery consultation where face the disease evolution was submitted surgical intervention.
Investigations
Laboratory tests (November 2012) did not reveal significant alterations having hepatic function conserved.
An abdominal echography (November 2012) showed multiple cystic formations in the left hepatic lobe with eccentric growth in an extension of about 16 cm, with the content globally homogeneous and without adjacent liquid or echographic signals of cystic rupture or haemorrhage. Remaining hepatic parenchyma was unchanged. Changes postcholecystectomy with surgical loca cleared. Intrahepatic and extrahepatic bile ducts and liver were not found enlarged. The pancreas was of normal size without enlargement of Wirsung, only with signs of diffuse lipomatosis. The spleen was not visualised by interposition of gas. There was no ascitis or collections in the upper abdomen. Large abdominal vessels were unchanged.
The abdominal CT (November 2012) revealed an extensive cystic conglomerate, which extended at least 19.9×14.9 cm and involved predominantly the left hepatic lobe segments IV, II and III, consisting of cysts, which by their number and dimensions extended to anterosuperior abdominal region and left flank, deviated spleen backwards and compressed the pancreas body and stomach wall. At the right lobe were found millimetric cysts, the largest in the segment VI with 16 mm, the remaining being infracentimetric and multiple cysts (figure 1).
Figure 1.

Contrast-enhanced CT scans of the abdomen revealed extensive cystic conglomerate involving predominantly the left hepatic lobe and compression of the stomach wall.
Differential diagnosis
PCLD represents a group of rare genetic diseases where cysts arise in the renal area (autosomal dominant polycystic kidney disease) or, less commonly, they occur only in the liver as autosomal dominant PCLD.1 The estimated prevalence of PCLD is 1:100 000. As far as we know, only two mutations have been reported, PRKCSH (locus 19p13.2) and SEC63 (locus 6q21) and they are responsible for 33–50% of cases.2
Treatment
On 6 February 2013, a left hepatectomy was performed by median supraumbilical laparotomy with right subcostal extension and elective section of vascular structures on the left and left suprahepatic vein with the placement of a Jackson-pratt drain.
Outcome and follow-up
Surgery and postoperative period had no intercurrences, so the patient was discharged on the third day after surgery. Histopathology (February 2013) confirmed the diagnosis of liver polycystic disease.
The patient was reassessed in consultation 1 month later and in August 2013 being verified an evident clinical improvement and no laboratorial alterations. An abdominal CT (March 2013) showed no valorisable changes in the liver or other organs (figure 2).
Figure 2.
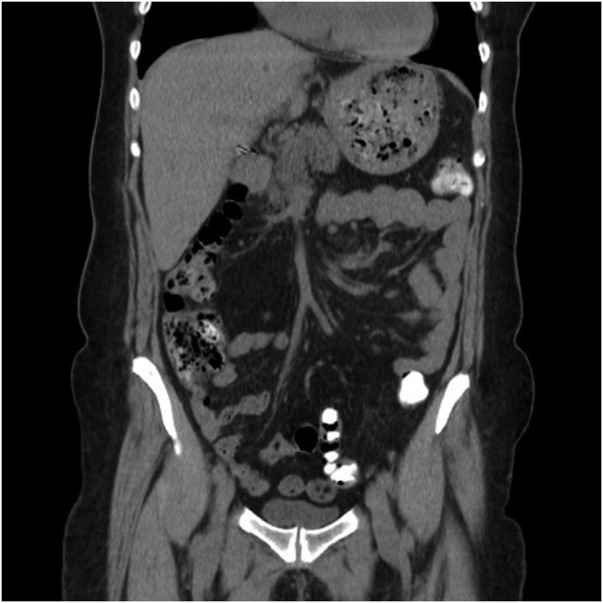
CT scan of the abdomen after left hepatectomy showed no valorisable abnormalities in the liver or other organs.
Owing to high recurrence risk, follow-up protocol includes analytical and imagiological (abdominal US or abdominal CT) studies twice yearly in the first year and then annually according with clinical evolution.
Discussion
PCLD reaches an advanced stage with an increasing age as occurred with our patient, which is related to the progressive increase in size and the number of cysts. Existing data indicate that cysts grow 0.9–3.2% annually. The hepatic cysts are rarely diagnosed before puberty and women are more frequently diagnosed, with symptomatic cysts, than men. Liver cysts are more common in multiparous women and those who underwent hormonal replacement therapy.3 4
Genetic studies confirmed that mutations in the genes PRKCSH and SEC63 are associated with defects in the function of the proteins hepatocystin and Sec63, respectively; however, the way they lead to hepatic cyst formation is not yet completely understood.2
The majority of the patients with hepatic cysts are asymptomatic. Only 3% of patients have abdominal symptoms and serious complications, due to liver expansion and vicinity organs compression. The massive hepatomegaly can cause abdominal distension, early satiety, gastro-oesophageal reflux and extreme malnutrition. Inferior vena cava or portal vein compression and Budd-Chiari syndrome are other important consequences of PCLD. Moreover, the cysts may cause infection, torsion, rupture or haemorrhage. Liver function is often normal (as we found in our patient). Patients with PCLD do not have a higher propensity for malignancy, and disorders such as mitral valve prolapse, inguinal hernia, diverticulosis or cerebral aneurysms occur rarely. The screening of these conditions is not recommended unless there is a clinical suspicion or indication for transplantation.1 5
The diagnosis is considered when we find >20 isolated cysts in the liver. If there is a familial history of PCLD, presence of more than four cysts is the criterion to diagnose.
Liver cysts are usually identified by abdominal US or abdominal CT scan, though MRI is the most sensitive technique.6 Laboratory tests are unspecific and hepatic transaminases, γ glutamyl transferase and alkaline phosphatase usually have normal values. Tumour markers such as CA 19.9 can be elevated in 45% of cases due to an increased production by biliary epithelium but without associated malignancy.7 8
Asymptomatic patients (majority of cases) only need clinical, laboratory and imagiological surveillance. In symptomatic patients palliative therapy is advocated and for prevention of recurrence of symptoms a surgical decreasing of liver volume is recommended. Surgical intervention is chosen based on the criteria defined by Gigot et al. Gigot classify PCLD taking into account the size, the number of hepatic cysts and the area of free hepatic parenchyma in three types: I—patients with less than 10 large cysts (with>10 cm); II—patients with multiple cysts of medium size but sparing large areas of the liver; III—massive spread of cysts affecting almost the entire liver. In types I and II, aspiration and sclerotherapy, laparoscopic fenestration or laparotomy and partial hepatectomy are indicated. In type III, these procedures are ineffective and are responsible for elevated morbidity, mortality and recurrence of the disease.9 Reports show perioperative morbidity in about 50% of the cases, the reason why surgeons operate only in cases where decompression of cysts is not effective. Hepatectomy can cause distortion of the biliary tree and intrahepatic vasculature, which can cause biliary leakage and bleeding. In severe clinical conditions affecting the quality of life, malnutrition or nutritional deficiencies, liver transplantation, which is the only curative treatment, should be considered.10 11
The recent findings suggest that octreotide and lanreotide (somatostatin analogues), sirolimus and everolimus (mammalian target of rapamycin (mTOR) inhibitors) are likely to prevent the cysts growth; however, there are no medical treatments approved for PCLD therapy. Octreotide and lanreotide have demonstrated effectiveness in reducing the liver volume. The long-term benefits and effective doses remain undefined and the importance of mTOR inhibitors is under investigation.12
Learning points.
Liver and kidney cysts are frequent findings in routine ultrasound examinations.
Polycystic liver disease (PCLD) is a rare disease diagnosed when more than 20 isolated liver cysts are present.
Symptoms appear late in the disease evolution and are caused by mass effect.
There is no effective medical treatment for PCLD. Surgery is the treatment of choice in symptomatic patients.
Liver transplantation is the only curative treatment.
Acknowledgments
The authors would like to express our deepest appreciation to Surgeon Emanuel Vigia who performed the patient's hepatobiliary surgery consultation and kindly provided the follow-up information also his expert opinion.
Footnotes
Contributors: All the authors of this article have directly participated in the planning, execution or analysis of this study and read and approved the final version submitted.
Competing interests: None.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Chandok N. Polycystic liver disease: a clinical review. Ann Hepatol 2012;11:819–26 [PubMed] [Google Scholar]
- 2.Temmerman F, Missiaen L, Bammens B, et al. Systematic review: the pathophysiology and management of polycystic liver disease. Alimen Pharmacol Therapeut 2011;34:702–13 [DOI] [PubMed] [Google Scholar]
- 3.Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med 2006;354:2122–30 [DOI] [PubMed] [Google Scholar]
- 4.Qian Q. Isolated polycystic liver disease. Adv Chronic Kidney Dis 2010;17:181–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoevenaren IA, Wester R, Schrier RW, et al. Polycystic liver: clinical characteristics of patients with isolated polycystic liver disease compared with patients with polycystic liver and autosomal dominant polycystic kidney disease. Liver Int 2008;28:264–70 [DOI] [PubMed] [Google Scholar]
- 6.Bae KT, Zhu F, Chapman AB, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the consortium for radiologic imaging studies of polycystic kidney disease cohort. Clin J Am Soc Nephrol 2006;1:64–9 [DOI] [PubMed] [Google Scholar]
- 7.Arnold HL, Harrison SA. New advances in evaluation and management of patients with polycystic liver disease. Am J Gastroenterol 2005;100:2569–82 [DOI] [PubMed] [Google Scholar]
- 8.Waanders E, van Keimpema L, Brouwer JT, et al. Carbohydrate antigen 19-9 is extremely elevated in polycystic liver disease. Liver Int 2009;29:1389–95 [DOI] [PubMed] [Google Scholar]
- 9.Gigot JF, Hubert C, Banice R, et al. Laparoscopic management of benign liver diseases: where are we? HPB 2004;6:197–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drenth JP, Chrispijn M, Nagorney DM, et al. Medical and surgical treatment options for polycystic liver disease. Hepatology 2010;52:2223–30 [DOI] [PubMed] [Google Scholar]
- 11.Armitage NC, Blumgart LH. Partial resection and fenestration in the treatment of polycystic liver disease. Bri J Surg 1984;71:242–4 [DOI] [PubMed] [Google Scholar]
- 12.Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol 2010;21:1052–61 [DOI] [PMC free article] [PubMed] [Google Scholar]