

Abstract
Purpose
Patients with Neurofibromatosis Type 1 (NF1) develop malignant peripheral nerve sheath tumors (MPNST) which are often inoperable and do not respond well to current chemotherapies or radiation. The goal of this study was to utilize comprehensive gene expression analysis to identify novel therapeutic targets.
Experimental Design
Nerve Schwann cells and/or their precursors are the tumorigenic cell types in MPNST due to the loss of the NF1 gene, which encodes the RasGAP protein neurofibromin. Therefore, we created a transgenic mouse model, CNP-HRas12V, expressing constitutively-active HRas in Schwann cells and defined a Ras-induced gene expression signature to drive a Bayesian factor regression model analysis of differentially expressed genes in mouse and human neurofibromas and MPNSTs. We tested functional significance of Aurora kinase over-expression in MPNST in vitro and in vivo using Aurora kinase shRNAs and compounds that inhibit Aurora kinase.
Results
We identified 2000 genes with probability of linkage to nerve Ras signaling of which 339 were significantly differentially expressed in mouse and human NF1-related tumor samples relative to normal nerves, including Aurora kinase A (AURKA). AURKA was dramatically over-expressed and genomically amplified in MPNSTs but not neurofibromas. Aurora kinase shRNAs and Aurora kinase inhibitors blocked MPNST cell growth in vitro. Furthermore, an AURKA selective inhibitor, MLN8237, stabilized tumor volume and significantly increased survival of mice with MPNST xenografts.
Conclusion
Integrative cross-species transcriptome analyses combined with preclinical testing has provided an effective method for identifying candidates for molecular-targeted therapeutics. Blocking Aurora kinases may be a viable treatment platform for MPNST.
Keywords: Neurofibroma, NF1, MPNST, Ras, Aurora Kinase
Introduction
MPNSTs are aggressive soft tissue sarcomas that frequently metastasize and rarely respond to chemotherapy. Patients with Neurofibromatosis type 1 (NF1) inherit mutations in the NF1 gene and are predisposed to developing MPNST, identified in approximately 10% of NF1 patients. MPNSTs, having a 20 – 50% five-year survival rate, are the major cause of mortality in adult NF1 patients (1). Approximately 50% of MPNST cases are sporadic, and some sporadic MPNSTs have mutations in the NF1 gene (2). The most common manifestation of NF1 is the development of benign peripheral nerve sheath tumors. Approximately 95% of NF1 patients harbor smaller benign dermal neurofibromas, and at least 30% of NF1 patients develop larger benign plexiform neurofibromas, typically associated with deeper nerve trunks. It is believed that plexiform neurofibromas can transform to malignant peripheral nerve sheath tumors (MPNSTs) (1)
The protein encoded by the NF1 gene, neurofibromin, is a RasGAP for all Ras isoforms, negatively regulating the Ras signal transduction pathway by accelerating the hydrolysis of active Ras-GTP to inactive Ras-GDP (1). Therefore, having loss of function mutations in NF1, cells isolated from neurofibromas (3) and NF1-derived MPNSTs (4, 5) have elevated levels of Ras-GTP. While in benign tumors, prolonged Ras activation can be associated with oncogene-induced senescence, in malignancy aberrant activation of Ras signaling generally leads to promotion of tumor cell proliferation and/or survival (6). It is unclear which Ras isoforms are essential in NF1 tumorigenesis, and the effects of specific isoforms appear to be cell type dependent (7).
Interestingly, NF1-associated MPNSTs and sporadic MPNSTs show similar pathology and share similar gene expression signatures, possibly indicating a convergence to similar pathways downstream of Ras signaling (8, 9). Multiple effector pathways lie downstream of Ras, many of which have been implicated in NF1 tumorigenesis (1). Neurofibromas and MPNSTs from NF1 patients and mouse models with Nf1 mutations have elevated levels of phosphorylated ERK (Jessen et al, in revision), the downstream effector of the canonical Ras/Raf/MEK/ERK pathway. While a MEK inhibitor reduced tumor volume in a neurofibroma mouse model, a more modest reduction in human MPNST cell survival in xenografts was observed (Jessen et al, In press). The mTOR signaling pathway is also activated in MPNST, but blocking mTOR signaling with rapamycin or its analogs only transiently delayed tumor growth (10, 11).
MPNSTs have a complex karyotype with amplifications and deletions of many alleles, some of which may represent therapeutic targets. Multiple studies have identified individual molecular alterations in addition to NF1 mutation in MPNST (1). Early alterations in premalignant tumors have included loss of the CDKN2A/B locus which normally encodes proteins that negatively regulate the cell cycle (12), and loss of function mutations in the common tumor suppressor gene TP53 have been frequently observed (1). Somatic inactivation of additional tumor suppressor genes has been used to model MPNSTs in mice. Null mutations in Nf1 and p53 in cis produced tumors in mice characteristic of human MPNST (GEM-PNST) (13, 14), and mice with targeted mutations at the Nf2 locus driven by P0B-Cre in conjunction with p53 or Nf1 mutations develop GEM-PNSTs (MG + MNK, unpublished). Amplification and/or overexpression of potential oncogenes, especially those encoding receptor tyrosine kinases, has also been implicated in NF1 tumorigenesis, including PDGFR, CKIT, EGFR (1), and IGF1R (15).
Despite these significant contributions to understanding the molecular etiology of NF1, thus far, no chemotherapeutic approach blocking any molecular target, including growth factor receptors upstream of Ras, Ras itself, Ras downstream effectors, or combinations of targets, has prevented or arrested neurofibroma formation or more than transiently delayed MPNST growth (16). However, a recent combinatorial study including rapamycin, an inhibitor of mTOR downstream of Ras, and an HSP90 inhibitor, enhancing proteotoxic stress, showed synergistic efficacy in the Nf1;p53 cis MPNST mouse model (17). The results of this study suggest that combining a Ras pathway inhibitor with a cytotoxic agent may be an effective treatment strategy for MPNST, an idea not yet tested in human clinical trials.
As additional candidates for MPNST chemotherapies are needed, we focused on molecular alterations downstream of H-Ras activation in Schwann cells, utilizing a Schwann cell-specific H-Ras gene expression signature derived from a novel transgenic mouse model to identify mechanisms contributing to tumorigenesis and potential therapeutic targets in NF1 tumors. We identify overexpression and amplification of a Ras target gene, AURKA, in MPNSTs. Furthermore, we show that blocking AURKA diminishes MPNST cell growth in vitro and in vivo.
Materials and Methods
CNP-HRas12V expression analysis
The CNP-HRas12V data set consisted of 8 control nerves encompassing 2 genotypes (4 Nf1 flox/flox; 4 P0CreB) and 6 CNP-HRas12V nerves. All statistical comparisons and data visualization were performed using GeneSpring GX v7.3.1 (Agilent Technologies). All statistical tests were corrected for multiple testing effects by applying the Benjamini and Hochberg false discovery rate correction. We used a T-Test and FDR ≤ 0.01 to compare groups.
Bayesian sparse latent class factor modeling
Gene orthologs of transcripts differentially expressed in CNP-HRas12V peripheral nerves were used as seeds in a Bayesian factor regression modeling analysis. The model includes 6 categorical responses: four species-specific tumor states (mouse neurofibroma, mouse MPNST, human neurofibroma, human MPNST) and two species-independent tumor states (neurofibroma, MPNST). By design, the 6 categorical factors are associated with the phenotypic outcome of NF1 tumor progression. Using a threshold of 0.85 for factor inclusion probability and 0.95 for variable inclusion probability, and constraining such that a minimum of 10 genes are required to exceed the factor threshold to include that factor in an expanded model, the analysis terminated with a model on 2000 gene orthologs distributed across k = 100 latent factors and 6 categorical response factors.
Gene interaction network
A total of 339 gene orthologs from the 2,000 genes linked to Ras signaling as determined by Bayesian factor regression modeling analysis were also identified as similarly regulated transcripts using frequentist statistics (Supplementary Fig. S1). We used GeneGo’s MetaCore to construct a gene interaction network using the direct interactions network building algorithm, which resulted in a single multi-node connected network of 85 gene orthologs.
AURKA locus copy number analysis
For SNP-array analysis, raw data from Illumina’s cnv370-duo and h660w-quad SNP array chips were processed using GenomeStudio (Ilumina). B-allele frequency (BAF) and Log Ratio (LRR) were calculated using the reference cluster file provided by Illumina. qPCR analysis of DNA, AURKA gene amplification was determined by quantitative PCR performed on a LightCycler® 480 Instrument (Roche Applied Science), using Universal Probe Library (UPL) technology. A total number of 37 samples were analyzed: 13 MPNSTs, 5 MPNST-derived cell lines, 8 neurofibromas and 11 normal samples (which presented a diploid status of AURKA and were used as controls). For additional details see Supplementary Materials and Methods.
Lentiviral shRNA infection
MPNST cell lines were cultured as described (8, 18). For lentiviral shRNA infection, MPNST cells at 50 – 60% confluence were infected with lentiviral particles containing shRNAs targeting AURKA, AURKB or shNon-targeting (SigmaAldrich; TRC library). The CCHMC Viral Vector Core produced virus using a 4-plasmid packaging system (http://www.cincinnatichildrens.org/research/div/exp-hematology/translational/vpf/vvc/default.htm). Lentiviral particles were incubated with MPNST cells (MOI ~ 10) in the presence of polybrene (8 μg/mL; Sigma) for 24 hours followed by selection in 2 μg/mL puromycin.
Quantitative real time PCR (QRT)-PCR
Total RNA was isolated from cells using the RNeasy kit (Qiagen) and used as a template for cDNA synthesis (High-Capacity cDNA archive kit, Applied Biosystems) and QRT-PCR (ABI 7500 Sequence Detection System) as described (8). Primers for AurkA were forward-TAGCCTTCAAGTGCCAGAGTGTGT and reverse-ACAAGACCCTGAATGGGTGTCACT and for AurkB forward-TGAGGAGGAAGACAATGTGTGGCA and reverse-AGGTCTCGTTGTGTGATGCACTCT.
Western Analysis
Cell lysates were created and Western blotting conducted as described (8), membranes were probed with several different antibodies including anti-AurkA antibodies (Cell Signalling Technology Inc., #3092), anti-AurkB antibody (Cell Signalling Technology Inc., #3094), anti-Histone 3 (Cell Signalling Technology Inc., #9715), anti-p-Histone 3 (Cell Signalling Technology Inc., #9701), anti-PARP (Cell Signalling Technology Inc., #9542) and then, stripped and re-probed with anti-β-actin antibodies (Cell Signaling Technology, Inc. #4967) as a loading control. Signals were detected using horseradish peroxidase-conjugated secondary antibodies (BioRad; Hercules, CA) and the ECL Plus developing system (Amersham Biosciences; Piscataway, NJ).
In vitro cytotoxicity assay
MPNST cell lines were plated in quadruplicate for each dose of Aurora kinase inhibitor on 96-well plates at 500 cells per well in serum-containing growth medium. Plates were incubated at 37°C and 5% CO2. 24hr after plating, cells were treated with carrier alone (10% fetal bovine serum (Hyclone) in DMEM) or inhibitors (LC laboratories, MA). MLN8237 compound was provided by Millennium Pharmaceuticals, Inc., Landsdowne Street, Cambridge, MA 02139. The amount of proliferation was quantified at 2 and 4 days post addition of drug by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay using Cell titer 96 proliferation kit (Promega). Absorbance was read at 490 nm in a Spectramax M2 plate reader (Molecular Devices).
MPNST xenograft
The subcutaneous NF1−/− S462-TY human MPNST xenograft model has been previously described (18). S462TY MPNST cells (1.8 × 106) from an NF1 patient were injected into four to five week old female nude mice (Harlan; Indianapolis, IN). Administration and dose of MLN8237 was determined by previous animal studies (19). Oral gavage of vehicle that constituted of 10% 2-hydroxypropyl-β-cyclodextrin (Fisher Scientific)/1% sodium bicarbonate (Sigma Aldrich) in sterile water or 20 mg/kg MLN8237 in vehicle was given twice daily Monday through Friday and once daily Saturday and Sunday beginning when tumors reached 250mm3. We measured tumors and weighed mice twice weekly. Tumor volume was calculated by: L * W2 (π/6), where L is the longest diameter and W is the width. Mice were treated until tumors reached 2500 mm3 or a maximum of 66 days. Control tumors (9/11) reached 2500mm3 by day 66 of treatment. The remaining 2 mice were >1800 mm3 and actively growing. Survival was analyzed by log-rank test using GraphPad Prism.
Pharmacokinetics
Blood was collected by cardiac puncture in non-tumor bearing mice. Blood was drawn at 2, 4, and 16 hours after dosing from 3 – 5 mice per time point. Plasma was separated by centrifugation at 2200 × g for 10 minutes and MLN8237 concentration quantified by LC/MS/MS (19, 20).
Immunohistochemistry
Teased sciatic nerves from adult wild type or CNP-HRAS12v mice were prepared for immune labeling as described (21). Schwann cells were labeled with anti-S100β antibodies (Dako, Carpenteria, CA) followed by FITC-conjugated secondary antibodies (green); expression of the HA-tagged CNP-HRAS12V transgene was detected with anti-HA antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) followed by TRITC-conjugated secondary antibodies (red).
Paraffin sections were de-paraffinized, hydrated and transferred to 0.1M citrate buffer (pH 6.0) for antigen retrieval. Slides were boiled for 10′ in citrate buffer, cooled at room temperature for 30 minutes, rinsed in water twice and in PBS 3 times. Sections were quenched with 3% hydrogen peroxide for 10 minutes, rinsed in PBS, and blocked in 10% normal goat serum with 0.3% Triton-X-100. Sections were incubated overnight in primary antibody; rabbit cleaved caspase-3 (1:8000, Cell Signaling, #9661), rabbit p-histone 3 (1:800, Cell Signaling, #9701), Rabbit Ki-67 (1:5000 NCL-Ki67-P; Novocastra), rabbit cyclinB1 (1:500, Cell Signaling, #4138). Sections were then incubated in biotinylated goat anti-rabbit secondary antibodies (Vector, at a dilution of 1:200). Sections were counterstained with Harris hemotoxylin and cover glassed in histo-mount. For nuclear staining, sections were incubated with propidium iodide/RNase staining solution (Cell Signaling, #4087) for 15 minutes at room temperature in the dark, rinsed in PBS 3 times and then cover glassed with Prolong® Gold Antifade Reagent (Life Technologies, #P36930). Sections were photographed on the Zeiss LSM 510 scanning confocal microscope equipped with an argon-ion multi line, HeNe 543, and HeNe 633 lasers. For nuclear staining with DAPI (Sigma), sections were incubated in DAPI (1:10,000) for 5 minutes, rinsed in PBS 3 times, and cover glassed in Flouromount G (EM Sciences, #17984-25) mounting medium.
Results
Bayesian Factor Model Analysis and Latent Structure Linked to Ras Signaling
Previous cross-species transcriptome analysis of mouse and human NF1 gene expression data suggested that NF1 loss results in up-regulation of genes that suppress Ras/Raf/MEK/ERK signaling (Jessen et al., In press), consistent with a negative feedback loop. It therefore seemed plausible that specific Ras targets would be re-expressed in the setting of MPNST to overcome this suppressive signaling. Ras signaling can be cell type dependent (7), and global transcriptional changes directly downstream of Ras activation specifically in Schwann cells or their precursors, the pathogenic cell type(s) in NF1 (22, 23), had not been reported previously. Therefore, to focus our analysis, we first identified genes in peripheral nerves associated with Ras-GTP expression in Schwann cells.
The starting point for our analysis was to generate a mouse expressing a constitutively active, oncogenic allele of Ras (HRAS12V) in Schwann cells, driven by the CNP promoter (24). The CNP promoter drives expression early in Schwann cell progenitors, and expression is maintained in mature Schwann cells (24). We chose to express H-Ras (in contrast to K- or N-Ras) as all three Ras proteins are expressed by mouse Schwann cells (25) yet the proliferation of Nf1 mutant Schwann cells in vitro is blocked by farnesyl protein transferase inhibitors, which affect H- but not N- or K-Ras (26). The expression of the HA-tagged HRAS12V transgene was validated in myelinating and non-myelinating Schwann cells using an anti-HA antibody by immunolabeling teased sciatic nerve preparations (Fig. 1A). These mice were viable and fertile, lived a normal lifespan, and did not form neurofibromas or MPNSTs. No peripheral nerve defects were noted on gross or pathologic examination. Analysis of CNP-HRAS12V expressing peripheral nerve by electron microscopy shows unmyelinated fiber bundles that are slightly disorganized (Fig. 1B) with a minor decrease in the number of axons per Remak bundle (Fig. 1C). The phenotype resembles a milder version of the nerve disruption observed in CNP-EGFR mice (27), and the phenotype was not enhanced in the Nf1+/− background (data not shown).
Figure 1. Schwann Cell-specific Ras-driven Gene Expression.

(A) Immunolabeling adult teased nerve fibers detected HRas (as detected by anti-HA, red) at the membrane of myelinated fibers (bottom: white arrow) and smaller unmyelinated fibers (top: white arrow). Schwann cell cytoplasm is counterstained with anti-S100 (green), which preferentially labels myelinating Schwann cells. (B) Electron micrographs of a cross-section through saphenous nerves from adult wild type or CNP-HRAS12V mice are shown. At high magnification, the unmyelinated axons with accompanying Schwann cell processes in the HRAS12V mutant showed normal ensheathment but few fibers per bundle. Groups of unmyelinated axons in sapphenous nerve contained diminished numbers of axons in the CNP-HRAS12V mutant. (C) At 1 year old, 13.0 +/− 2.2 axons were grouped by a single non-myelin forming Schwann cell in wild type mice, but only 6.4 +/− 0.9 in CNP-V12Ras mutants (P ≤ 0.004). Black arrows point to small groups of unmyelinated axons in mutant. The phenotype did not worsen with age (not shown). (D) Schwann-cell specific Ras signature in CNP-HRAS12V transgenic mouse model.
We identified 308 genes differentially expressed between mouse nerves from the CNP-HRas12V and normal control mouse nerves (FDR ≤ 0.01) (Fig. 1D). We did not observe significant functional enrichment of genes involved in Ras-MAPK (Raf/MEK/ERK) signaling or senescence after querying the 154 orthologs in the DAVID database or the GATACA knowledgebase.
Transcriptional changes in CNP-HRas12V nerves compared to wild type nerves downstream should result (directly or indirectly) from altered Ras signaling in Schwann cells. These Schwann cell-specific Ras pathway gene orthologs were used as seeds in a Bayesian factor regression modeling analysis of an orthologous transcriptome dataset including mouse and human benign neurofibromas and malignant peripheral nerve sheath tumors (MPNST) (28). The model identified 2000 genes with probability of linkage to Ras signaling (Fig. 2A).
Figure 2. AURKA is Overexpressed and Amplified in MPNST.

(A) Bayesian latent factor regression modeling of microarray gene expression data identified 2,000 gene orthologs linked to the Ras pathway in mouse and human NF1 tumors. (B) AURKA and AURKB were expressed at high levels in MPNST cell lines and primary tumors relative to neurofibromas and normal nerves. Each sample is represented by a vertical line and is directly under the sample type categories represented in (A). Sample types categories are separated by an open space. The vertical dashed line divides mouse samples (left) and human samples (right). Expression level of AURKA (top) or AURKB (bottom) was normalized to mouse or human normal nerve, respectively, and represented as 1.00 on a log scale. The red line represents the average expression level among samples within each sample type (C) Example of SNP-array analysis of an MPNST. This figure shows the log R ratio (LRR) of chromosome 20 for tumor MPNST K (see Supplementary Table S1). A blue line denotes the running average of LRR values. A value of 0 indicates the presence of two copies of each locus analyzed. A mean value above 0 indicates a copy number gain of the locus interrogated. A vertical yellow line localizes the AURKA locus.
To determine which, if any, of the 2000 gene orthologs show significant de-regulated expression in NF1 samples, we filtered the 2000 gene orthologs linked to Ras signaling across genes up-regulated or down-regulated in mouse or human neurofibroma or MPNST relative to respective normal nerve controls, as determined in previous studies (Jessen et al., In Press). A total of 339 gene orthologs showed increased or decreased expression in mouse model and human neurofibromas or mouse model and human MPNSTs (Supplementary Fig. S1).
We used the GeneGo MetaCore Systems Biology Analysis Platform MetaCore to explore the connections between the 339 genes and identified a major network (Supplementary Fig. S2). This network contained the over-expressed AURKA gene and its over-expressed downstream targets PLK1, Cyclin B1, TPX2, HEC and TACC3. Upstream activators of AURKA, E2F3 and FOXM1, as well as Securin (PTTG1), which regulates AURKA phosphorylation of Histone H3 within pericentromeric heterochromatin early in the G2 phase (29) and interacts with AURKA to regulate cellular responses to anti-neoplastic drugs (30), were also over-expressed genes within the network. Another prominent theme was decreased expression of PPAR-gamma and its downstream targets Leptin, CD36, Cytochrome B, RBP7, MGST, PREG3 and SCD. A decrease in gene copy number at the PPAR-gamma locus was not detected by SNP-array analysis (data not shown).
Notably, dramatic over-expression of AURKA (up-regulated 7.9-fold) was observed in the filtered mouse and human MPNST sample data as compared to normal nerve (Fig. 2B) as well as within the Ras-driven latent factor search results (Fig. 2A) and the original K-means clustering of the cross-species NF1 dataset (Jessen Miller et al., In press). Expression of AURKB was moderately increased in human MPNST (1.9-fold; Fig. 2B), and AURKC was not differentially expressed in either mouse or human tumor samples.
To assess the mechanism underlying high expression of AURKA, we determined copy number of the AURKA locus using SNP-array and qPCR analyses on DNA from dermal neurofibromas, MPNSTs and MPNST cell lines (Fig. 2C; Supplementary Fig S3, Tables S1 and S2). MPNSTs and MPNST cell lines but not neurofibromas showed copy number gains in the AURKA locus. All (5/5) MPNST cell lines tested, each previously analyzed by gene expression microarrays, and 3 of 14 (21%) primary MPNSTs (one analyzed by gene expression microarray, MPNST #19) exhibited ≥3n copies of the AURKA locus. The SNP-array technique can underestimate gene amplification in tumor cells (31) due to the presence of normal cells. Therefore, we used qPCR-DNA analysis to detect copy number gains with more sensitivity. Using this technique we confirmed copy number gains in 5/5 MPNST cell lines and detected copy number gains in 8 of 13 (62%) primary MPNSTs (data not shown). Increases in copy number were also identified in 1 of 8 neurofibroma samples, consistent with the low number of structural alterations in the AURKA locus in 19 dermal neurofibromas (32). These data suggest that amplification of AURKA could be an early event in progression to malignancy. Indeed, AURKA gene expression was elevated in some human neurofibroma samples (Fig. 2B).
Copy number alteration at the Aurora Kinase A locus correlated with variations in mRNA expression levels across tumor samples (Fig. 2B, C), indicating that gene amplification may contribute to AURKA overexpression in MPNST. This supports the idea that increased AURKA expression relative to AURKB and the other mitotically-regulated genes listed above is not simply due to comparing proliferating cells to non-proliferating normal nerve cells leading to a proliferative signature, but that AURKA is distinctly upregulated.
Reducing AuroraKinase Gene Expression Inhibits MPNST Cell Survival in vitro
To test whether Aurora kinases play a functional role in MPNST cells, AURKA or AURKB expression was blocked with lentiviral short hairpin RNAs (shRNAs). Three days after lentiviral infection of MPNST cells, shAURKA reduced AURKA mRNA expression 3.7-fold relative to a non-targeting shRNA (shNT); shAURKB reduced AURKB mRNA expression 5.8-fold (Fig. 3A). Down-regulated expression of each gene was specific with respect to the other. Expression of Aurora kinases A and B were confirmed at the protein level by western blotting in MPNST cells three days after lentiviral shRNA infection (Fig. 3B). Inhibiting either AURKA or AURKB expression with shRNA diminished expression of the specific aurora kinase (A or B) proteins respectively. Knocking down expression of either AURKA or AURKB reduced MPNST cell accumulation relative to an shNT control as analyzed at 5 days after lentiviral infection using phase contrast microscopy (Fig. 3C) or an MTS assay (Fig. 3D).
Figure 3. Blocking AURK gene expression reduces MPNST cell survival in vitro MLN8237 reduces AURKA activity and MPNST cell survival in vitro.

(A) AURKA expression was diminished by AURKA shRNA; AURKB expression was diminished by AURKB shRNA. Blocking each AURK expression at the RNA level was specific as determined by qRT-PCR. (B) Blocking expression of either AURK reduced protein levels of both as detected by Western blot. (C) Phase contrast images of MPNST cells showed a dramatic reduction in cell number after treating with shAURKA or shAURKB. (D) Quantification of cell accumulation by MTS assay showed decreased survival in response to shAURKA and shAURKB. (E) MPNST cells became senescent 48hrs post treatment with MLN8237 as compared to vehicle treated cells as seen in these phase contract images. (F) MLN8237 Aurora kinase A inhibitor reduced survival of all 5 MPNST cell lines tested at an average IC50 of 164nm. (G) Protein levels for AurkA, p-Histone3, total Histone 3, PARP and loading control β-actin for MPNST cells (S462TY) treated with 100nM, 300nM and 1μM of MLN8237 collected at 24hr and 48hr post treatment with drug. Untreated and vehicle treated were used as a negative control and shAURKA treated MPNST cells collected at 4 days post virus treatment serve as a positive control.
MLN8237 reduces AURKA activity and MPNST cell survival in vitro
These AURK gene expression knock down data suggested a role for aurora kinases in MPNST cell survival. To verify this finding, human MPNST cells were separately treated with three different aurora kinase inhibitors. VX-680 and ZM447439 have been shown to block Aurora kinase A, B and C at nanomolar concentrations (33). At concentrations defined for each inhibitor, both compounds effectively decreased cell survival (Supplementary Fig. S4A,B). In a 4-day assay, the IC50 values for VX-680 ranged from 40 nM (T265) to 110 nM (26T) (Supplementary Fig. S4A); the IC50 values for ZM447439 ranged from 450nM (T265) to 2000 nM (S462TY) (Supplementary Fig. S4B). A compound selectively targeting Aurora kinase A, MLN8237 (19), also significantly reduced MPNST cell survival with IC50 values ranging from 100 nM (S462) to 210 nM (ST8814) and averaging 164 nM (Fig. 3E). MPNST cells showed signs of senescence including enlargement in cell size and flattening of the cell body at 48hrs post treatment with 100nM MLN8237, as compared to vehicle treated cells (Fig. 3F). Previous studies reported an average IC50 of 32 nM for a panel of Ewing’s sarcoma and neuroblastoma cell lines (20). These values are well below the average trough concentration of 1uM reported in human phase I studies using the recommended phase 2 dose of 50 mg twice daily (34).
Inhibiting Aurora kinase A results in mitotic spindle defects, mitotic delay, and eventually apoptosis (35). Therefore, pharmacodynamic analysis of AURKA inhibitors often includes measuring markers of proliferation and apoptosis, including p-Histone 3 and Poly (ADP-ribose) polymerase (PARP), respectively (36). Since phosphorylation of the serine 10 residue of the N-terminal tail of histone H3 is crucial for chromosome condensation and cell-cycle progression (37), protein levels for p-Histone-3 were measured at 24hr and 48hr post treatment with MLN8237, in vitro. Although there was no change in total Histone 3, there was significant reduction in p-Histone 3 levels at 300nM and 1μM treated MPNSTs at both time points (Fig. 3G). Similar reduction in p-Histone 3 was observed for cells treated with lentiviral shRNA against AURKA (Fig. 3G), indicating a decrease in mitotic cells. In contrast, there was no measurable increase in cell death at either time point, as indicated by an absence of cleaved PARP, which was clearly visible after staurosporin exposure. Post treatment with MLN8237, AurkA protein levels increased as compared to vehicle-treated MPNST cell doses over 300nM at 24hrs post treatment and for all the doses at the 48hr post treatment time point. Increase in AurkA protein is likely due to the accumulation of cells in the G2/M phase post-treatment with the drug (Fig. 3G).
MLN8237 Inhibits MPNST Cell Growth In vivo
To test the effect of Aurora kinase inhibition on MPNST in vivo, S462TY MPNST cells derived from an NF1 patient were implanted into nu/nu mice and treated with MLN8237 (n=11) or vehicle control (n=11), at 20 mg/kg/dose, a dose 50% of the MTD in most mouse strains. Inhibiting Aurora kinase with MLN8237 diminished tumor volume (Fig. 4A, B, C) and increased survival (p=0.0005) (Fig. 4D). Notably only one of 11 mice treated with MLN8237 required sacrifice within a prolonged (66 day) treatment period (Fig. 4B).
Figure 4. MLN8237 reduces Tumor Volume and Increases Survival of Mice with MPNST Xenografts.

(A – C) Inhibiting Aurora kinase A diminished tumor volume. (D) Kaplan-Meier curve shows increased survival of mice with MPNST xenografts in response to MLN8237 (p=0.0005); median survival for vehicle-treated mice was 41 days; median survival for MLN8237-treated mice was undefined. (E) Pharmacokinetic analysis of MLN8237 in MPNST xenografts. Average blood plasma concentrations of MLN8237 (nM) in 3 control nude mice at 2, 4, and 16 hours after dosing are plotted on a log scale. Dashed line represents dose (1μM) required for Aurora kinase inhibition in vivo.
To confirm MLN8237 concentrations that inhibited MPNST xenograft growth were clinically relevant, pharmacokinetic analysis of MLN8237 was conducted (Fig. 4E). Mice were administered MLN8237 at 20 mg/kg b.i.d. for 5 days and blood plasma was collected at 2, 4, and 16 hours after dosing. Cmax was reached by 4 hours and measured 2.9μM; blood plasma levels reached 2.77 by 2 hours. The blood plasma levels measured at 2 and 4 hours are well above the 1μM dosage necessary to successfully inhibit Aurora Kinase A in vivo, as previously reported (19, 20).
MLN8237 Induces Cytomegaly in MPNST Xenografts
To define cellular effects that may contribute to MLN8237-induced tumor stasis, we examined cell cycle markers using immunohistochemistry on paraffin sections from MPNST xenografts. There was a marked decrease in the number of cells positively labeled for Ki67 in the MLN8237-tissue as compared to vehicle-tissue samples (Fig. 5A) indicating a reduction in actively proliferating cells. No difference was observed in number of cells labeled for cleaved caspase-3 in vehicle versus MLN8237-treated xenografts (Fig. 5A) indicating no effect on apoptosis due to MLN8237. In spite of a decrease in Ki67, there was no change in percent of cells immune-positive for phistone-3 in the vehicle versus MLN8237 treated samples, indicating possible arrest in the G2/M phase of the cell cycle. CyclinB1 was used as a marker of G2/M. There was an increase in cyclinB1 in the nucleus and cytoplasm of cells from MLN8237 treated xenograft samples as compared to vehicle, supporting the idea that the AURKA inhibitor caused tumor cells to stall in G2/M. Consistent with G2/M arrest xenografts treated with MLN8327 contained numerous enlarged cells with multiple nuclei, as observed with propidium iodide (Fig. 5B). Quantification of this effect indicated that 4 or 16 hours after the last dose of MLN8237, the population of xenograft tumor cells with multiple nuclei was significantly increased (Fig. 5B). As Aurora-A is a mitotic kinase that regulates mitotic spindle formation and segregation, the enlarged multinucleated cells indicate arrest in G2/M phase.
Figure 5. MLN8237 induces cytomegaly in MPNST Xenografts.
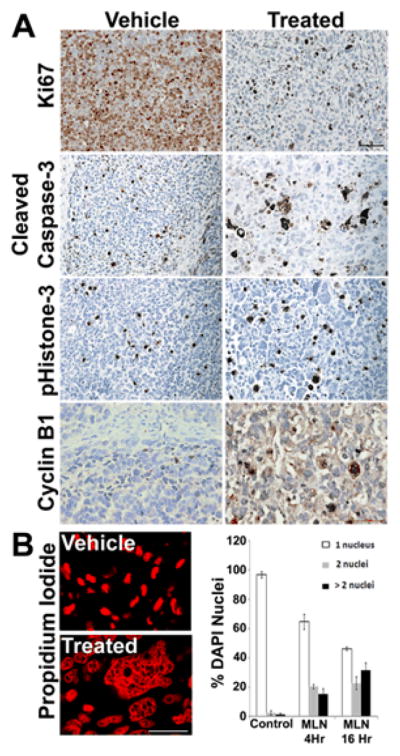
(A) MPNST xenografts treated with vehicle control or MLN8237 and assayed for Ki67, a marker of cell proliferation, p-Histone 3, a marker of proliferation, cleaved caspase 3, a marker for apoptosis, or cyclinB1, a marker of cell in the G2/M phase of the cell cycle. Vehicle and MLN8237-treated mice showed similar levels of p-histone 3 and cleaved caspase 3. Noticeably fewer cells were positively labeled for Ki67 in the MLN-8237 treated tissue samples as compared to Vehicle. There was a marked increase in the positive staining for CyclinB1 in the MLN8237 treated tissue samples compared to vehicle treated samples. (B) Many multinucleated cells were detected in MLN8237-treated xenografts, as visualized with propidium iodide and quantified by counting DAPI-stained nuclei per cell at 4 hours and 16 hours.
Discussion
The purpose of this study was to identify molecular alterations downstream of Ras signaling in Schwann cells relevant to NF1 tumorigenesis. We utilized a Schwann cell-specific Ras signature to drive Bayesian factor analysis, and gene expression data derived from human NF1 tumors and mouse Nf1 GEM models to identify Ras target genes that may lead to tumor progression. Gene amplification of the downstream MAPK1/ERK2 target AURKA resulted in AURKA over-expression in MPNST. MPNST cells were dependent on AURKA over-expression for survival, as inhibition of AURKA reduced cell survival in vitro and caused tumor stasis in vivo. The cumulative results of these experiments suggest that AURKA amplification contributes to NF1 mutant Schwann cell progression to malignancy. Our preclinical data support advancement of the AURKA specific inhibitor, MLN8237, as a molecular-targeted clinical candidate for the treatment of MPNST. Finally, our approach supports investigation of additional over-expressed Ras targets in NF1 tumorigenesis.
We identified 339 potential Ras target genes differentially expressed in mouse and human neurofibromas and/or MPNSTs relative to normal nerve. We hypothesized that expression of some or all of these specific Ras target genes might enhance the effects of the already activated Ras pathway, promoting tumorigenesis. Validating this idea, AURKA, a known transcriptional target of Ras-MAPK signaling (38), was identified by Bayesian analysis as a Schwann cell H-Ras target overexpressed in mouse and human MPNSTs but to a much lesser extent in neurofibromas.
Notably, AURKA can potentiate HRAS-mediated transformation correlating with increased phosphorylation of MEK and ERK (39, 40). Indeed, phosphorylated (p)-ERK is increased in MPNSTs relative to neurofibromas (Jessen et al., In press). While the exact mechanism by which AURKA and Ras pathways co-operate is unknown, it is intriguing that aurora kinases can directly interact with RasGAP, which is, like neurofibromin, an off signal for Ras proteins (41, 42). AURK genes encode Aurora kinases that regulate mitosis and promote cell cycle progression (43). AURKA, AURKB, and AURKC have each been implicated as oncogenes in multiple types of cancer (43), and AURKA was robustly over-expressed in our mouse and human MPNST datasets.
AURKA gene amplification provides a mechanism to increase AURKA expression, and AURKA amplification is often observed in cancer (43). The level of AURKA mRNA overexpression (10-fold) in MPNST is greater than the copy number gains (2 – 5 fold) detected at the AURKA locus, suggesting that additional mechanisms, perhaps including Ras/Raf/MEK/ERK signaling and/or the overexpression of AURKA upstream activators, contribute to elevated AURKA expression in MPNST. In some studies, AURKA gene amplification showed an inverse relationship with response to MLN8237 (48). In contrast, MLN8237 exhibited significant antitumor activity in our NF1−/− MPNST model with AURKA amplification. This model differed from those previously reported, which showed increased pHis-3 in response to MLN8237 and lacked the cytomegaly (48).
Aurora kinase inhibitors, like many anti-mitotic compounds, kill a variety of tumor cells, including some types of sarcomas (44, 45). MLN8237 has successfully treated leukemias that become resistant to the tyrosine kinase inhibitor, nilotinib (46) and enhanced cisplatin-induced cell death in esophageal cancer xenografts (47) and medulloblastoma cell lines (48). However, cells can become resistant to Aurora kinase inhibitors (44). Our preclinical studies using Aurora kinase inhibitors to diminish MPNST cell growth in vitro and in vivo provide evidence supporting investigation into effectiveness of Aurora kinase inhibitors in NF1 treatment. However, Aurora kinase inhibitors had a static effect on MPNST in vivo, and the addition of cytotoxic agents may be required. Importantly, clinical trials are ongoing using Aurora kinase inhibitors in the treatment of solid tumors (43). As Ras-driven tumors are often non-responsive to conventional chemotherapeutics, the development of combinatorial therapeutic regimens is essential (17). Combining Aurora kinase inhibitors with other therapeutics, including MEK inhibitors or chemo-sensitizing agents, may be useful. Given the cell cycle arrest in G2/M phase, compounds that target the G2 checkpoint may be attractive candidates.
In summary, our results support a molecular model in which amplification of AURKA drives cell cycle progression in NF1 mutant Schwann cells, promoting malignant transformation. Several lines of evidence make AURKA and attractive candidate for molecular-targeted MPNST therapeutics. AURKA expression is downstream of Ras signaling, and AURKA and several of its substrates are overexpressed in MPNST. Furthermore, several Aurora kinase inhibitors have shown efficacy in preclinical studies and clinical trials for a variety of tumors. The recent development and assessment of the AURKA-specific inhibitor, MLN8237, is highly effective against leukemias and solid tumors in a panel of preclinical models of pediatric cancers and has proceeded to clinical trials (20, 49). In our preclinical model, MLN8237 increases survival of mice with MPNST xenografts. The results of this study support further investigation of Aurora kinase inhibitors, alone or in combination, in the treatment of MPNST.
Supplementary Material
Statement of Translational Relevance.
Malignant peripheral nerve sheath tumors (MPNST) are aggressive sarcomas that arise sporadically or in the context of Neurofibromatosis Type 1 (NF1), especially in NF1 patients with benign plexiform neurofibromas. MPNSTs respond poorly to chemotherapy or radiation and are a major source of mortality in NF1 patients. No effective treatments for MPNST are proven, and molecular events contributing to malignant transformation of NF1 tumors are incompletely understood. However, identification of tumorigenic signaling pathways may facilitate the development of effective treatments. Using a combined gene expression database of mouse and human neurofibromas and MPNSTs, we identified overexpression of Aurora Kinases in MPNSTs. Furthermore, we provide in vitro and in vivo evidence supporting the use of Aurora kinase inhibitors in MPNST treatment strategies. The combination of cross-species transcriptome analyses with in vitro and in vivo MPNST models for preclinical testing provides a powerful translational pipeline for candidate molecular-targeted therapeutics into human clinical trials.
Acknowledgments
Financial Support: DAMD Program on Neurofibromatosis supported the NF1 Microarray Consortium (DODW81XWH- 09-1-0135 to NR); Children’s Tumor Foundation supported the NF1 Preclinical Consortium (TC and NR); WJJ was supported in part by an ARRA supplement to NIH grant R01-NS28840.
We thank Robert Hennigan for assistance with confocal microscopy. We also thank Jianqiang Wu and Edwin Jousma for assisting with administration of MLN8237 to mice and Michelle Meadows for assistance with MPNST cell culture.
Grant Support
We gratefully acknowledge support from the DAMD Program on Neurofibromatosis for the NF1 Microarray Consortium DODW81XWH- 09-1-0135. WJJ was supported in part by an ARRA supplement to NIH grant R01-NS28840 (to NR).
Footnotes
Conflicts of Interest: None
References
- 1.Katz D, Lazar A, Lev D. Malignant peripheral nerve sheath tumour (MPNST): the clinical implications of cellular signalling pathways. Expert Rev Mol Med. 2009;11:e30. doi: 10.1017/S1462399409001227. [DOI] [PubMed] [Google Scholar]
- 2.Bottillo I, Ahlquist T, Brekke H, Danielsen SA, van den Berg E, Mertens F, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol. 2009;217:693–701. doi: 10.1002/path.2494. [DOI] [PubMed] [Google Scholar]
- 3.Sherman LS, Atit R, Rosenbaum T, Cox AD, Ratner N. Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. Journal of Biological Chemistry. 2000;275:30740–30745. doi: 10.1074/jbc.M001702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:663–664. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 5.DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, et al. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (Type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- 6.Narita M, Lowe SW. Senescence comes of age. Nat Med. 2005;11:920–2. doi: 10.1038/nm0905-920. [DOI] [PubMed] [Google Scholar]
- 7.Carroll SL, Ratner N. How does the Schwann cell lineage form tumors in NF1? Glia. 2008;56:1590–605. doi: 10.1002/glia.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller SJ, Rangwala F, Williams J, Ackerman P, Kong S, Jegga AG, et al. Large-scale molecular comparison of human schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 2006;66:2584–91. doi: 10.1158/0008-5472.CAN-05-3330. [DOI] [PubMed] [Google Scholar]
- 9.Watson MA, Perry A, Tihan T, Prayson RA, Guha A, Bridge J, et al. Gene expression profiling reveals unique molecular subtypes of Neurofibromatosis Type I-associated and sporadic malignant peripheral nerve sheath tumors. Brain Pathol. 2004;14:297–303. doi: 10.1111/j.1750-3639.2004.tb00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansson G, Mahller YY, Collins MH, Kim MO, Nobukuni T, Perentesis J, et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol Cancer Ther. 2008;7:1237–45. doi: 10.1158/1535-7163.MCT-07-2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johannessen CM, Johnson BW, Williams SM, Chan AW, Reczek EE, Lynch RC, et al. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 12.Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Schoenaers J, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–32. doi: 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- 13.Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, et al. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- 14.Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–2179. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang J, Ylipaa A, Sun Y, Zheng H, Chen K, Nykter M, et al. Genomic and molecular characterization of malignant peripheral nerve sheath tumor identifies the IGF1R pathway as a primary target for treatment. Clin Cancer Res. 2011;17:7563–73. doi: 10.1158/1078-0432.CCR-11-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalamarides M, Acosta MT, Babovic-Vuksanovic D, Carpen O, Cichowski K, Gareth Evans D, et al. Neurofibromatosis 2011: a report of the Children’s Tumor Foundation Annual Meeting. Acta Neuropathol. 2011 doi: 10.1007/s00401-011-0905-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–13. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahller YY, Vaikunth SS, Currier MA, Miller SJ, Ripberger MC, Hsu YH, et al. Oncolytic HSV and erlotinib inhibit tumor growth and angiogenesis in a novel malignant peripheral nerve sheath tumor xenograft model. Mol Ther. 2007;15:279–86. doi: 10.1038/sj.mt.6300038. [DOI] [PubMed] [Google Scholar]
- 19.Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011;17:7614–24. doi: 10.1158/1078-0432.CCR-11-1536. [DOI] [PubMed] [Google Scholar]
- 20.Carol H, Boehm I, Reynolds CP, Kang MH, Maris JM, Morton CL, et al. Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer. Cancer Chemother Pharmacol. 2011 doi: 10.1007/s00280-011-1618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron. 1992;8:415–28. doi: 10.1016/0896-6273(92)90270-n. [DOI] [PubMed] [Google Scholar]
- 22.Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, et al. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–16. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng H, Chang L, Patel N, Yang J, Lowe L, Burns DK, et al. Induction of abnormal proliferation by nonmyelinating schwann cells triggers neurofibroma formation. Cancer Cell. 2008;13:117–28. doi: 10.1016/j.ccr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 24.Chandross KJ, Cohen RI, Paras P, Gravel M, Braun PE, Hudson LD. Identifcation and characterization of early glial progenitors using a transgenic selection strategy. Journal of Neuroscience. 1999;19:759–774. doi: 10.1523/JNEUROSCI.19-02-00759.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Y, Rangwala F, Fulkerson PC, Ling B, Reed E, Cox AD, et al. Role of TC21/R-Ras2 in enhanced migration of neurofibromin-deficient Schwann cells. Oncogene. 2004;23:368–78. doi: 10.1038/sj.onc.1207075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HA, Ling B, Ratner N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate : reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Molecular and Cellular Biology. 1997;17:862–872. doi: 10.1128/mcb.17.2.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ling BC, Wu J, Miller SJ, Monk KR, Shamekh R, Rizvi TA, et al. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell. 2005;7:65–75. doi: 10.1016/j.ccr.2004.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang JT, Carvalho C, Mori S, Bild AH, Gatza ML, Wang Q, et al. A genomic strategy to elucidate modules of oncogenic pathway signaling networks. Mol Cell. 2009;34:104–14. doi: 10.1016/j.molcel.2009.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H, et al. Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol. 2002;22:874–85. doi: 10.1128/MCB.22.3.874-885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tong Y, Ben-Shlomo A, Zhou C, Wawrowsky K, Melmed S. Pituitary tumor transforming gene 1 regulates Aurora kinase A activity. Oncogene. 2008;27:6385–95. doi: 10.1038/onc.2008.234. [DOI] [PubMed] [Google Scholar]
- 31.Assie G, LaFramboise T, Platzer P, Bertherat J, Stratakis CA, Eng C. SNP arrays in heterogeneous tissue: highly accurate collection of both germline and somatic genetic information from unpaired single tumor samples. Am J Hum Genet. 2008;82:903–15. doi: 10.1016/j.ajhg.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia-Linares C, Fernandez-Rodriguez J, Terribas E, Mercade J, Pros E, Benito L, et al. Dissecting loss of heterozygosity (LOH) in neurofibromatosis type 1-associated neurofibromas: Importance of copy neutral LOH. Hum Mutat. 2011;32:78–90. doi: 10.1002/humu.21387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agnese V, Bazan V, Fiorentino FP, Fanale D, Badalamenti G, Colucci G, et al. The role of Aurora-A inhibitors in cancer therapy. Ann Oncol. 2007;18(Suppl 6):vi47–52. doi: 10.1093/annonc/mdm224. [DOI] [PubMed] [Google Scholar]
- 34.Dees EC, Infante JR, Cohen RB, O’Neil BH, Jones S, von Mehren M, et al. Phase 1 study of MLN8054, a selective inhibitor of Aurora A kinase in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2011;67:945–54. doi: 10.1007/s00280-010-1377-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hirota T, Kunitoku N, Sasayama T, Marumoto T, Zhang D, Nitta M, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114:585–98. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 36.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 37.Nowak SJ, Corces VG. Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004;20:214–20. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 38.Furukawa T, Kanai N, Shiwaku HO, Soga N, Uehara A, Horii A. AURKA is one of the downstream targets of MAPK1/ERK2 in pancreatic cancer. Oncogene. 2006;25:4831–9. doi: 10.1038/sj.onc.1209494. [DOI] [PubMed] [Google Scholar]
- 39.Tatsuka M, Sato S, Kitajima S, Suto S, Kawai H, Miyauchi M, et al. Overexpression of Aurora-A potentiates HRAS-mediated oncogenic transformation and is implicated in oral carcinogenesis. Oncogene. 2005;24:1122–7. doi: 10.1038/sj.onc.1208293. [DOI] [PubMed] [Google Scholar]
- 40.Huang HY, Chang HY, Chou CH, Tseng CP, Ho SY, Yang CD, et al. sRNAMap: genomic maps for small non-coding RNAs, their regulators and their targets in microbial genomes. Nucleic Acids Res. 2009;37:D150–4. doi: 10.1093/nar/gkn852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gigoux V, L’Hoste S, Raynaud F, Camonis J, Garbay C. Identification of Aurora kinases as RasGAP Src homology 3 domain-binding proteins. J Biol Chem. 2002;277:23742–6. doi: 10.1074/jbc.C200121200. [DOI] [PubMed] [Google Scholar]
- 42.Pamonsinlapatham P, Hadj-Slimane R, Raynaud F, Bickle M, Corneloup C, Barthelaix A, et al. A RasGAP SH3 peptide aptamer inhibits RasGAP-Aurora interaction and induces caspase-independent tumor cell death. PLoS One. 2008;3:e2902. doi: 10.1371/journal.pone.0002902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Jr, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639–48. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 44.Dreier MR, Grabovich AZ, Katusin JD, Taylor WR. Short and long-term tumor cell responses to Aurora kinase inhibitors. Exp Cell Res. 2009;315:1085–99. doi: 10.1016/j.yexcr.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 45.Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatr Blood Cancer. 2010;55:26–34. doi: 10.1002/pbc.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelly KR, Ecsedy J, Medina E, Mahalingam D, Padmanabhan S, Nawrocki ST, et al. The novel Aurora A kinase inhibitor MLN8237 is active in resistant chronic myeloid leukemia and significantly increases the efficacy of nilotinib. J Cell Mol Med. 2010 doi: 10.1111/j.1582-4934.2010.01218.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sehdev V, Peng D, Soutto M, Washington MK, Revetta F, Ecsedy JA, et al. The Aurora kinase A inhibitor MLN8237 Enhances Cisplatin-induced Cell Death in Esophageal Adenocarcinoma Cells. Mol Cancer Ther. 2012 doi: 10.1158/1535-7163.MCT-11-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.El-Sheikh A, Fan R, Birks D, Donson A, Foreman NK, Vibhakar R. Inhibition of Aurora Kinase A enhances chemosensitivity of medulloblastoma cell lines. Pediatr Blood Cancer. 2010;55:35–41. doi: 10.1002/pbc.22465. [DOI] [PubMed] [Google Scholar]
- 49.Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, et al. Initial testing of the aurora kinase a inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP) Pediatr Blood Cancer. 2010 doi: 10.1002/pbc.22430. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.