

Abstract
DNA demethylation has been primarily studied in the context of development biology, cell fate, and cancer, with less attention on inflammation. Herein, we investigate the association between DNA methylation and production of the chemoattractant cytokine eotaxin-3 in the tissue of patients with allergic disease. Regions of the human eotaxin-3 promoter were found to be hypomethylated in primary epithelial cells obtained from allergic tissue compared with normal control tissue (CTL). The demethylation of a specific CpG site (designated CpG 2), which is juxtaposed to a key cyclic AMP-responsive element (CRE) site, was significantly demethylated in patient-derived compared to CTL-derived epithelial cells. Levels of methylation at CpG 2 inversely correlated with basal and IL-13–induced eotaxin-3 gene expression. Conversely, global inhibition of methylation with 5-azacytidine (5-AzaC) promoted eotaxin-3 production in association with decreasing CpG 2 methylation. In addition, the basal and IL-13-induced eotaxin-3 transcriptional activity was suppressed by promoter methylation using a methylation-free in vitro system. Further, electrophoretic mobility shift assays (EMSA) demonstrated that the attachment of CREB binding protein (CBP) and activating transcription factor 2 (ATF-2) to the CRE site was methylation dependent. Taken together, these data identify a contributory role for DNA methylation in regulating eotaxin-3 production in human allergic inflammation.
Introduction
Although inheritance, susceptibility, and phenotype of particular diseases are seemingly dictated by variations in DNA sequence, DNA sequence is often not the primary driver of disease phenotype, as evidenced from identical twin studies, which often reveal low concordance between twins (1, 2). Substantial evidence links the interplay between genetics and the environment in fully explaining disease susceptibility and phenotype. However, the mechanisms by which environmental factors specifically regulate the abnormal gene expression associated with particular diseases are not well understood (3). Epigenetics, the study of a heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence (4, 5), has been identified as a key contributor to disease manifestations. In somatic cells, external signals can induce epigenetic modifications such as modification of DNA methylation which causes changes in chromosomal structure and gene transcription (6).
Epigenetic mechanisms are likely key contributory processes to inflammatory diseases (7); yet, they have received relatively little attention compared to the contribution of primary DNA sequence variations. Allergic inflammatory diseases are particularly likely to involve epigenetic mechanisms as these diseases are constantly responding to environmental stimulants (e.g. allergens). In an effort to understand epigenetic mechanisms that are involved in human allergic inflammation, we focused our attention on eosinophilic esophagitis (EoE), an emerging chronic esophageal inflammatory disease that is triggered by immune hypersensitivity to food and results in an intense eosinophil infiltration of the esophageal epithelium (8-10). Unlike other allergic diseases, EoE provides an opportunity to directly examine operational mechanisms, as the diseased tissue is readily procured by routine endoscopic biopsy, facilitating detailed molecular assessment of human inflammatory processes. Microarray analysis of esophageal biopsy specimens has defined an EoE transcriptome that contains a number of genes highly inducible by the TH2 cytokine IL-13 in human primary esophageal epithelial cells (11, 12). In allergic inflammation, epithelial cells regulate the recruitment of eosinophils into the mucosa as TH2 cell–derived IL-13 drives the release of eosinophil-activating chemokines, especially eotaxin-3, from epithelial cells, thereby contributing to various aspects of EoE (9, 13-15). In support of a key role of esophageal epithelial cells in EoE disease pathogenesis, genetic analysis of EoE has identified susceptibility loci in the regions that contain candidate genes that are expressed in epithelial cells and strongly implicated in regulating immune responses such as innate immune stimuli (TSLP, thymic stromal lymphopoietin and TSLP receptor (16, 17)), inflammatory cell recruitment and activation (CCL26, eotaxin-3 (11, 13, 14)) and epithelial barrier function (FLG, filaggrin (12)).
Regulation of eotaxin-3 gene expression involves both transcriptional and posttranscriptional mechanisms. Binding of transcription factors (e.g. signal transducer and activator of transcription 6 (STAT-6) and CREB-binding protein (CBP)) to the promoter regulates eotaxin-3 gene expression by induction of histone 3 acetylation (18). The CBP histone acetyltransferase complex functions as a coactivator for specific transcription factors at proximal and distal regulatory elements, driving epigenetic programs involved in cellular proliferation, apoptosis, differentiation, and DNA stability (19). Indeed, the level of histone 3 acetylation is increased in esophageal tissue from patients with EoE compared with normal controls (18). In addition to epigenetic regulation of histone proteins, DNA methylation is thought to negatively regulate target gene expression by interfering with the binding of transcription factors and by facilitating formation of an unfavorable chromatin structure. In general, the density of DNA methylation is low in transcriptionally active genes and high in silenced genes (20).
Herein, we aimed to examine the DNA methylation of human primary esophageal epithelial cells producing eotaxin-3 and its relevance to allergic inflammation. Accordingly, we compared the level of eotaxin-3 promoter DNA methylation in human esophageal epithelial cells from patients with EoE (EoE-derived esophageal epithelial cells) and from normal controls (CTL-derived esophageal epithelial cells). After discovering that a specific CpG site (designated CpG 2) showed a lower level of methylation in EoE-derived esophageal epithelial cells, we demonstrated that higher eotaxin-3 mRNA levels were associated with lower methylation levels at the same CpG site. Conversely, demethylation by 5-azacytidine (5-AzaC) promoted eotaxin-3 gene expression in esophageal epithelial cells. To examine the direct contribution of methylation of the eotaxin-3 promoter, we used a methylation-free promoter-reporter system (pCpGL) that allowed us to specifically regulate the degree of eotaxin-3 promoter methylation. Overall, methylation resulted in reduced promoter activity. Direct demethylation of the CpG 2 site increased gene expression and allowed the recruitment of CBP and activating transcription factor-2 (ATF-2) to the CRE consensus site in the eotaxin-3 promoter. We propose a novel link between the epigenetic state of epithelial cells and inflammatory cytokine production in human allergic inflammation.
Materials and Methods
Primary epithelial cell culture and cell lines
The Institutional Review Board of the Cincinnati Children's Hospital Medical Center approved this study. Informed consent was obtained to examine biopsy specimens during a routine endoscopy performed for clinical indications; the population included 18 male subjects. Samples were divided into two groups; normal controls (n = 7) had no history of EoE, 0 eosinophils per high-powered field (hpf) in the esophagus at the time of biopsy, and no concomitant swallowed glucocorticoid treatment; these patients are referred to as normal controls (CTL). Patients with EoE (n = 11) had > 15 esophageal eosinophils/hpf at the time of biopsy and no concomitant swallowed glucocorticoid treatment (Table 1). The distal esophageal biopsy specimens were digested with trypsin. As previously described, esophageal epithelial cells were cultured for 2 weeks, and fibroblasts were depleted by means of differential trypsinization (11). Human esophageal epithelial cell lines (TE-7, TE-1, TE-13; a kind gift of Dr. Hainault, International Agency for Research on Cancer, Lyon, France) were cultured in RPMI 1640 medium supplemented with 5% fetal bovine serum and 1% penicillin and streptomycin (28, 29). The human esophageal epithelial cell line (EPC2), a kind gift of Anil Rusti (21) was cultured in Keratinocyte Serum Free Medium (KSFM, Invitrogen) with 0.09 mM CaCl2, 1 ng/ml EGF, and 25 μg/ml of BPE. Recombinant human IL-13 was obtained from PeproTech, Inc. (Rocky Hill, NJ). 5-AzaC refers to an equimolar mix of 5-azacytidine (5-AzaC-R, A2385, Sigma) and 5-aza-2-deoxycytidine (5-AzaC-dR, A3636, Sigma).
Table 1. Patients' clinical characteristics.
| Normal control | Patients with EoE | |
|---|---|---|
| No. of patients | 7 | 11 |
| Male sex, no. (%) | 7 (100) | 11 (100) |
| Age (y)(mean range) | 8.71 (5.81-12.07) | 4.59 (1.51-14.07) |
| * PPI, no. (%) | 6 (85) | 6 (54) |
| Steroids, no. (%) | 0 | 0 |
Proton-Pump Inhibitor (PPI)
Pyrosequencing of the eotaxin-3 promoter
Genomic DNA was purified from cell lines and primary esophageal epithelial cells with MasterPureTM DNA purification Kit from Epicentre (Madison, WI) as recommended by the manufacturer. 500 ng of genomic DNA from each sample was treated with bisulfite using an EZ DNA methylation-Gold Kit (ZYMO research) according to the manufacturer's specifications (22). All primers were designed to be specific for bisulfite-converted DNA. The bisulfite-treated genomic DNA was amplified by PCR using unbiased nested primers. The first round of PCR amplification in 25 μl was done using a 58°C annealing temperature for 35 cycles and the primer pair site 1-5 long forward (5′-AAGTGATTTTTTTGTTTTATTTTTAG-3′) and site 1-5 long reverse (5′-ATCAAACCCTTCTCAAATTTCTC-3′). A 4-μl aliquot of the material obtained in the first round was further amplified in the second round in 50 μl, using a 58°C annealing temperature for 40 cycles and the primer pair site 1-5 nested forward (5′-GGAGTAGTTGGGATTATAGTTGTTTGT-3′) and site 1-5 nested reverse (5′-TCCTACCTAATCCCCTTTTATAAAACTA-3′). DNA methylation was measured by quantitative pyrosequencing using a PSQ HS96 (Biotage). The DNA methylation percentage at each CpG site was determined using Q-CpG methylation software (Biotage). Human genomic DNA was amplified using the REPLI-g kit (Qiagen) and GenomePlex Complete Whole Genome Amplification (WGA) Kit (GPlex2, Sigma-Aldrich). All procedures were performed according to the manufacturer's instructions. Half of the amplified human genomic DNA was used as the 0% methylated DNA control, and half of the amplified human genomic DNA was treated by M.SssI methyltransferase and used as the 100% methylation control.
Real-time quantitative RT-PCR
Total RNA was isolated from esophageal epithelial cells using the RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. Total RNA was reversed transcribed using the iScriptTM cDNA Synthesis kit (Bio-Rad). The levels of mRNA expression were determined using an iQ5 real-time PCR detection system (Bio-Rad) with iQTMSYBR Green Supermix. Expression of the gene of interest was normalized to GAPDH. Relative expression was calculated using the comparative Ct method as described previously (34). The cDNAs were amplified using the following primers:
Eotaxin-3:
Forward: 5′-AACTCCGAAACAATTGTACTCAGCTG-3′,
Reverse: 5′-GTAACTCTGGGAGGAAACACCCTCTCC-3′;
GAPDH:
Forward: 5′-TGGAAATCCCATCACCATCT-3′,
Reverse: 5′-GTCTTCTGGGTGGCAGTGAT-3′.
Transfections and luciferase reporter assay
The luciferase reporter plasmid lacking CpGs in the backbone (pCpGL; a kind gift of M. Rehli, University Hospital, Resensberg, Germany (23)), was used to clone proximal promoters of housekeeping and tissue-specific genes, allowing evaluation of the effect of CpG methylation in the cloned region on reporter gene activity without any confounding affects of CpG methylation in the backbone (23). A reporter plasmid (pCpGL-Eox-3) containing the human eotaxin-3 gene promoter was prepared by PCR amplification of pGL3-basic-Eotaxin-3 as previously described (18). The pCpGL-Eox-3 was cloned into the Spe I and Hind III sites of pCpGL. The resulting plasmids (pCpGL-Eox-3) were grown in the E. coli PIR 1 strain using Zeocin as a selection marker. Each plasmid was confirmed by DNA sequence analysis using the sequencing primers 5′-tgaataaaaaattattagca-3′ and 5′-tgtgcagttgctcaccagca-3′. The plasmids with and without CpG methylation were transfected into 293 cells. Methylated CpG constructs were prepared by incubating 10 μg of DNA with 20 units of M.SssI in presence of 0.32 mM S-adenosylmethionine and NEB 2 buffer in a 100-μl reaction volume at 37°C overnight followed by 15 min incubation at 65°C. The methylation status of the plasmid DNA was confirmed by restriction digestion. An equal number of cells were seeded in 12-well plates and grown for 24 hrs at 37°C. The next day, HEK293 cells were co-transfected with 500 ng/well of pCpGL with a cloned unmethylated or methylated wild type (WT) or mutant (MUT) promoter region of the eotaxin-3 gene or 500 ng/well of a STAT-6 expression vector and 5 ng/well of renilla luciferase plasmid as an internal control in 100 μl of serum-free S-MEM medium and 3 μl/well TransIT-LT1 transfection reagents (Mirus). After 24 hrs, cells were lysed in passive lysis buffer (Promega), and the lysate was assayed for firefly and renilla luciferase activity using dual-luciferase reporter assay system (Promega) in a Synergy HT Multi-Mode Microplate Reader (BioTek Instruments, Winooski, VT). The relative luciferase activity was calculated from four replicates, and fold increase over the unmethylated reporter activity was determined.
Chromatin immunoprecipitation (ChIP)-qPCR
Esophageal epithelial cells (TE-1, TE-7) were plated in 15-cm dishes and grown to ∼80-90% confluence. Cells were treated with 100 ng/ml of IL-13 in medium for 48 hrs. Cells were cross-linked on the plates with 1% formaldehyde, and chromatin was prepared essentially as previously described (24) with some modifications. Sonication was performed using a Covaris S2 sonicator for 60 seconds with the following setting: eight cycles for Duty cycle 10%, Intensity 5, Cycles per Burst 200. Chromatin fragments were determined to be less than 500 bp in size. STAT-6 antibody for ChIP assay was used from Santa Cruz Biotechnology (sc-621). In parallel reactions, an equivalent concentration of rabbit IgG was used as a negative control. The SX-8G IP-Star™ system (Diagenode) performed automated ChIP assay using the iPure 200 CHIP protocol. After reversing cross-links, DNA was extracted using phenol-chloroform and then precipitated with ethanol. The pellet was resuspended in H2O and purified using the QIAquick PCR purification kit. Then it was eluted in 50 μl of the elution buffer provided with the kit. For quantitative PCR, equal amounts of starting input DNA was used in parallel ChIP experiments. Each experiment included two technical replicates and was repeated with two biological replicates. 2 μl of ChIP or 2 μl of input DNA from corresponding samples were used for the qPCR. For the calibration curve, 2 μl of serially diluted input DNA (stock 10 ng/μl) was used in the TaqMan PCR reaction using PCR master mix (Applied Biosystems). 500 nM of primers and 125 nM of TaqMan probe were used in 10 μl of PCR reaction. Primers and dual-labeled TaqMan probes designed using Beacon Designer (PREMIER Biosoft International) are as follows:
Eotaxin-3 (STAT-6-1):
Forward: 5′-TGTTCCCAACCACAGAATTCTCT-3′,
Reverse: 5′-TCCTGCCTGATCCCCTTTT-3′
TaqMan probe: 5′6-FAM-AATTGTTTTCAGGGCCGTCTCAGTCTCA-ZEN-BHQ1-3′
The negative control locus (4 kb upstream from the eotaxin-3 transcription start site):
Forward: 5′-CAAAAGACACACACTAGAATATTCATAGCA-3′
Reverse: 5′-TCTCACTGCGGAATAGTATTGTATTATGT -3′
TaqMan probe: 5′6-FAM-AGCTCCAAACTGGAAATGACCCTCATGTC-ZEN-BHQ1-3′
Ct values were generated for each primer set using the auto cut off of SDS2.3 Software (Applied Biosystems). Quantities of DNA in the ChIP and input DNA samples were calculated using linear interpolation between Ct values and logarithms of DNA quantities in the calibration curve.
Electrophoretic mobility shift assays (EMSA)
Nuclear extracts from TE-7 cells were prepared as described previously (18). All steps were performed at 4°C. Synthetic 5′-biotinylated complementary oligonucleotides were purchased from IDT (USA). Equimolar labeled oligonucleotides were annealed by heating the mixture in annealing buffer at 65°C for 15 min and snap cooling on ice for 2 min followed by incubation at room temperature for 15 min. The following oligonucleotides and its compliment were used: Eotaxin-3 CpG 2, 5′-CCAGGCTGATCTTGAACTCCTGACCTCAAGGGATCmeCGCCCACCT-3′
The CRE-like sequence is underlined. The cytosine in the bold single CpG in these oligonucleotides was either unmethylated or contained a 5′-methyl group on both strands. In a 20-μl sample volume, 20 fmol of labeled oligonucleotide probes containing the DNA element with CRE-like sequence was added to 3-6 μg of nuclear extract and incubated in binding buffer (10 mM HEPES, 80 mM KCl, 0.05 mM EDTA, 6% glycerol, 1 mM DTT, 1 mM MgCl2) at room temperature for 30 min. The competition experiment was performed in the presence of unlabeled competitor DNA fragments at the indicated molar excess. Samples were separated on a 4-12% native PAGE gel in 0.5 × TBE at 150 Volts for 1.5 hrs and quantified with a LightShift chemiluminescent EMSA kit (Pierce, Rockford, IL) (25). The DNA-protein complex bands were supershifted by the inclusion of either 2 μg rabbit IgG, anti-ATF-2 (Santa Cruz Biotechnology sc-187), or anti-CBP (Santa Cruz Biotechnology, sc-369) antibodies.
Results
Human primary esophageal epithelial cells from patients with EoE have site-specific demethylation of the eotaxin-3 promoter
We have previously identified that the basal and IL-13–inducible eotaxin-3 promoter activity is regulated within 929 bp 5′ of the transcription start site (18). Analysis of this region did not identify a CpG island, but 5 CpG sites were identified at position -307 CG, -225 CG, -216 CG, -176 CG, -43 CG bp in the promoter, and we refer to these as CpG 1-5 sites, respectively (Fig. 1, top panel). We analyzed the methylation status of each individual CpG sequence by bisulfite pyrosequencing. CpG sites had methylation levels that varied between 1.4% and 42% (Fig. 1, bottom panel). Notably, the overall percentage of methylation of the CpG 1 site and the CpG 2 site were significantly decreased in EoE-derived esophageal epithelial cells (n = 11) compared to CTL-derived esophageal epithelial cells (n = 7) (Table 1). For example, CpG 2 methylation was 14.6 ± 1.8% versus 42.9 ± 3.8% in EoE- and CTL-derived esophageal epithelial cells, respectively (mean ± SEM, p < 0.0001). Of the 5 CpG sites examined in the eotaxin-3 promoter, the greatest difference in methylated CpG abundance between EoE-derived esophageal epithelial cells and CTL-derived epithelial cells was observed at CpG2 (Fig. 1, A).
Figure 1.
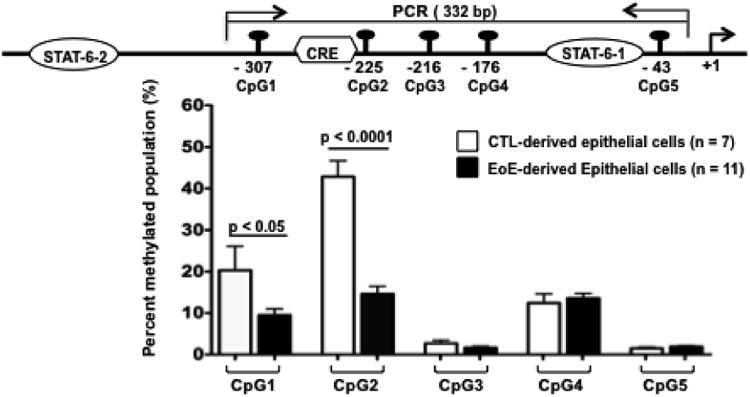
Methylation status of specific eotaxin-3 CpG sites in primary esophageal epithelial cells from normal control tissue (CTL) and eosinophilic esophagus (EoE). Diagram of the regulatory elements within the eotaxin-3 promoter. The CpG number indicates the position of the CpG dinucleotides along the promoter in relation to the transcription start site. The CpG 2 is juxtaposed to a key cyclic AMP-responsive element (CRE) site (top panel). The bottom panel shows methylation level as determined by bisulfite-pyrosequencing analysis (bottom panel). The specific DNA methylation for each position is analyzed by consecutive dispensation and luciferase based visualization of T and C dNTP incorporation (according to the C/T polymorphism in CpGs after bisulfite conversion). The methylation values are calculated by estimating the ratio of T to C peaks. Quantitative methylation was determined at 5 CpG sites by PyroMark analysis software using the equation: % methylation = (C peak height × 100/C peak height + T peak height). Data are represented as the mean ± SEM percentage of methylation at the 5 CpG sites in primary esophageal epithelial cells from 7 CTL individuals and 11 EoE patients.
Low methylation level at CpG 2 site associates with transcriptional activation of eotaxin-3
To investigate whether the methylation status of the CpG 2 site correlated with eotaxin-3 transcription, we examined eotaxin-3 DNA methylation level in several esophageal epithelial cell lines (EPC2, TE-1, TE-7, and TE-13). We found that low methylation level at the CpG 2 site correlated with basal eotaxin-3 gene expression in the different esophageal epithelial cell lines. High levels of methylated CpG 2 in TE-1 cells correlated with low eotaxin-3 gene expression and low levels of methylated CpG 2 in TE-7 cells correlated with high eotaxin-3 gene expression (Fig. 2, A). Overall, the promoter CpG sites in TE-1 cells were highly methylated compared to the promoter CpG sites in TE-7 cells (Fig. 2, B). In contrast, methylation at the CpG 5 site did not correlate with eotaxin-3 production in TE-1 or TE-7 cells. These results imply that the eotaxin-3 expression level is associated with low methylation level at CpG 2 site. To test whether epithelial cells with hypo- (TE-7) or hyper-methylated (TE-1) eotaxin-3 have a different propensity to respond to IL-13, cells were treated with IL-13. The fold change of eotaxin-3 mRNA expression and eotaxin-3 protein secretion with IL-13 at 100 ng/ml in TE-7 cells was significantly higher than in TE-1 cells (Fig. 2, C).
Figure 2.
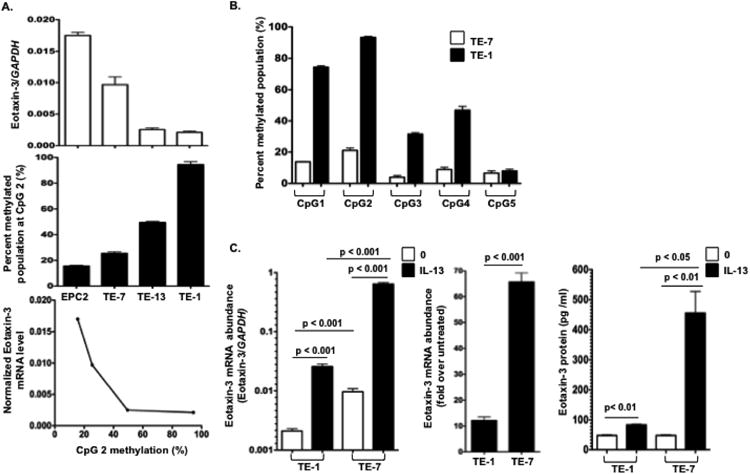
Methylation status of a single, specific CpG site in the eotaxin-3 promoter correlates with transcription. A. Total RNA and genomic DNA were purified from TE-1, TE-7, TE-13, and EPC cells. Expression levels of eotaxin-3 mRNA were determined by real-time PCR and normalized to GAPDH expression (top panel). The methylation status of the CpG 2 site was determined by bisulfite pyrosequencing anlaysis (middle panel). The correlation between the methylation status of the CpG 2 site in the eotaxin-3 promoter and gene expression (bottom panel) was analyzed. B. Quantitative methylation in TE-1 and TE-7 cells was determined at 5 CpG sites in the eotaxin-3 promoter. C. TE-1 and TE-7 cells were stimulated with IL-13 (100 ng/ml) for 48 hrs. Relative eotaxin-3 mRNA levels (left panel) and protein levels (right panel) were measured by quantitative real-time PCR and ELISA, respectively. Data are given as mean ± SEM; n = 3 replicates per group per experiment. Data shown are a representative experiment of three independent experiments.
Previous studies have demonstrated that STAT-6 regulates eotaxin-3 through its interactions with the STAT-6 sites located at positions -89 and -98 of the promoter (Fig. 1, top panel) (11, 14, 18). Therefore, we performed anti-STAT-6 ChIP analysis using TE-1 and TE-7 cells with or without IL-13 to determine the binding to STAT-6 as a function of the methylation level of the eotaxin-3 promoter. Anti-STAT-6 ChIP analysis showed that STAT-6 preferentially bound to the eotaxin-3 promoter in IL-13–treated TE-7 cells compared to IL-13–treated TE-1 cells (Fig. 3, A, Right panel). As a negative control, IL-13 did not affect STAT-6 recruitment to the 4 kb upstream from the eotaxin-3 promoter (Fig. 3, A, Right panel). Western blot confirmed that similar amounts of STAT-6 protein expression are expressed in TE-1 and TE-7 cells (Fig. 3, A, Left panel). After IL-13 stimulation in TE-7 cells, CpG 2 was less methylated by 30%, whereas no effect was seen at the other CpG sites (Fig. 3, B). These data reveal dynamic methylation of the eotaxin-3 promoter, particularly at CpG 2, by IL-13.
Figure 3.
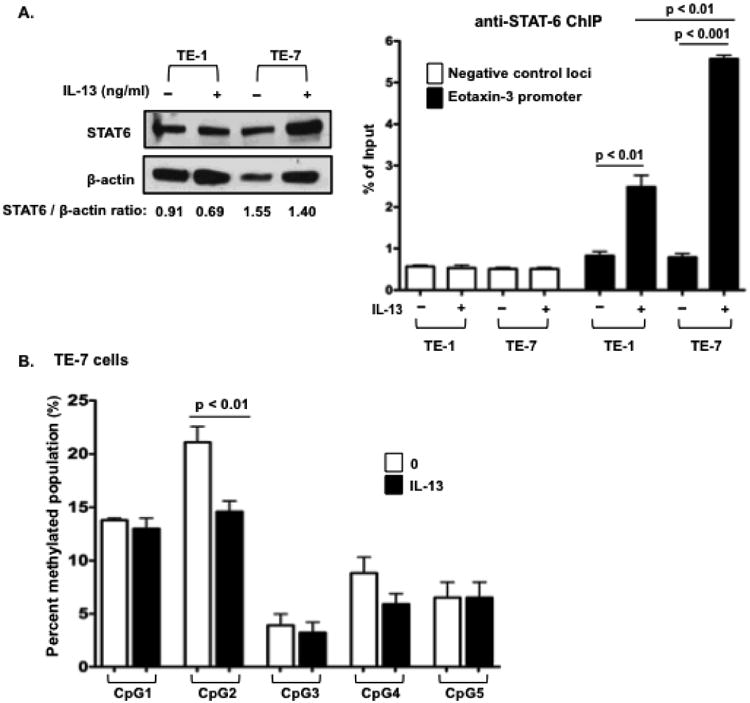
IL-13 influences eotaxin-3 expression and promoter methylation pattern. A. ChIP analysis with anti-STAT-6 antibody in untreated or IL-13–treated TE-1 and TE-7 cells was performed to determine the binding of STAT-6 to the eotaxin-3 promoter. Quantitative ChIP analysis was performed using real-time PCR. A ChIP primer set to a negative control locus within the 4 kb upstream from transcription start site of eotaxin-3 was used to measure background binding. The results shown are the mean values (± SEM, A, Right panel). TE-1 and TE-7 cells were assessed by immunoblot for STAT-6 and β-actin (A, Left panel). The protein levels were quantified by densitometric analysis. B. To investigate the effect of IL-13 on methylation pattern of eotaxin-3, esophageal epithelial cells were treated with IL-13 (100 ng/ml) for one doubling time. Eotaxin-3 DNA methylation levels in TE-7 cells were analyzed by pyrosequencing. Data are given as mean ± SEM; n = 3 replicates per group per experiment. Data shown are a representative experiment of at least three independent experiments.
Induced demethylation of the eotaxin-3 promoter in esophageal epithelial cells increases eotaxin-3 gene expression
DNA methylation can be pharmacologically manipulated by induction of DNA replication in the presence of DNA methyltransferase inhibitors, such as 5-azacytidine. To further investigate demethylation of the CpG sites of the eotaxin-3 promoter, we exposed cell lines expressing various levels of eotaxin-3 to 5-AzaC. TE-1 (Fig. 4, A) and TE-7 cells (Fig. 4, B) were treated with 5-AzaC for one population doubling. In both TE-1 and TE-7 cells, global levels of methylation were reduced (data not shown), and eotaxin-3 transcription was significantly upregulated in TE-1 and TE-7 cells exposed to 5-AzaC relative to control cells (Fig. 4, A-B, left panel). In both esophageal cell lines, increased expression of eotaxin-3 in cells treated with 5-AzaC was accompanied by demethylation of the eotaxin-3 CpG sites relative to control cells (Fig. 4, A-B, right panel). These data demonstrate that 5-AzaC treatment results in CpG site demethylation of the eotaxin-3 promoter with a concomitant increase in gene expression.
Figure 4.
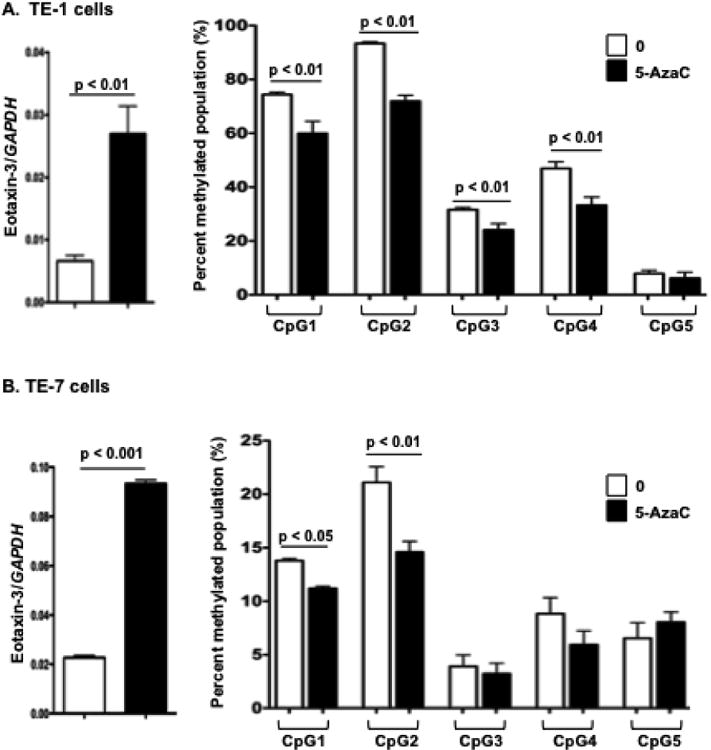
5-AzaC-induced demethylation of CpG sites increases eotaxin-3 gene expression. The mRNA expression and methylation status of eotaxin-3 in TE-1 (A) and TE-7 (B) cells that were treated with 5-AzaC (0.5 μM). Data are given as mean ± SEM; n = 3 replicates per group per experiment. Data shown are a representative experiment of at least three independent experiments.
Suppression of eotaxin-3 promoter activity by methylation of the CpG 2 site
The data presented thus far demonstrate strong associations between CpG 2 site methylation and eotaxin-3 expression. We aimed to extend these findings by proving causality between these events. Accordingly, we examined the direct effect of methylation on the transcription of eotaxin-3 using a transient reporter assay in HEK293 cells in the presence or absence of IL-13. The indicated eotaxin-3 promoters were methylated in vitro with the enzyme M.SssI and its substrate S-adenosylmethionine (SAM) and inserted into an unmethylated plasmid containing the luciferase gene (pCpGL) (23). The CpGs in the pCpGL-eotaxin-3 plasmid insert were enzymatically methylated before transfection (Fig. 5, A). The methylated and unmethylated eotaxin-3 promoter reporters were cotransfected with STAT-6 expression vector (Fig. 5, B, left panel) into HEK293 cells, where they maintained their particular CpG methylation status for 30 hrs (Fig. 5, B, right panel). A transient reporter assay showed 31.3 ± 6.9% (mean ± SEM, p < 0.05) and 51.6 ± 5.0% (mean ± SEM, p < 0.001) reduction in luciferase activity following the methylation of constructs in the absence or presence of IL-13, respectively (Fig. 5, C). To further prove the direct effect of CpG 2 site methylation on eotaxin-3 promoter activity, we generated eotaxin-3 promoter constructs in which the cytosine present in each CpG site was individually mutated to an adenine, preventing methylation at each specific locus (Fig. 5, D). Whereas mutation of CpG sites 3, 4, and 5 did not alter the repressive effect of methylation, mutation of the cytosine at the CpG 2 site completely abolished the ability of methylation to repress eotaxin-3 promoter activity (Fig. 5, E).
Figure 5.
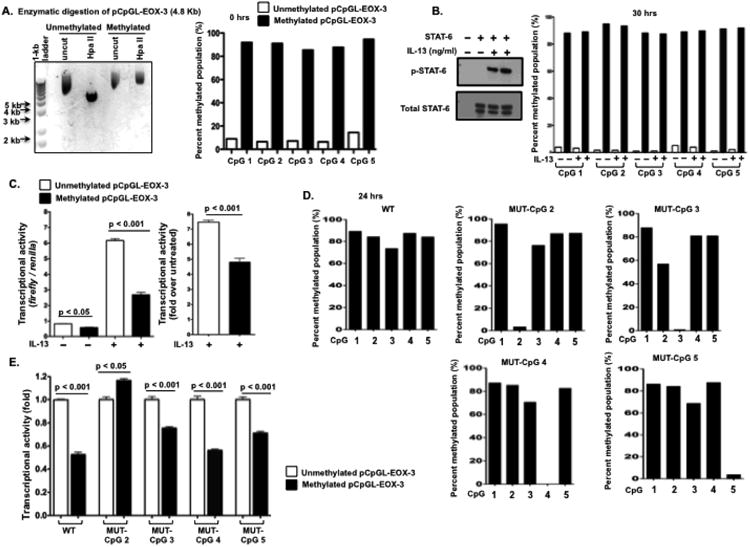
Methylation of the CpG 2 site of the eotaxin-3 promoter inhibits transcriptional activity and abrogates the transcription factor binding. A-C. Expression vectors of the transcription factor STAT-6 were cotransfected with pCpGL-eotaxin-3 methylated and unmethylated promoter vectors to activate promoter luciferase activity in HEK293 cells. A. Enzymatic methylation of the CpGs in the pCpGL-eotaxin-3 plasmid using M. SssI CpG methyltransferase was confirmed by enzymatic digestion of the plasmid followed by gel electrophoresis. Unmethylated (first two lanes) or methylated (last two lanes) pCpGLeotaxin-3 were either undigested or digested with Hpa II alone. Hpa II specifically digests the unmethylated CCGG that is in the central 4 bases of the CpG 1 site. The gel shows that Hpa II does not digest the plasmid when the CpGs in the eotaxin-3 were enzymatically methylated (A, left panel). Bisulfite pyrosequencing of transfected pCpGL-eotaxin-3 reporter plasmid shows that the enzymatically methylated plasmid retains CpG methylation before (A, right panel) and after transfection for 30 hrs (B, left panel) in HEK293 cells. After transfected HEK293 cells had been cultured for 24 hrs, they were treated with IL-13 for 6 hrs (B and C). C. Cellular proteins from unstimulated and IL-13–stimulated, pCpGL-eotax-3/STAT-6 transfected HEK293 cells were only observed upon ectopic expression of functional STAT-6 protein as assessed by immunoblot for tyrosine-phosphorylated STAT-6 (pSTAT-6), STAT-6 (B, left panel). The cells were lysed and the luciferase activity in the cell lysates was determined. Relative expression of unmethylated (open bars) and methylated (solid bars) eotaxin-3 promoter constructs after normalization to renilla activity (C, left panel) and fold change (C, right panel) over untreated. D-E. Effect of methylation of eotaxin-3 promoter on wild type (WT) and mutated (MUT) eotaxin-3 promoter activity. The pCpGL-eotaxin-3 WT or other MUT constructs with individual cytosines mutated to adenosines at the position CpG 2, CpG 3, CpG 4, or CpG 5 (D). E. Relative expression of unmethylated (open bars) and methylated (solid bars) eotaxin-3 promoter constructs after normalization to renilla activity.
In vitro methylation of the CpG 2 site abrogates the binding of ATF-2 and CBP
To investigate why methylation of the CpG 2 site inhibits transcription, an EMSA was performed to examine the effects of CpG methylation on the binding of transcription factors. Previous observations have shown that CBP cooperates with STAT-6 as a key regulator of eotaxin-3 transcription (14, 18). Therefore, we performed EMSA with an oligonucleotide containing the CBP- and ATF-2-binding site (CRE) and the CpG 2 site of eotaxin-3 promoter (CpG 2 probe). This identified a specific retarded band when nuclear extracts from TE-7 cells were assayed (Fig. 6A, lane 2). Preincubation of nuclear extract with antibodies against CBP or ATF-2 prevented appearance of the retarded band, showing that CBP and ATF-2 specifically bound to the CpG 2 probe (Fig. 6B, lane 4-5). Notably, this band was not observed when a methylated CpG 2 probe was used (Fig. 6B, lane 5-6). IL-13 increased the binding of both CBP and ATF-2 to the CpG 2 probe compared to untreated cells (Fig. 6C, lane 2). These results indicate that the methylation of the CpG 2 site abrogates the binding of both CBP and ATF-2 transcription factors to the eotaxin-3 promoter.
Figure 6.
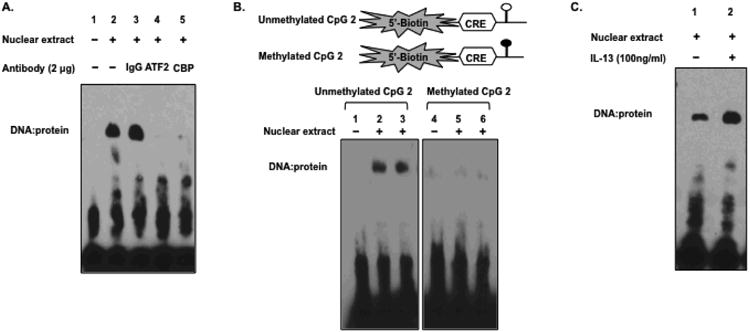
Identification of proteins interacting with the CpG 2 site using EMSA. A. Nuclear extracts prepared from TE-7 cells were incubated using antibodies to CBP and ATF-2 protein. B. CpG 2 site methylation inhibits the binding of factors to the CpG 2 site and the cyclic AMP-responsive element (CRE) site as measured by EMSA. Nuclear extracts were incubated with double-stranded oligonucleotide probes containing either an unmethylated cytosine or methylated cytosine at the CpG 2 site. C. The effect of IL-13 on binding proteins to the CpG2 site.
Discussion
Collectively herein, we have demonstrated that methylation of the eotaxin-3 promoter, specifically at the CpG 2 site, has a key role in basal and IL-13–induced eotaxin-3 production in esophageal epithelial cells. We elucidated epigenetic characteristics of human primary epithelial cells derived from human allergic inflammatory tissue. In particular, methylation of the CpG 2 site was significantly decreased in patient-derived epithelial cells compared with normal control epithelial cells. Levels of methylation at the CpG 2 site inversely correlated with basal and IL-13–induced eotaxin-3 gene expression in epithelial cell lines (TE-1, TE-7). Conversely, global inhibition of methylation with 5-AzaC promoted eotaxin-3 production. In addition, the basal and IL-13–induced eotaxin-3 transcriptional activity was suppressed in vitro by DNA methylation of the eotaxin-3 promoter. Furthermore, we identified that CBP and ATF-2 binding to the CRE site eotaxin-3 promoter was methylation dependent.
One of the hallmarks of allergic inflammation is tissue eosinophilia. The eosinophil chemoattractant eotaxin-3 is well known to be up-regulated in a variety of allergic inflammatory states. Herein, we investigated whether differential DNA methylation may be involved in the acquisition of eotaxin-3 expression in primary esophageal epithelial cells. Our results are compatible with the notion that methylation is important in the control of eotaxin-3 production and could contribute to the altered production of this chemokine as seen in the esophagus of patients with EoE. The methylation difference identified between patients with EoE and normal controls could be a primary phenomenon, causing an increased risk of developing allergic inflammation. Alternatively, the difference could be secondary to other processes triggered by the inflammation. We have analyzed the methylation on eotaxin-3 promoter in other cell types using the ENCODE database (26). We did not find eotaxin-3 promoter methylation in other cell types, but we found three CpG sites (cg113038391, cg18243003, cg23298782) in non-promoter regions. The methylation of cg11308391 was variable in different cells (data not shown).
The CpG 2 site is hypomethylated in TE-7 cells, which have with high basal eotaxin-3 mRNA levels, and hypermethylated in TE-1 cells, which have low basal eotaxin-3 mRNA levels. Demethylation by treatment with the DNA methyltransferase inhibitor 5-AzaC in TE-1 and TE-7 cells resulted in an increased eotaxin-3 gene expression. In addition, epithelial cell lines with eotaxin-3 hypomethylation (TE-7, 25% at the CpG 2 site) or hypermethylation (TE-1, 90% at the CpG 2 site) were shown to have different propensities to respond to IL-13. The IL-13–induced fold change of eotaxin-3 mRNA expression and protein was significantly higher in TE-7 cells than in TE-1 cells. Moreover, ChIP showed that STAT-6 preferentially binds to the eotaxin-3 promoter in IL-13–treated TE-7 cells compared to IL-13–treated TE-1 cells. After IL-13 stimulation, the CpG 2 site was less methylated by 30% in TE-7 cells. Thus, eotaxin-3 gene expression was likely dependent on the CpG 2 site demethylation of the eotaxin-3 promoter. We have measured levels of DNMT1, DNMT3A and DNMT3B mRNA, as these gene products are involved in the maintenance of DNA methylation. DNMT1 and DNMT3A mRNA levels were significantly decreased in TE-7 cells in comparison to TE-1 cells (data not shown), consistent with the methylation of the CpG2 site in these cells.
A CpG-free reporter system was used to assess the direct effect of differential DNA promoter methylation on the transcriptional activity of eotaxin-3. We generated a series of specific CpG-mutated constructs, and only mutation at the CpG 2 site prevented the downregulation of eotaxin-3 promoter activity by DNA methylation. It is important to point out that the methylation status of CpG 2 site (-225 C) in the native gene in the context of chromatin may behave differently than that in a transiently transfected (episomal) plasmid. Evidence of a functional interaction between CBP and ATF-2 and the eotaxin-3 promoter at the CpG 2 site was demonstrated by EMSA. Notably, this interaction occurred in a methylation-dependent manner and relied on the demethylation status of the CpG 2 site. The ability of specific CpG site low methylation level to govern gene expression by allowing CBP binding in promoter regions was demonstrated by using differentially methylated and unmethylated CRE-containing probes in EMSA (27). Methylation of this site inhibited the binding of CBP and ATF-2 to the eotaxin-3 promoter's CRE site and inhibited eotaxin-3 expression. ATF-2, a member of the leucine zipper family of DNA-binding proteins, is known to bind to the CRE site and to stimulate CRE-dependent transcription (28).In summary, our data highlight the role of DNA methylation in the regulation of eotaxin-3 in esophageal epithelial cells. We have identified a particular CpG site that is subjected to differential methylation and has a regulatory role in mediating eotaxin-3 expression. Mechanistically, CpG 2 methylation likely regulates binding of ATF-2 and CBP, which mediate IL-13–induced STAT-6–dependent eotaxin-3 production. Thus, changes in DNA methylation may be an early event that enhances transcription of eotaxin-3, leading to development of allergic inflammation.
Acknowledgments
The authors would like to thank Dr. Artem Barski for his insight, advice, and review of the data and manuscript; Dr. Matthew Weirauch for assistance with transcription factor binding analysis; and Shawna Hottinger for editorial assistance.
This work was supported in part by National Institutes of Health grants [2U19 AI066738, U19 AI070235, R01 DK076893, R37 AI1045898]; PHS Grant [P30 DK078392]; the Department of Defense; the Food Allergy Research & Education Foundation; the Buckeye Foundation; and the Campaign Urging Research for Eosinophilic Disease (CURED) Foundation.
Footnotes
Disclosures: The authors have no financial conflicts of interest
References
- 1.Ballestar E. Epigenetics lessons from twins: prospects for autoimmune disease. Clin Rev Allergy Immunol. 2010;39:30–41. doi: 10.1007/s12016-009-8168-4. [DOI] [PubMed] [Google Scholar]
- 2.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wallace DC. Bioenergetics and the epigenome: interface between the environment and genes in common diseases. Dev Disabil Res Rev. 2010;16:114–119. doi: 10.1002/ddrr.113. [DOI] [PubMed] [Google Scholar]
- 4.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Javierre BM, Hernando H, Ballestar E. Environmental triggers and epigenetic deregulation in autoimmune disease. Discov Med. 2011;12:535–545. [PubMed] [Google Scholar]
- 7.Ho SM. Environmental epigenetics of asthma: an update. J Allergy Clin Immunol. 2010;126:453–465. doi: 10.1016/j.jaci.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abonia JP, Rothenberg ME. Eosinophilic esophagitis: rapidly advancing insights. Annu Rev Med. 2012;63:421–434. doi: 10.1146/annurev-med-041610-134138. [DOI] [PubMed] [Google Scholar]
- 9.Blanchard C, Wang N, Rothenberg ME. Eosinophilic esophagitis: pathogenesis, genetics, and therapy. J Allergy Clin Immunol. 2006;118:1054–1059. doi: 10.1016/j.jaci.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 10.Noel RJ, Rothenberg ME. Eosinophilic esophagitis. Curr Opin Pediatr. 2005;17:690–694. doi: 10.1097/01.mop.0000184291.34654.be. [DOI] [PubMed] [Google Scholar]
- 11.Blanchard C, Mingler MK, Vicario M, Abonia JP, Wu YY, Lu TX, Collins MH, Putnam PE, Wells SI, Rothenberg ME. IL-13 involvement in eosinophilic esophagitis: transcriptome analysis and reversibility with glucocorticoids. J Allergy Clin Immunol. 2007;120:1292–1300. doi: 10.1016/j.jaci.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 12.Blanchard C, Stucke EM, Burwinkel K, Caldwell JM, Collins MH, Ahrens A, Buckmeier BK, Jameson SC, Greenberg A, Kaul A, Franciosi JP, Kushner JP, Martin LJ, Putnam PE, Abonia JP, Wells SI, Rothenberg ME. Coordinate interaction between IL-13 and epithelial differentiation cluster genes in eosinophilic esophagitis. Journal of immunology. 2010;184:4033–4041. doi: 10.4049/jimmunol.0903069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blanchard C, Wang N, Stringer KF, Mishra A, Fulkerson PC, Abonia JP, Jameson SC, Kirby C, Konikoff MR, Collins MH, Cohen MB, Akers R, Hogan SP, Assa'ad AH, Putnam PE, Aronow BJ, Rothenberg ME. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest. 2006;116:536–547. doi: 10.1172/JCI26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanchard C, Durual S, Estienne M, Emami S, Vasseur S, Cuber JC. Eotaxin-3/CCL26 gene expression in intestinal epithelial cells is up-regulated by interleukin-4 and interleukin-13 via the signal transducer and activator of transcription 6. Int J Biochem Cell Biol. 2005;37:2559–2573. doi: 10.1016/j.biocel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 15.Konikoff MR, Blanchard C, Kirby C, Buckmeier BK, Cohen MB, Heubi JE, Putnam PE, Rothenberg ME. Potential of blood eosinophils, eosinophil-derived neurotoxin, and eotaxin-3 as biomarkers of eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006;4:1328–1336. doi: 10.1016/j.cgh.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 16.Rothenberg ME, Spergel JM, Sherrill JD, Annaiah K, Martin LJ, Cianferoni A, Gober L, Kim C, Glessner J, Frackelton E, Thomas K, Blanchard C, Liacouras C, Verma R, Aceves S, Collins MH, Brown-Whitehorn T, Putnam PE, Franciosi JP, Chiavacci RM, Grant SF, Abonia JP, Sleiman PM, Hakonarson H. Common variants at 5q22 associate with pediatric eosinophilic esophagitis. Nat Genet. 2010;42:289–291. doi: 10.1038/ng.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherrill JD, Rothenberg ME. Genetic dissection of eosinophilic esophagitis provides insight into disease pathogenesis and treatment strategies. J Allergy Clin Immunol. 2011;128:23–32. doi: 10.1016/j.jaci.2011.03.046. quiz 33-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim EJ, Lu TX, Blanchard C, Rothenberg ME. Epigenetic regulation of the IL-13-induced human eotaxin-3 gene by CREB-binding protein-mediated histone 3 acetylation. J Biol Chem. 2011;286:13193–13204. doi: 10.1074/jbc.M110.210724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 21.Andl CD, Mizushima T, Nakagawa H, Oyama K, Harada H, Chruma K, Herlyn M, Rustgi AK. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem. 2003;278:1824–1830. doi: 10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 22.Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, Rossi DJ, Inlay MA, Serwold T, Karsunky H, Ho L, Daley GQ, Weissman IL, Feinberg AP. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–342. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klug M, Rehli M. Functional analysis of promoter CpG methylation using a CpG-free luciferase reporter vector. Epigenetics. 2006;1:127–130. doi: 10.4161/epi.1.3.3327. [DOI] [PubMed] [Google Scholar]
- 24.Ravnskjaer K, Kester H, Liu Y, Zhang X, Lee D, Yates JR, 3rd, Montminy M. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. 2007;26:2880–2889. doi: 10.1038/sj.emboj.7601715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lim EJ, Smart EJ, Toborek M, Hennig B. The role of caveolin-1 in PCB77-induced eNOS phosphorylation in human-derived endothelial cells. Am J Physiol Heart Circ Physiol. 2007;293:H3340–3347. doi: 10.1152/ajpheart.00921.2007. [DOI] [PubMed] [Google Scholar]
- 26.Consortium, E.P. A user's guide to the encyclopedia of DNA elements (ENCODE) PLoS biology. 2011;9:e1001046. doi: 10.1371/journal.pbio.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bui C, Barter MJ, Scott JL, Xu Y, Galler M, Reynard LN, Rowan AD, Young DA. cAMP response element-binding (CREB) recruitment following a specific CpG demethylation leads to the elevated expression of the matrix metalloproteinase 13 in human articular chondrocytes and osteoarthritis. FASEB J. 2012;26:3000–3011. doi: 10.1096/fj.12-206367. [DOI] [PubMed] [Google Scholar]
- 28.Seong KH, Maekawa T, Ishii S. Inheritance and memory of stress-induced epigenome change: roles played by the ATF-2 family of transcription factors. Genes Cells. 2012;17:249–263. doi: 10.1111/j.1365-2443.2012.01587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]