

Abstract
Non-small cell lung cancer (NSCLC) not amenable to surgical resection has a high mortality rate, due to the ineffectiveness and toxicity of chemotherapy. Thus, there remains an urgent need of efficacious drugs that can combat this disease. In this study, we show that targeting the formation of pro-angiogenic epoxyeicosatrienoic acids (EETs) by the cytochrome P450 arachidonic acid epoxygenases (Cyp2c) represents a new and safe mechanism to treat NSCLC growth and progression. In the transgenic murine K-Ras model and human orthotopic models of NSCLC, we found that Cyp2c44 could be downregulated by activating the transcription factor PPARα with the ligands bezafibrate and Wyeth-14,643. Notably, both treatments reduced primary and metastatic NSCLC growth, tumor angiogenesis, endothelial Cyp2c44 expression and circulating EET levels. These beneficial effects were independent of the time of administration, whether before or after the onset of primary NSCLC, and they persisted after drug withdrawal, suggesting the benefits were durable. Our findings suggest that strategies to downregulate Cyp2c expression and/or its enzymatic activity may provide a safer and effective strategy to treat NSCLC. Moreover, as bezafibrate is a well-tolerated clinically approved drug used for managing lipidemia, our findings provide an immediate cue for clinical studies to evaluate the utility of PPARα ligands as safe agents for the treatment of lung cancer in humans.
Keywords: Cytochrome P450 epoxygenases, epoxyeicosatrienoic acids, PPARα ligands, KRas, orthotopic models, tumorigenesis, metastasis
INTRODUCTION
Non-small cell lung cancer (NSCLC) accounts for ~85% of all lung cancer cases and has a 5-year survival rate of only 15% (1). Early-stage NSCLC is potentially curable by surgery, but its long-term survival after resection is limited by local or distal recurrences. The best therapy available for inoperable and/or late-stage NSCLC is radiation with or without chemotherapy (2, 3). In addition, anti-angiogenic therapy targeting VEGF prolongs survival in lung cancer patients (4). However, the outcomes of targeting VEGF are dependent on cancer type and stage, and often result in the development of resistance (5), hypertension and proteinuria (6, 7), and death associated to primary or metastatic disease. Thus, there is the need for new, more effective, and safer anti-tumorigenic and/or anti-angiogenic therapies.
The cytochrome P450 arachidonic acid epoxygenases (CYP2C) oxidize arachidonic acid to epoxyeicosatrienoic acids (EETs). EETs stimulate hormonal signaling (8–10) and have anti-hypertensive and vasodilatory actions (11, 12). In addition, we and others characterized the EETs as pro-angiogenic lipids both in vitro and in vivo (13–15) and identified the murine Cyp2c44 as a pro-angiogenic epoxygenase (16). Disruption of the Cyp2c44 gene, or downregulation of its expression via activation of the peroxisomal proliferator activating receptor (PPAR)α, reduces endothelial cell function in vitro and primary tumor growth in vivo (17).
Like Cyp2c44, its functional homologue human CYP2C9, is expressed in endothelial cells and its downregulation reduces EET biosynthesis and endothelial cell function (17). Interestingly, CYP2C9 is upregulated in the vasculature of human NSCLC (17), making this epoxygenase an attractive target for anti-angiogenic therapy.
PPARs regulate the transcription of genes involved in lipid and glucose homeostasis (18). Upon ligand binding, the three PPAR isotypes PPARα, -β/δ, and -γ form heterodimers with the retinoic acid receptor and, in the presence of specific co-activators, bind to response elements in the promoter region of their target genes. The PPARα ligands derivatives of fibric acid are used for the treatment of hyperlipidemia and their safety, tolerance, and minimal side effects are well documented (19, 20). In addition, the PPARα ligands Bezafibrate and Wyeth-14,643 decrease in vivo hepatic Cyp2c expression and hepatic and plasma EET levels (16). Wyeth-14,643 decreases Cyp2c44 and CYP2C9 expression and EET biosynthesis in mouse and human endothelial cells (16, 17). PPARα ligands exhibit also pro- and anti-tumorigenic effects. In rodents, extended exposure to fibrates causes PPARα-mediated hepatocarcinoma (21–23) which is not observed in humans even after extended exposure to PPARα ligands (24). In contrast, PPARα ligands decrease intestinal polyp formation in ApcMin/+ mice, inhibit vascular smooth muscle hyperplasia, induce apoptosis, and prevent tumor cell invasion (25–27). We showed that Wyeth-14,643 inhibits primary growth of human NSCLC cells, its effects require a functional host PPARα gene and are associated with PPARα-mediated reduction in endothelial Cyp2c expression and EET levels (16). Thus, PPARα acts as an anti-angiogenic and anti-tumorigenic receptor.
A limitation of the primary model of human NSCLC cells showing beneficial effects of PPARα activation is that the tumors do not metastasize and do not recapitulate the steps of tumor progression in human NSCLC (16). In this study, we used the KRasLA2 mouse model of spontaneous NSCLC to analyze tumor initiation and growth (28), and a human orthotopic model of NSCLC to analyze tumor progression and metastasis (29). We show that treatment with selective PPARα ligands inhibits NSCLC primary and metastatic growth and that their beneficial effects are associated with downregulation of Cyp2c expression in tumor-associated vasculature and EET biosynthesis. This study, together with the fact that PPARα ligands are in clinical use as hypolipidemic drugs, suggests that they should be also safe and well-tolerated drugs for the prevention and treatment of NSCLC.
MATERIALS AND METHODS
EET analogs
The EET analogs 2-(13-(3-pentylureido)tridec-8(Z)-enamido)succinic acid (EET Analog 1) and N-isopropyl-N-(5-((2-pivalamidobenzo[d]thiazol-4-yl)oxy)pentyl)heptanamide (EET Analog 2) were generated based on 14,15-EET. The labile epoxide characteristic of EETs was replaced by urea and amide bioisosteres, respectively (30). The carboxylate was replaced by bioisosteres and most of the olefins were removed to minimize auto-oxidation. EET analogs with these types of modifications have been described (31). These two EET analogs can ameliorate cisplatin nephrotoxicity and hypertension-mediated kidney injury in vivo (32, 33).
Cell Culture and Assays
Pulmonary microvascular endothelial cells were freshly isolated from wild type and Cyp2c44-null (Cyp2c44KO) mice and cultured as described (17). The large T antigen/ras/Myc-transformed mouse fibroblast p60.5 cells, lung tumor cells from KrasLA2 mice, and the human NSCLC A549 cells transfected with pEGFP-N1 (GFP-A549) were previously generated (16, 28, 29). Cells were authenticated before use by CD31 positive staining, ability to form tumors in vivo, or GFP intensity. Cells were cultured for one month from freshly isolated organs or low-passage frozen stocks. For proliferation, cells were plated (5×103 /well; 96-well plate) in serum-free media with or without EETs (1 μM), or the EET Analogs 1 and 2 (0.125–1 μM) (34). In some experiments, cells were cultured in 2.5% FCS with or without different concentrations of Wyeth-14,643. Two days after, the medium was replaced with medium containing [3H]thymidine (1μCi/well), the cells incubated for 48 hours, and proliferation was determined as described (16, 17). Two-three independent experiments were performed in quadruplicate.
In vivo treatment with EET analogs
Animal protocols were approved by the Vanderbilt University Institutional Animal Care and Use Committee. 129SvJ male (12 weeks old, n=9) wild type and Cyp2c44KO mice [generated as described, (17)] were given two dorsal subcutaneous injections of syngeneic p60.5 cells (2×105 in 200 μl PBS) (17). Cyp2c44KO mice were left untreated or administered the EET Analog 1 or 2 (10 mg/Kg in drinking water) four days before tumor injection and throughout the duration of the experiment. The animals were sacrificed 2 weeks after cell injection and tumor volume and weight were evaluated. Tumor volumes were calculated using the formula: tumor volume (mm3) = (length [mm] × width [mm]2)/2 (35). The dose of EET analogs selected was chosen as it did not significantly enhance tumor volume in p60.5 tumor-bearing wild type mice after 18 days of treatment.
The KrasLA2 mouse model of lung cancer
Age- and gender-matched 129SvJ KrasLA2 heterozygotes [described in (36) and here referred as to KrasLA2] mice were divided into water-treated and sacrificed at 1 month (group 1), 3 months (group 2), and 5 months (group 4) of age)] or Wyeth-14,643-treated [0.02% in drinking water (16, 17)]. Among the Wyeth-14,643-treated mice, one group received treatment from 1 to 3 months of age and then immediately sacrificed (group 3, early treatment). Another group received treatment from 3 to 5 months of age and then immediately sacrificed (group 5, late treatment), while a third group received treatment from 1 to 3 months of age and sacrificed at 5 months of age (group 6, treatment withdrawal). At sacrifice, the number of tumors visible on the lung surface was counted. Tumors were divided into three groups: 0–2 mm, 2–5 mm, and >5 mm tumor diameter. Lungs containing tumors were fixed in 4% paraformaldehyde or frozen in OCT and processed for histology or immunofluorescence. Livers and plasma were collected at sacrifice for evaluation of liver/body weight ratio and analysis of circulating levels of EETs.
Orthotopic model of NSCLC
Human GFP-A549 cells [1×106 in 20 μl PBS containing 20 μl Matrigel (BD Pharmingen)] were injected into the left lung parenchyma of 12 weeks old athymic nude mice as described (29). Mice were then were divided into groups of water-treated and sacrificed 5 weeks after injection (group a) or Wyeth-14,643-treated [0.02% in the drinking water] or Bezafibrate (0.3% in drinking water). Treated mice were divided into 3 groups. One group was treated immediately after cell injection for 5 weeks and then sacrificed (group b, prevention group). Another group was treated immediately after cell injection for 2 weeks and then sacrificed 3 weeks later (group c, treatment withdraw). A third group received treatment for 3 weeks starting 2 weeks after injection and then immediately sacrificed (group d, late treatment). Two weeks after tumor cell injection was chosen as at this time point GFP-A549 cells form only visible primary (left lung) but not metastatic (right lung and liver) tumors. All mice were sacrificed 5 weeks after tumor cell injection as at this time point GFP-A549 cells generate both primary and metastatic tumors (29). At sacrifice, organs (left and right lungs, liver) were removed, placed under a fluorescence microscope, and the number of tumors on the surface and within the organs was counted. Images of GFP positive tumors were recorded and processed with Scion Image software as described (29). Tumor area was expressed as percentage of area occupied by GFP positive structures per microscopic field. Organs containing tumors were fixed in 4% paraformaldehyde and processed for histology or immunohistochemistry. Liver and plasma were collected for evaluation of liver/body weight ratio and analysis of circulating levels of EETs.
Analysis and Quantification of plasma Epoxygenase Metabolites
Mouse plasma was obtained from blood collected in EDTA. Plasma EETs and dihydroxyeicosatrienoic acids (DHETs) were extracted in the presence of equimolar mixtures of 2H3-labeled EETs and 2H8-labeled DHETs (5 ng each). After saponification and purification (37), EETs and DHETS were quantified by ultra high pressure liquid chromatography/tandem mass spectrometry and the isotope ratio method (38).
Western Blot analysis
Microsomal fractions from livers or A549 cells were isolated by differential centrifugation as described (39). Microsomal proteins (20 μg/well) were resolved by SDS-PAGE and transferred to Immobilon-P membranes (Millipore), and the membranes were incubated with anti-Cyp2c44 (17) or rabbit anti-human CYP2C9 (Abcam) antibodies. Proteins were visualized using peroxidase-conjugated anti-rabbit antibodies and an ECL kit (Pierce). Membranes were stained with Ponceau S Stain to verify equal loading.
Immunofluorescence
Frozen sections (5 μm) of lungs from KrasLA mice, or tumors grown in wild type and Cyp2c44KO mice, were stained with anti-mouse CD31 antibodies (1:400, BD Pharmingen) with or without anti-mouse Cyp2c44 antibodies (1:1000) followed by the appropriate fluorescence labeled secondary antibodies (1:200, Jackson ImmunoResearch). Vascularization, expressed as percentage of area occupied by CD31-positive structures/microscopic field, was evaluated using Scion Imaging Software (Frederick, MD) (16), using 5–6 images/tumor and 5 tumors/genotype.
RT-PCR analysis
Total RNAs were isolated from GFP-A549 cells or human liver with the TRIzol Reagent (Invitrogen) and 2 μg of RNA were reverse-transcribed using SuperScript™ III (Invitrogen) and Oligo (dT) 20. RNA incubated without RT-PCR SuperScript™ III was used as negative control. RT-PCR was performed using primers for human β-actin (5′-CAGGATGCAGAAGGAGATCAC-3′, 5′-TGTCAAGAAAGGGTGTAACGC-3′), PPARα (5′-TGGTAGCGTATGGAAATGGG-3′, 5′-GTGGAGTCTGAGCACATGTAC-3′) and CYP2C9 (5′-TTGCTTCTCCTTTCACTCTGG-3′, 5′-TCCTTCACTGCTTCATATCCATG-3′) using the following program: 94°C/3 min; 40 cycles of 94°C/1 min, 57°C/30 sec, 72°C/1 min; 72°C/10 min.
Statistical analysis
Student’s t test was used for comparisons between two groups (unpaired, 2-tailed), and analysis of variance using Sigma-Stat software was used to analyze statistical differences between multiple groups. p≤0.05 was considered statistically significant.
RESULTS
EET analogs promote tumorigenesis and angiogenesis
Disruption of Cyp2c44, the gene that codes for the murine Cyp2c44 and the major EET generating epoxygenase, leads to decreased endothelial EET biosynthesis and primary tumor growth and vascularization by (17). To determine whether the decreased tumorigenesis in the Cyp2c44KO mice was mainly due to reduced production and levels of EETs, we tested two synthetic EET functional analogs, namely EET Analogs 1 and 2 (see Methods for details). Cyp2c44KO mice received water or one of the two EET analogs. Four days later, mice were subcutaneous injected with p60.5 cells and the EET analog treatment continued for 2 weeks. Mice were then sacrificed and tumor number, weight, and volume were evaluated. As reported (17), tumors in Cyp2c44KO mice were smaller and paler compared to those in untreated wild type mice (Figs. 1A–C). In contrast, EET analog-treated Cyp2c44KO mice grew bigger and more vascularized tumors when compared to untreated Cyp2c44KO mice, and their size and weight were similar to those in untreated wild type mice (Figs. 1A–C). In contrast to treated Cyp2c44KO mice, EET analog-treated wild type mice did not grow significantly bigger p60.5-derived tumors when compared to untreated wild type mice (not shown).
Figure 1. EET analogs promote tumorigenesis.
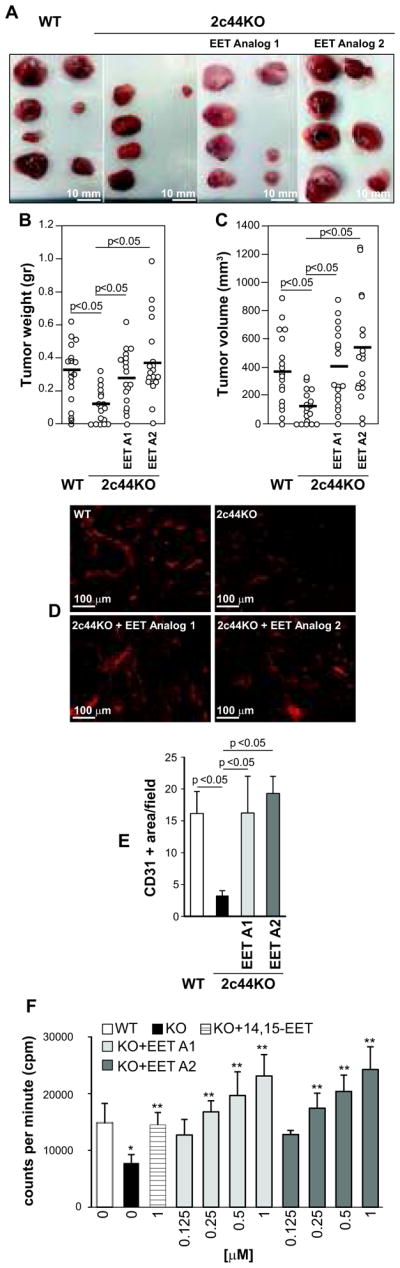
(A) Images of large T antigen/ras/Myc-transformed mouse fibroblast p60.5 cell-derived tumors grown in the mice indicated. Mice were left untreated or administered the EET analogs 2-(13-(3-pentylureido)tridec-8(Z)-enamido)succinic acid (EET Analog 1) or N-isopropyl-N-(5-((2-pivalamidobenzo[d]thiazol-4-yl)oxy)pentyl)heptanamide (EET Analog 2) for 4 days prior injection with tumor cells. Mice were further treated for 2 weeks and then sacrificed. (B, C) Weight (B) and volume (C) of tumors grown in the mice indicated (n=9). Circles show individual values, while bars show mean values. (D, E) Frozen sections of the tumors grown in the mice indicated were stained with anti-mouse CD31 antibodies and their vascularization was quantified as described in Materials and Methods. The values are means ± S.D. of 10 tumors/group with 2 images/tumor analyzed. (F) Primary lung endothelial cells were cultured with or without 14,15-EET or the EET analogs (EET A1 and EET A2) at the indicated concentrations. Cell proliferation was then evaluated as described in Materials and Methods. Values are means ± S.D. of 3 experiments performed in quadruplicate. p<0.05 between untreated WT vs. Cyp2c44KO (*) or untreated vs. treated Cyp2c44KO (**) cells.
As Cyp2c44KO mice show decreased tumor vascularization (17), tumor sections were stained with anti-CD31 antibody. Tumors in Cyp2c44KO mice were significantly less vascularized than those in wild type mice, while tumors from EET analog-treated Cyp2c44KO mice showed similar levels of vascularization as tumors from untreated wild type mice (Figs. 1D, 1E).
We next determined whether the increased tumorigenesis and angiogenesis in EET analog-treated mice was because the analogs stimulated proliferation of endothelial and/or p60.5 cells. At baseline, Cyp2c44KO endothelial cells grew significantly less than wild type cells, and their growth was significantly stimulated by the two EET analogs in a dose dependent manner (Fig. 1F). In contrast, both EET analogs failed to promote p60.5 cell proliferation (Suppl. Fig. 1A). Thus, EET Analogs 1 and 2, at a dose that does not significantly affect tumor growth in wild type mice, potentiate primary tumor growth in Cyp2c44KO mice by stimulating endothelial cell proliferation in vitro and angiogenesis in vivo.
Activation of PPARα decreases primary lung cancer growth
The PPARα ligand Wyeth-14,643 downregulates endothelial Cyp2c44 expression and EET biosynthesis in a mouse model of subcutaneously injected NSCLC A549 tumor cells, which leads to reduced primary tumor growth and vascularization (16). As this mouse model is based on the subcutaneous injection of tumor cells, it fails to recapitulate the steps of NCSLC development seen in humans. To define the role of EETs and the effects of PPARα ligands to NCSLC initiation and growth, we used the KRasLA2 mouse model of spontaneous primary NSCLC (36). As described in Figure 2A, three groups of mice received vehicle (water) and were sacrificed at 1 month (group 1), 3 months (group 2), and 5 months (group 4) of age. Three other groups received Wyeth-14,643. Group 3 (early treatment) received treatment from 1 to 3 months of age and then sacrificed; group 5 (late treatment) received treatment from 3 to 5 months of age and then sacrificed; and group 6 (treatment withdrawal) received treatment from 1 to 3 months of age and sacrificed at 5 months of age.
Figure 2. Treatment of KRasLA2 mice with Wyeth-14,643.

(A) Diagram showing the Wyeth-14,643 treatment protocol used in KRasLA2 mice. The number of mice/group is indicated in parenthesis. (B) Liver/body weight ratio of the KRasLA2 mice described in (A). Circles show individual values, while bars show mean values (C). Levels of EETs and DHETs in the plasma of the KRasLA2 mice described in (A). Values are averages ± SD of the mice indicated. (D) Hepatic microsomal fractions (20 μg/lane) of the KRasLA2 mice indicated were analyzed by Western blot for expression of Cyp2c44. Ponceau staining shows equal loading.
As PPARα activation leads to liver hypertrophy in rodents (16), we analyzed liver weight of untreated and Wyeth-14,643-treated mice. Significant liver hypertrophy was observed in mice in groups 3 and 5. Interestingly, mice in group 6 (treatment withdrawal) showed a liver/body weight ratio comparable to that of mice receiving water (Fig. 2B). As Wyeth-16,463 reduces hepatic Cyp2c44 expression and plasma EETs (16), we analyzed these parameters in KrasLA2 mice. Both early and late Wyeth-14,643 administration (groups 3 and 5) resulted in marked reductions in plasma EET levels and hepatic Cyp2c44 expression (Figs. 2C, D). Interestingly, mice in the Wyeth-14,643 withdrawal protocol (group 6) showed similar levels of plasma EETs and hepatic Cyp2c44 to those of untreated mice (Figs. 2C, D). Thus, the effects of Wyeth-14,643 on liver hypertrophy, hepatic Cyp2c44 expression, and plasma EET levels are reversible.
We next analyzed whether early Wyeth-14,643 treatment reduced the number and size of lung tumors. One-month-old untreated mice (group 1) presented with a few small tumors on their lung surface (Figs. 3A–D). The number and size of lung tumors increased significantly when mice reached 3 months of age (group 2) (Figs. 3A–D). By contrast, 3 months old mice treated with Wyeth-14,643 for 2 months starting at 1 month of age (group 3) showed lung tumor lesions and numbers comparable to those of group 1 (Figs 3A–C). Wyeth-14,643 treatment also decreased tumor size in this group (Fig. 3D).
Figure 3. Wyeth-14,643 reduces primary NSCLC growth.
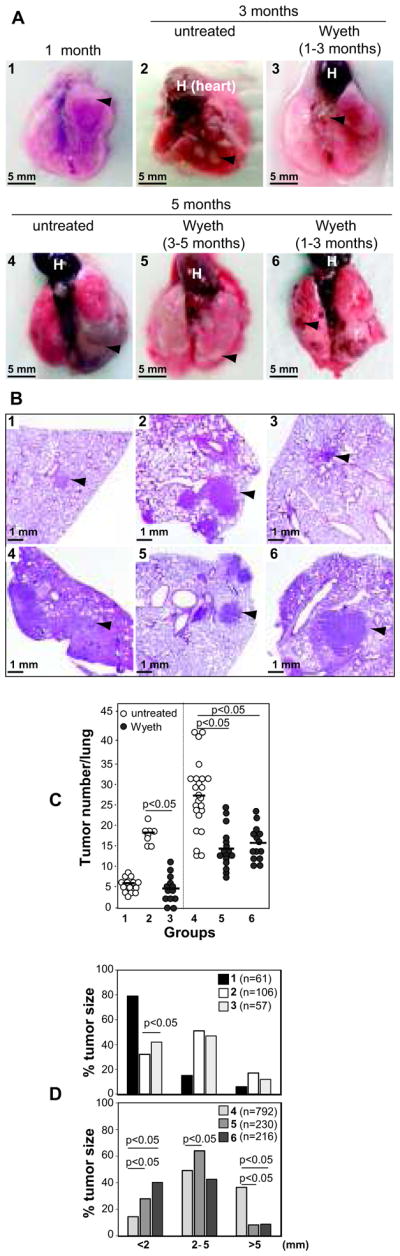
Images (A) and hematoxylin and eosin staining (B) of lungs and lung paraffin sections derived from the KRasLA2 mice indicated. At sacrifice, the number (C) and size (D) of tumors (arrowhead) on the lung surface were evaluated. (C) Circles show individual values, while bars show mean values. (D) Distribution of tumor size (diameter) in the KRasLA2 mice indicated (groups 1–3, upper panel; groups 4–6, lower panel). The number in parenthesis indicates tumors analyzed.
We next evaluated the beneficial effects of late treatment and/or the long lasting effects of Wyeth-14,643. Five-month-old untreated mice (group 4) presented with the highest number and biggest tumors on their lung surface (Figs. 3A–C–D). Tumors in groups 5 and 6 were significantly fewer and smaller when compared to those of group 4 (Figs 3A–D). Thus, Wyeth-14,643 reduces the number and size of primary lung tumors, and its beneficial anti-tumorigenic effects are long lasting and independent of treatment time.
Activation of PPARα decreases primary lung cancer vascularization
Activation of PPARα decreases endothelial cell function by downregulating Cyp2c expression and EET biosynthesis (16). Therefore, we evaluated vascularization in lung tumors from the various groups of KrasLA2 mice. A significant decrease in vascularization was evident in tumors of KrasLA2-treated mice compared to untreated mice and this effect was independent of the length of treatment (Figs. 4A, B). Double immunostaining with anti-CD31 and anti-Cyp2c44 antibodies revealed that in the untreated mice, Cyp2c44 was expressed in the endothelium and tumor tissue, whereas in the tumors grown in Wyeth-14,643-treated mice, Cyp2c44 expression was restricted mostly to tumor cells (Figs. 4C, D). Since Wyeth-14,643 does not affect the in vitro proliferation of lung tumor cells from KrasLA2 mice (Suppl. Fig. 1B), we concluded that Wyeth-14,643 only affects endothelial Cyp2c44 expression with no measurable effects on the proliferation of KrasLA2 lung tumor cells.
Figure 4. Wyeth-14,643 reduces NSCLC angiogenesis and endothelial Cyp2c44 expression.

(A, B) Frozen sections of lung tumors from the KrasLA2 mice indicated were stained with anti-mouse CD31 antibodies. Tumor vascularization was quantified as described in the Materials and Methods. The values are means ± SD of 10 tumors/group with 2 images/tumor analyzed. (C, D) Frozen sections of lung tumors from KrasLA2 mice in groups 2 and 3 (C) or groups 4–6 (D) were co-stained with anti-mouse CD31 and anti-mouse Cyp2c44 antibodies. Images were merged to visualize endothelial expression of Cyp2c44. T, tumor; V, vasculature.
Activation of PPARα reduces lung cancer metastasis
Tumors in the KRasLA2 mice rarely metastasize, thus making the analysis of interventional therapy in lung cancer progression impossible to perform. To determine whether activation of PPARα is beneficial in cancer progression, we used a lung orthotopic model in which human NSCLC A549 cells are injected directly into the lung parenchyma of immunodeficient mice (29). Upon injection into the primary site (left lung), primary tumors form and metastasize to secondary sites (right lung and liver) (29). We used NSCLC A549 transfected with GFP (GFP-A549 cells) since: a) they metastasize within 5 weeks of injection (29); b) they express PPARα, but lack CYP2C9, the functional homologue of Cyp2c44 (Suppl. Figs. 2A, 2B); c) they do not produce EETs (Suppl. Fig. 2C); and d) their growth is not affected by Wyeth-14,643 (Suppl. Fig. 2D). Thus, in contrast to endothelial cells, PPARα ligands do not affect A549 cell growth.
As described in Figure 5A, injected mice were divided into one control group (water) and sacrificed 5 weeks after tumor cell injection (group a), or three Wyeth-14,643-treated groups. Group b (prevention group) was treated for 5 weeks immediately after tumor cell injection and then sacrificed; group c (treatment withdraw) was treated for 2 weeks immediately after tumor cell injection and sacrificed after 5 weeks; and group d (late treatment) was treated for 3 weeks starting 2 weeks after tumor cell injection and then sacrificed.
Figure 5. Treatment of athymic mice orthotopically injected with human GFP-A549 cells with Wyeth-14,643.
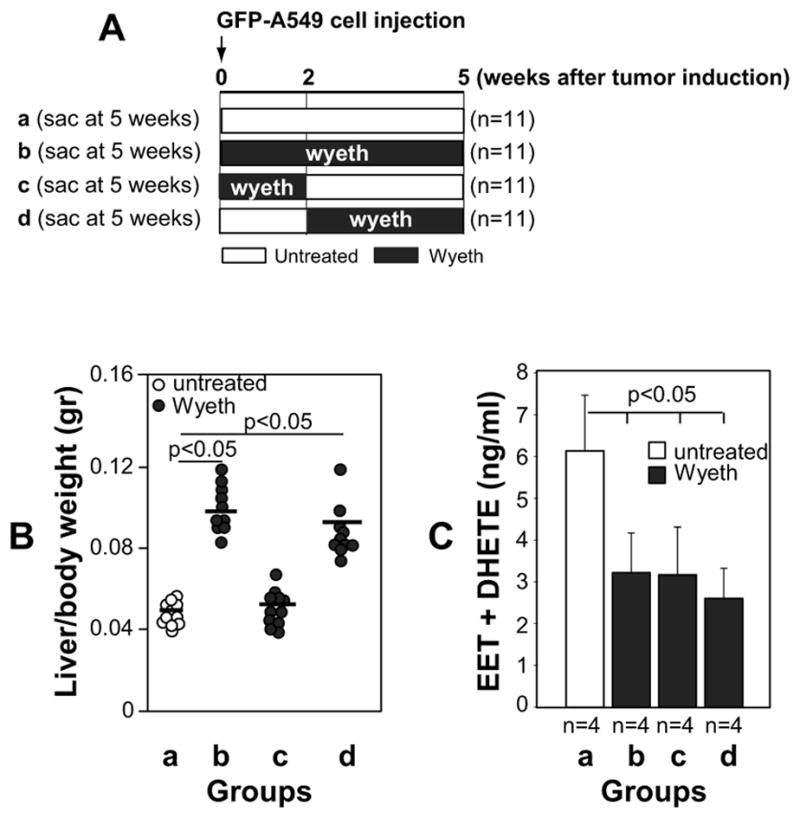
(A) Diagram showing the Wyeth-14,643 treatment protocol used in athymic mice. The number in parenthesis indicates mice used in each group. (B) Liver/body weight ratio of the mice described in (A). The circles show individual values, while bars show mean values. (C) Levels of EETs and DHETs in the plasma of the athymic mice described in (A). Values are averages ± SD of the mice indicated.
Analysis of liver hypertrophy revealed that while Wyeth-14,643 increased liver/body weight ratio in groups b and d, its effects were no longer evident when the ligand was withdrawn after 2 weeks treatment (group c) (Fig. 5B). In contrast, plasma EET levels in Wyeth-14,643-treated mice remained significantly lower compared to untreated mice regardless of the length and time of treatment (Fig. 5C). The number and size of primary (left lung) and metastatic (right lung and liver) GFP-A549-derived tumors were significantly reduced in Wyeth-14,643-treated mice and these effects were independent of the time of treatment initiation or its duration (Figs. 6 and 7). Thus, activation of the host PPARα with Wyeth-14,643 reduces both primary and metastatic growth of human tumor cells.
Figure 6. Wyeth-14,643 reduces the number of primary and metastatic GFP-A549 cell-derived tumors.

(A) Lungs from athymic mice untreated (a) or treated with Wyeth-14,643 (b–d) isolated 5 weeks after GFP-A549 cell injection. (B) Number of GFP-positive primary (left lung) and metastatic (right lung and liver) tumors detected in freshly isolated organs 5 weeks after tumor cell injection. Values represent the mean +/− S.D. of the organs indicated.
Figure 7. Wyeth-14,643 reduces the size of primary and metastatic GFP-A549 cell-derived tumors.

(A) Images of primary and metastatic tumors visible in the organs of athymic mice untreated (a) or treated with Wyeth-14,643 (b–d) 5 weeks after GFP-A549 cell injection. (B) Tumor area expressed as percentage of area occupied by GFP-positive structures per microscopic field. Values represent the mean +/− s.e.m. of the organs indicated.
To explore the generality and potential clinical relevance of the above results, we performed orthotopic models of NSCLC using Bezafibrate, a PPARα ligand used clinically as a hypolipidemic drug with limited toxicity and excellent tolerance (40). Early and late treatment with Bezafibrate decreased the numbers of primary and metastatic GFP-A549-derived lung tumors and, as with Wyeth-14,643, these effects were accompanied by liver hypertrophy and decreased plasma EET levels (Suppl. Fig. 3A–F). Thus, Bezafibrate inhibits lung tumor progression irrespective of whether it is administered at early and/or late stages of cancer progression.
DISCUSSION
The goal of this study was to determine whether activation of PPARα plays a beneficial role in NSCLC progression, based on the findings that PPARα ligands reduce primary growth of subcutaneously injected human NSCLC (16) and are safely used clinically for the treatment of dyslipidemia (40). We show using two models of NSCLC that PPARα ligands inhibit both primary and metastatic lung cancer growth, and these effects are independent of the time of treatment (before or after onset of primary lesions) or whether the drug is withdrawn. We further show that the anti-tumorigenic and anti-angiogenic activity of PPARα ligands reside in their ability to downregulate endothelial cell Cyp2c expression and EET biosynthesis. Taken together, these results suggest the PPARα/Cyp2c axis is an attractive target for treating NSCLC.
PPARs regulate different cellular functions including proliferation, thus making PPAR agonists promising drugs for the treatment of cancer (41). Published data suggest a beneficial role for PPARγ ligands in lung cancer (42). PPARγ is upregulated in human lung cancer (43) and activation of PPARγ by troglitazone inhibits lung cancer cell growth (44). Thiazolidinediones, a group of PPARγ activating drugs, are associated with a lower risk of developing lung cancer (45), prevent smoke-induced lung cancer in mice, and together with glucocorticoids prevent carcinogen-induced lung cancer in rodents (46, 47). In contrast to these findings, expression of PPARγ in multidrug resistant NSCLC cells protects them from cytokine-mediated mitoinhibition (48) and the PPARγ ligand pioglitazone increases the rate of lung cancer metastasis by promoting recruitment of anti-inflammatory macrophages in mice (49). Finally, the observation that some of the effects of PPARγ ligands do not require the expression of this nuclear receptor (50), makes PPARγ an ambiguous target for the treatment of NSCLC.
Unlike PPARγ, the significance of PPARα ligands to lung cancer development and progression has received less attention. Fenofibrate and Wyeth-14,643 inhibit A549 cell proliferation in vitro, suggesting that PPARα has anti-tumorigenic activity (51). However, these effects were evident only when the ligands were used at high concentration questioning PPARα dependent vs. cytotoxic effects. The in vivo anti-tumorigenic properties of Wyeth-14,643 and fenofibrate have been reported (16, 52) and we showed that they are PPARα-dependent and the result of reduced endothelial cell proliferation and tumor vascularization (16). The beneficial effects of PPARα ligands seem to reside in their ability to promote the generation of anti-angiogenic factors (52), or to decrease endothelial Cyp2c44 expression and EET biosynthesis (16). The finding that Cyp2c44KO mice show decreased tumor growth and vascularization which are no further reduced by Wyeth-14,643 treatment (17), points to the Cyp2c44 as a pro-tumorigenic gene and suggests that reduction in Cyp2c44 expression is a major mechanism responsible for the beneficial anti-tumorigenic effects of PPARα ligands. Finally, the present finding that when used at concentrations known to inhibit endothelial cell growth, Wyeth-14,643 failed to alter the growth of human and mouse tumor cells, indicates that its anti-tumorigenic properties are primarily endothelial and not tumor cell-mediated.
The finding that synthetic EET functional analogs promote tumor growth in Cyp2c44KO mice and stimulate tumor angiogenesis and growth to levels comparable to wild type mice, supports a central role for epoxygenases and their EET metabolites in tumorigenesis and angiogenesis. Several mechanisms have been proposed for the pro-angiogenic action of EETs (53), including potentiation of the VEGF-activated signal transduction cascade. In this context, EETs induce VEGF synthesis (54); VEGF promotes CYP2C expression and EET biosynthesis (55); and disruption of the Cyp2c44 gene (17) and/or chemical inhibition of epoxygenase activity (56) prevent VEGF-mediated endothelial cell function.
Tumor metastasis is a serious clinical challenge due to limited treatment strategies, severe treatment-associated side effects, and negative outcomes. The two models of NSCLC used in this study show that PPARα-mediated downregulation of endothelial Cyp2c epoxygenase expression and EET biosynthesis might be viewed as a beneficial and safe strategy for the prevention and treatment of NSCLC. Consistent with a role for the endothelial Cyp2c/EET axis in tumor growth and metastasis, mice expressing the human CYP2C8 in the endothelium develop faster and bigger primary and metastatic tumors compared to wild type mice (57). Moreover, EET administration promotes exit from tumor dormancy and stimulates metastasis by enhancing tumor-associated angiogenesis (57).
Our finding that Wyeth-14,643 reduces primary and metastatic NSCLC growth whether administered before or after the onset of primary growth, and its anti-metastatic effects persisted for at least three weeks after cessation of treatment clearly identifies new avenues for cancer treatment and management. The potential clinical usefulness of these results is further highlighted by the demonstration that Bezafibrate, a safe and well-tolerated drug for the treatment of dyslipidemia (40), inhibits primary and metastatic NSCLC tumor growth whether it is administered at early and/or late stages of lung cancer development. In addition to PPARα, Bezafibrate can also target PPARβ/δ and PPARγ. Interestingly, activation of PPARβ/δ and PPARγ in lung cancer cells inhibits their proliferation (44, 58). Thus, Bezafibrate may be beneficial for the treatment of lung cancer, since it can target both tumor and endothelial cells and it does not have the severe side effects often associated with classic anti-tumorigenic and/or anti-angiogenic therapies (5–7).
In conclusion, in this study we identified two different, yet interlinked participants to the biology of lung tumor progression, namely Cyp2c epoxygenases and PPARα. We propose that, in addition to its well-documented role in the metabolism of carcinogens and anti-cancer drugs (59), cytochrome P450 is a key participant in the development and progression of lung cancer and a potentially valuable target for anti-cancer drug development. Furthermore, as PPARα ligands are used clinically as effective and safe hypolipidemic drugs (19, 20), our study could set the basis for future studies designed to validate in patients the usefulness of PPARα ligands as new, safer and well-tolerated anti-angiogenic and anti-tumorigenic agents. Targeting PPARα, endothelial Cyp2c epoxygenase, and EETs with new drugs, designed to interact with these targets either individually or in combination, could provide new, potent, and effective tools for the treatment of cancer, including lung cancer.
Supplementary Material
Acknowledgments
This work was supported by the VA Merit Review 1I01BX002025 (AP) and 1I01BX002196 (RZ); the NIH Grants R01-CA162433 (AP), R01-DK095761 (AP), RO1-DK083187 (RZ), RO1-DK075594 (RZ), R01-DK383069221 (RZ); P01-DK38226 (AP, JRF, JCH) and NCI SPORE in Lung Cancer 5P50 CA-090949 (Lung Cancer Pilot Project to AP); and the Vanderbilt University Mass Spectrometry Center, supported in part by Cancer Center Grant CA-68485. RZ is an American Heart Association Established Investigator.
References
- 1.Walker S. Updates in non-small cell lung cancer. Clin J Oncol Nurs. 2008;12:587–96. doi: 10.1188/08.CJON.587-596. [DOI] [PubMed] [Google Scholar]
- 2.Berman AT, Rengan R. New approaches to radiotherapy as definitive treatment for inoperable lung cancer. Semin Thorac Cardiovasc Surg. 2008;20:188–97. doi: 10.1053/j.semtcvs.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 3.Patel S, Sanborn RE, Thomas CR., Jr Definitive chemoradiotherapy for non-small cell lung cancer with N2 disease. Thorac Surg Clin. 2008;18:393–401. doi: 10.1016/j.thorsurg.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Cabebe E, Wakelee H. Role of anti-angiogenesis agents in treating NSCLC: focus on bevacizumab and VEGFR tyrosine kinase inhibitors. Curr Treat Options Oncol. 2007;8:15–27. doi: 10.1007/s11864-007-0022-4. [DOI] [PubMed] [Google Scholar]
- 5.Fernando NT, Koch M, Rothrock C, Gollogly LK, D’Amore PA, Ryeom S, et al. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin Cancer Res. 2008;14:1529–39. doi: 10.1158/1078-0432.CCR-07-4126. [DOI] [PubMed] [Google Scholar]
- 6.Chung EK, Stadler WM. Vascular endothelial growth factor pathway- targeted therapy as initial systemic treatment of patients with renal cancer. Clin Genitourin Cancer. 2008;6:s22–8. doi: 10.3816/cgc.2008.s.004. [DOI] [PubMed] [Google Scholar]
- 7.Obhrai JS, Patel TV, Humphreys BD. The case / progressive hypertension and proteinuria on anti-angiogenic therapy. Kidney Int. 2008;74:685–6. doi: 10.1038/ki.2008.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capdevila J, Chacos N, Falck JR, Manna S, Negro-Vilar A, Ojeda SR. Novel hypothalamic arachidonate products stimulate somatostatin release from the median eminence. Endocrinology. 1983;113:421–3. doi: 10.1210/endo-113-1-421. [DOI] [PubMed] [Google Scholar]
- 9.Falck JR, Manna S, Moltz J, Chacos N, Capdevila J. Epoxyeicosatrienoic acids stimulate glucagon and insulin release from isolated rat pancreatic islets. Biochem Biophys Res Commun. 1983;114:743–9. doi: 10.1016/0006-291x(83)90843-4. [DOI] [PubMed] [Google Scholar]
- 10.Michaelis UR, Fleming I. From endothelium-derived hyperpolarizing factor (EDHF) to angiogenesis: Epoxyeicosatrienoic acids (EETs) and cell signaling. Pharmacol Ther. 2006;111:584–95. doi: 10.1016/j.pharmthera.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 11.Imig JD. Eicosanoids and renal vascular function in diseases. Clin Sci (Lond) 2006;111:21–34. doi: 10.1042/CS20050251. [DOI] [PubMed] [Google Scholar]
- 12.Campbell WB, Gauthier KM. What is new in endothelium-derived hyperpolarizing factors? Curr Opin Nephrol Hypertens. 2002;11:177–83. doi: 10.1097/00041552-200203000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Potente M, Michaelis UR, Fisslthaler B, Busse R, Fleming I. Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J Biol Chem. 2002;277:15671–6. doi: 10.1074/jbc.M110806200. [DOI] [PubMed] [Google Scholar]
- 14.Pozzi A, Macias-Perez I, Abair T, Wei S, Su Y, Zent R, et al. Characterization of 5,6- and 8,9-epoxyeicosatrienoic acids (5,6- and 8,9-EET) as potent in vivo angiogenic lipids. J Biol Chem. 2005;280:27138–46. doi: 10.1074/jbc.M501730200. [DOI] [PubMed] [Google Scholar]
- 15.Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR) Faseb J. 2003;17:770–2. doi: 10.1096/fj.02-0640fje. [DOI] [PubMed] [Google Scholar]
- 16.Pozzi A, Ibanez MR, Gatica AE, Yang S, Wei S, Mei S, et al. Peroxisomal proliferator-activated receptor-alpha-dependent inhibition of endothelial cell proliferation and tumorigenesis. J Biol Chem. 2007;282:17685–95. doi: 10.1074/jbc.M701429200. [DOI] [PubMed] [Google Scholar]
- 17.Pozzi A, Popescu V, Yang S, Mei S, Shi M, Puolitaival SM, et al. The anti-tumorigenic properties of peroxisomal proliferator-activated receptor alpha are arachidonic acid epoxygenase-mediated. J Biol Chem. 2010;285:12840–50. doi: 10.1074/jbc.M109.081554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–4. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 19.Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med. 1999;341:410–8. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 20.Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317:1237–45. doi: 10.1056/NEJM198711123172001. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez FJ, Shah YM. PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246:2–8. doi: 10.1016/j.tox.2007.09.030. [DOI] [PubMed] [Google Scholar]
- 22.Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol Med. 2005;83:774–85. doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- 23.Corton JC, Lapinskas PJ, Gonzalez FJ. Central role of PPARalpha in the mechanism of action of hepatocarcinogenic peroxisome proliferators. Mutat Res. 2000;448:139–51. doi: 10.1016/s0027-5107(99)00232-8. [DOI] [PubMed] [Google Scholar]
- 24.Bonovas S, Nikolopoulos GK, Bagos PG. Use of fibrates and cancer risk: a systematic review and meta-analysis of 17 long-term randomized placebo-controlled trials. PLoS ONE. 2012;7:e45259. doi: 10.1371/journal.pone.0045259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niho N, Takahashi M, Kitamura T, Shoji Y, Itoh M, Noda T, et al. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res. 2003;63:6090–5. [PubMed] [Google Scholar]
- 26.Gizard F, Amant C, Barbier O, Bellosta S, Robillard R, Percevault F, et al. PPAR alpha inhibits vascular smooth muscle cell proliferation underlying intimal hyperplasia by inducing the tumor suppressor p16INK4a. J Clin Invest. 2005;115:3228–38. doi: 10.1172/JCI22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grabacka M, Plonka PM, Urbanska K, Reiss K. Peroxisome proliferator-activated receptor alpha activation decreases metastatic potential of melanoma cells in vitro via down-regulation of Akt. Clin Cancer Res. 2006;12:3028–36. doi: 10.1158/1078-0432.CCR-05-2556. [DOI] [PubMed] [Google Scholar]
- 28.Macias-Perez I, Borza C, Chen X, Yan X, Ibanez R, Mernaugh G, et al. Loss of integrin alpha1beta1 ameliorates Kras-induced lung cancer. Cancer Res. 2008;68:6127–35. doi: 10.1158/0008-5472.CAN-08-1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Su Y, Fingleton B, Acuff H, Matrisian LM, Zent R, et al. An orthotopic model of lung cancer to analyze primary and metastatic NSCLC growth in integrin alpha1-null mice. Clin Exp Metastasis. 2005;22:185–93. doi: 10.1007/s10585-005-7453-8. [DOI] [PubMed] [Google Scholar]
- 30.Falck JR, Kodela R, Manne R, Atcha KR, Puli N, Dubasi N, et al. 14,15-Epoxyeicosa-5,8,11-trienoic acid (14,15-EET) surrogates containing epoxide bioisosteres: influence upon vascular relaxation and soluble epoxide hydrolase inhibition. J Med Chem. 2009;52:5069–75. doi: 10.1021/jm900634w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falck JR, Krishna UM, Reddy YK, Kumar PS, Reddy KM, Hittner SB, et al. Comparison of vasodilatory properties of 14,15-EET analogs: structural requirements for dilation. Am J Physiol Heart Circ Physiol. 2003;284:H337–49. doi: 10.1152/ajpheart.00831.2001. [DOI] [PubMed] [Google Scholar]
- 32.Khan MA, Liu J, Kumar G, Skapek SX, Falck JR, Imig JD. Novel orally active epoxyeicosatrienoic acid (EET) analogs attenuate cisplatin nephrotoxicity. Faseb J. 2013;27:2946–56. doi: 10.1096/fj.12-218040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khan MA, Neckar J, Manthati V, Errabelli R, Pavlov TS, Staruschenko A, et al. Orally Active Epoxyeicosatrienoic Acid Analog Attenuates Kidney Injury in Hypertensive Dahl Salt-Sensitive Rat. Hypertension. 2013 doi: 10.1161/HYPERTENSIONAHA.113.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Imig JD. Epoxides and soluble epoxide hydrolase in cardiovascular physiology. Physiol Rev. 2012;92:101–30. doi: 10.1152/physrev.00021.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X, Su Y, Fingleton B, Acuff H, Matrisian LM, Zent R, et al. Increased plasma MMP9 in integrin alpha1-null mice enhances lung metastasis of colon carcinoma cells. Int J Cancer. 2005 doi: 10.1002/ijc.20997. [DOI] [PubMed] [Google Scholar]
- 36.Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–6. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 37.Karara A, Wei S, Spady D, Swift L, Capdevila JH, Falck JR. Arachidonic acid epoxygenase: structural characterization and quantification of epoxyeicosatrienoates in plasma. Biochem Biophys Res Commun. 1992;182:1320–5. doi: 10.1016/0006-291x(92)91877-s. [DOI] [PubMed] [Google Scholar]
- 38.Capdevila JH, Dishman E, Karara A, Falck JR. Cytochrome P450 arachidonic acid epoxygenase: stereochemical characterization of epoxyeicosatrienoic acids. Methods Enzymol. 1991;206:441–53. doi: 10.1016/0076-6879(91)06113-h. [DOI] [PubMed] [Google Scholar]
- 39.Capdevila JH, Falck JR, Dishman E, Karara A. Cytochrome P-450 arachidonate oxygenase. Methods Enzymol. 1990;187:385–94. doi: 10.1016/0076-6879(90)87045-5. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton-Craig I, Kostner KM, Woodhouse S, Colquhoun D. Use of fibrates in clinical practice: Queensland Lipid Group consensus recommendations. International journal of evidence-based healthcare. 2012;10:181–90. doi: 10.1111/j.1744-1609.2012.00275.x. [DOI] [PubMed] [Google Scholar]
- 41.Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. Journal of advanced pharmaceutical technology & research. 2011;2:236–40. doi: 10.4103/2231-4040.90879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reka AK, Goswami MT, Krishnapuram R, Standiford TJ, Keshamouni VG. Molecular cross-regulation between PPAR-gamma and other signaling pathways: implications for lung cancer therapy. Lung Cancer. 2011;72:154–9. doi: 10.1016/j.lungcan.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Giaginis C, Politi E, Alexandrou P, Sfiniadakis J, Kouraklis G, Theocharis S. Expression of peroxisome proliferator activated receptor-gamma (PPAR-gamma) in human non-small cell lung carcinoma: correlation with clinicopathological parameters, proliferation and apoptosis related molecules and patients’ survival. Pathol Oncol Res. 2012;18:875–83. doi: 10.1007/s12253-012-9517-9. [DOI] [PubMed] [Google Scholar]
- 44.Jeong Y, Xie Y, Lee W, Bookout AL, Girard L, Raso G, et al. Research resource: Diagnostic and therapeutic potential of nuclear receptor expression in lung cancer. Mol Endocrinol. 2012;26:1443–54. doi: 10.1210/me.2011-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, Kim L, et al. Thiazolidinediones and the risk of lung, prostate, and colon cancer in patients with diabetes. J Clin Oncol. 2007;25:1476–81. doi: 10.1200/JCO.2006.07.2777. [DOI] [PubMed] [Google Scholar]
- 46.Li MY, Kong AW, Yuan H, Ma LT, Hsin MK, Wan IY, et al. Pioglitazone prevents smoking carcinogen-induced lung tumor development in mice. Curr Cancer Drug Targets. 2012;12:597–606. doi: 10.2174/156800912801784848. [DOI] [PubMed] [Google Scholar]
- 47.Fu H, Zhang J, Pan J, Zhang Q, Lu Y, Wen W, et al. Chemoprevention of lung carcinogenesis by the combination of aerosolized budesonide and oral pioglitazone in A/J mice. Mol Carcinog. 2011;50:913–21. doi: 10.1002/mc.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin LC, Hsu SL, Wu CL, Liu WC, Hsueh CM. Peroxisome proliferator-activated receptor gamma (PPARgamma) plays a critical role in the development of TGFbeta resistance of H460 cell. Cell Signal. 2011;23:1640–50. doi: 10.1016/j.cellsig.2011.05.018. [DOI] [PubMed] [Google Scholar]
- 49.Li H, Sorenson AL, Poczobutt J, Amin J, Joyal T, Sullivan T, et al. Activation of PPARgamma in myeloid cells promotes lung cancer progression and metastasis. PLoS ONE. 2011;6:e28133. doi: 10.1371/journal.pone.0028133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kulkarni AA, Woeller CF, Thatcher TH, Ramon S, Phipps RP, Sime PJ. Emerging PPARgamma-Independent Role of PPARgamma Ligands in Lung Diseases. PPAR Res. 2012;2012:705352. doi: 10.1155/2012/705352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang H, Kowalczyk P, Junco JJ, Klug-De Santiago HL, Malik G, Wei SJ, et al. Differential Effects on Lung Cancer Cell Proliferation by Agonists of Glucocorticoid and PPARalpha Receptors. Mol Carcinog. 2013 doi: 10.1002/mc.22029. [DOI] [PubMed] [Google Scholar]
- 52.Panigrahy D, Kaipainen A, Huang S, Butterfield CE, Barnes CM, Fannon M, et al. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci U S A. 2008;105:985–90. doi: 10.1073/pnas.0711281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC. EET signaling in cancer. Cancer Metastasis Rev. 2011 doi: 10.1007/s10555-011-9315-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki S, Oguro A, Osada-Oka M, Funae Y, Imaoka S. Epoxyeicosatrienoic acids and/or their metabolites promote hypoxic response of cells. Journal of pharmacological sciences. 2008;108:79–88. doi: 10.1254/jphs.08122fp. [DOI] [PubMed] [Google Scholar]
- 55.Webler AC, Michaelis UR, Popp R, Barbosa-Sicard E, Murugan A, Falck JR, et al. Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis. Am J Physiol Cell Physiol. 2008;295:C1292–301. doi: 10.1152/ajpcell.00230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang S, Wei S, Pozzi A, Capdevila JH. The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Arch Biochem Biophys. 2009;489:82–91. doi: 10.1016/j.abb.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest. 2012;122:178–91. doi: 10.1172/JCI58128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukumoto K, Yano Y, Virgona N, Hagiwara H, Sato H, Senba H, et al. Peroxisome proliferator-activated receptor delta as a molecular target to regulate lung cancer cell growth. FEBS Lett. 2005;579:3829–36. doi: 10.1016/j.febslet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 59.Oyama T, Uramoto H, Kagawa N, Yoshimatsu T, Osaki T, Nakanishi R, et al. Cytochrome P450 in non-small cell lung cancer related to exogenous chemical metabolism. Front Biosci (Schol Ed) 2012;4:1539–46. doi: 10.2741/s350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.