


Abstract
HT1080 cells stably expressing green fluorescent protein (GFP) linked to a 3′ terminal AU-rich element (ARE) proved to be a convenient system to study the dynamics of mRNA stability, as changes in mRNA levels are reflected in increased or decreased fluorescence intensity. This study examined whether mRNA stability can be regulated by small interfering RNAs (siRNAs) targeted to AU-binding proteins (AUBPs), which in turn should reveal their intrinsic role as stabilizers or destabilizers of ARE-mRNAs. Indeed, siRNAs targeting HuR or BRF1 decreased or increased fluorescence, respectively. This effect was abolished if cells were treated with both siRNAs, thus indicating antagonistic control of ARE-mRNA stability. Unexpectedly, downregulation of all four AUF1 isoforms by targeting common exons did not affect fluorescence whereas selective downregulation of p40AUF1/p45AUF1 strongly increased fluorescence by stabilizing the GFP–ARE reporter mRNA. This observation was fully confirmed by the finding that only selective reduction of p40AUF1/p45AUF1 induced the production of GM-CSF, an endogenous target of AUF1. These data suggest that the relative levels of individual isoforms, rather than the absolute amount of AUF1, determine the net mRNA stability of ARE-containing transcripts, consistent with the differing ARE-binding capacities of the isoforms.
INTRODUCTION
Regulation of mRNA turnover is a key mechanism determining the intracellular abundance of transcripts, thus contributing to the controlled production of the encoded gene products (1–3). So far, the best characterized cis-elements mediating control of mRNA stability are the AU-rich elements (ARE) contained in the 3′ untranslated region (3′UTR) of cytokines, growth factors and proto-oncogenes (4,5). Presence of an ARE induces rapid shortening of the poly-A tail followed by exosomal degradation of the mRNA body (6–8). Transcripts containing an ARE usually exhibit rapid decay in resting cells, but are readily stabilized upon exposure to exogenous signals. Importantly, defective ARE function causing increased transcript stability has been reported to be a pathogenic factor in human malignancies (9–11), experimental tumors (12) and inflammatory disorders (13,14).
Several proteins binding to the ARE, AU-rich element binding proteins (AUBPs) have been identified and shown to exert a defined role in ARE-mRNA turnover. HuR, a member of the embryonic lethal abnormal vision (ELAV) RNA binding protein family has been shown to stabilize ARE-mRNAs (15,16). Two closely related proteins also containing RNA recognition motifs (RRM), TIA-1 (T-cell internal antigen-1) and TIAR (TIA-1-related protein), bind the ARE and act as translational silencers (17). Tristetraprolin (TTP, zfp-36) the prototype of a family of zinc-finger proteins of the unusual Cys-Cys-Cys-His (CCCH) class promotes degradation of tumor necrosis factor α (TNFα) (18,19), granulocyte-macrophage colony stimulating factor (GM-CSF) (20) and interleukin-3 (IL-3) mRNA (21). Butyrate response factor 1 (BRF1, zfp-36l1), a second member of the same class of zinc finger proteins, has been identified using functional genetic screening (22) and was shown to mediate rapid decay of various cytokine mRNAs, including IL-3, IL-6, GM-CSF and TNFα (23). Finally, AUF1 was first identified as a promoter of rapid ARE-mRNA decay in extracts of K562 cells (24). However, upon purification and cloning, the recombinant protein lacked decay promoting activity (25). The analysis of AUF1 has been further complicated by the existence of four isoforms that arise by differential splicing (26). This family of proteins distinguishes a 37 kDa (p37AUF1) core protein, a 40 kDa protein (p40AUF1) containing an N-terminal 19 amino acid insertion (exon 2), a 42 kDa protein (p42AUF1) exhibiting a C-terminal 49 amino acids insertion (exon 7), and a 45 kDa protein (p45AUF1) with insertions of both exon 2 and exon 7. Presence or absence of these alternatively spliced exons confers distinct biological properties to individual AUF1 isoforms. Presence of exon 7 not only affects nucleo-cytoplasmic distribution (27,28), but also blocks ubiquitination of p42AUF1 and p45AUF1 (29). In contrast, the lack of exon 7 targets p37AUF1 and p40AUF1 to the ubiquitin–proteasome pathway, which results in rapid decay of ARE-mRNAs (30,31). Importantly, absence of exon 2 in 37AUF1 and p42AUF1 is associated with high affinity binding of these isoforms (26). In addition to its role in ARE-mRNA turnover, AUF1 has been proposed to be involved in multiple cellular processes such as telomere maintenance (32) and transcriptional activation (33). The fact that AUF1 was identified to be a component of the α-globin mRNA stability complex (34) and to participate in mRNA decay directed by the c-fos major coding determinant (35) points to a more general function of this protein in mRNA turnover. With respect to ARE-mRNA turnover, the role of AUF1 has been extensively studied, but results were conflicting. Increased expression of AUF1 has been associated with rapid ARE-mRNA decay in peripheral blood mononuclear cells (PBMC) (36,37), smooth muscle cells (38) and monocytes (39). These data, together with the finding that forced expression of p37AUF1 and p42AUF1 antagonized stabilization of ARE-mRNAs accompanying hemin-induced erythroid differentiation of K562 cells (40) argue for an intrinsic destabilizing activity of AUF1. However, overexpression of all four AUF1 isoforms in NIH3T3 cells stabilized ARE-containing transcripts (41). It has not been resolved whether these disparities were due to the different cell types examined or changes in experimental conditions. Analysis of AUF1 knockout mice or studies using small interfering RNAs (siRNAs) targeted to AUF1 should help to clarify these issues.
The present study examined whether single or combined treatment of cells with siRNAs targeting HuR, BRF1 and AUF1 would affect ARE-mediated mRNA turnover. Our data indicate that BRF1 and HuR control ARE-mRNA stability in an antagonistic fashion. Furthermore, we present evidence that effects of AUF1 on ARE-containing transcripts depend on the expression level of individual AUF1 isoforms.
MATERIALS AND METHODS
Cell culture and transfection
The previously described HT1080-derived reporter cell line HTwt16 (21) was cultured in Iscove’s Modified Dulbecco Medium containing 10% fetal calf serum (FCS), 50 µM 2-mercaptoethanol, 2 mM glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C/7% CO2. In addition, NIH 3T3 cells stably expressing the GFP–ARE (3T3GFP–ARE) were generated and cultured as described above.
For transfections, 1.5 × 105 cells were plated per well of a 6-well plate and cultured overnight. Thereafter, siRNAs were transfected at a final concentration of 240 nM in the presence of serum free medium (OptiMEM, Invitrogen) using Oligofectamine (Invitrogen) as the carrier. Analyses were performed 48–72 h after transfection.
Oligoribonucleotides
The following 19mer oligoribonucleotide duplexes containing a 2-nt deoxythymidine overhang at each 3′ end were purchased from Xeragon. BRF1, 5′ CAA GAU GCU CAA CUA UAG U; HuR (oligo A), 5′ UAA AGU AGC AGG ACA CAG C; HuR (oligo B), 5′ CAC GCU GAA CGG CUU GAG G (42); AUF1 (exon 1), 5′ GAU UGA CGC CAG UAA GAA C; AUF1 (exon 2/oligo A), 5′ ACG AGG AGG AUG AAG GCC A (the first 16 nt are located in exon 1, the last 3 nt in exon 2); AUF1 (exon 2/oligo B), 5′ GCA GCG ACG GCA CAG CGG G; AUF1 (exon 3), 5′ GAU CCU AUC ACA GGG CGA U; AUF1 (exon 7), 5′ CUG GAA CCA GGG AUA UAG U; β-globin, 5′ CAA GAA AGU GCU CGG UGC C.
Sequences were derived from the human sequences of HuR mRNA (accession number BC003376), BRF1 mRNA (accession number NM_007564), AUF1 mRNA (accession number NM_031370) and β-globin mRNA (accession number V00497.1). Blast search was performed on all the sequences to avoid cross-reaction with other genes.
Flow cytometry
FACS analysis of 1–2 × 104 cells was performed using a FACScan (Becton Dickinson) and the Cellquest software. GFP fluorescence was excited at 488 nm and emission was measured at 510 nm. To analyze the difference in fluorescence between medium and siRNA treated cells, Kolmogorow–Smirnow statistics calculating the greatest difference (D) in fluorescence between these two treatments were performed. The mean ± standard deviation (SD) of D for 4–9 independent experiments was calculated and Student’s t-test comparing D of β-globin and specific siRNA-treated cells was performed.
Northern blot analysis
To analyze mRNA levels in cells treated with specific siRNAs, total RNA was harvested 72 h after transfection using Trizol® (Invitrogen). Total RNA (10–15 µg) was resolved on 1% agarose–formaldehyde gels, which were subsequently blotted onto Hybond-N+ membranes (Amersham) using 20× SSC. To detect HuR, filters were hybridized in the presence of 50% formamide (43) using a [α-32P]GTP-labeled SP6 transcribed probe spanning nt 121–488 of the murine HuR cDNA (accession number NM_010485). To detect BRF1, control β-actin and G3PDH, PCR fragments of the respective cDNAs (BRF1, nt 524–1019, accession number X99404; G3PDH, nt 589–1246, accession number M33197; murine β-actin, nt 554–815, accession number X03765) were labelled with [α-32P]dCTP using Klenow enzyme (New England Biolabs) and the gene specific 3′ primers. Filters were hybridized overnight at 65°C in 0.5 M sodium phosphate buffer pH 7.2 containing 1% BSA (Fraction V, Sigma), 7% SDS and 5 mM EDTA, and subsequently washed at 55°C each in 2× SSC/0.2% SDS and 0.2× SSC/0.2% SDS.
To analyze mRNA stability of the GFP–ARE reporter, cells were treated 72 h after transfection with 5 µg/ml of actinomycin D. Cytoplasmic RNA was harvested at the indicated time-points (44) and northern blot analysis performed using an SP6 transcribed [α-32P]GTP-labeled probe spanning a XbaI–EcoRI fragment containing the murine IL-3 3′UTR as described.
Signal intensities were quantified using a Personal Molecular Imager® FX (Bio-Rad) and the Quantity One™ software (Bio-Rad). To normalize for loading differences, signal intensities were normalized to the β-actin or G3PDH control.
Western blot analysis
Western blot analysis was performed as recently described (21). Briefly, 30–50 µg of total cell lysates in RIPA buffer (BRF1 and AUF1) or 15 µg of cytoplasmic cell lysate (HuR), prepared with the NE-PER® nuclear cytoplasmic extraction reagents (Pierce), were resolved on 4–20% gradient polyacrylamide gels (Anamed Electrophorese GmbH) and blotted onto Immobilon-P (Millipore) membranes. To detect BRF1, a polyclonal antibody was generated in rabbits immunized with a C-terminal BRF1 peptide (SDSPTLDNSRRLPIFSRLSISDD; Neosystem) covalently linked to KLH. Antibodies were subsequently affinity purified using the BRF1 peptides employed in immunization. Monoclonal mouse-anti human HuR (Santa Cruz), polyclonal rabbit-anti human AUF1 (Upstate Biotechnology) and monoclonal mouse-anti bovine α-tubulin antibodies (Molecular Probes) served to detect the respective genes. An alkaline phosphatase-coupled goat-anti-rabbit IgG (Southern Biotechnology Associates Inc.) or a horseradish-peroxidase-coupled goat-anti-mouse IgG (DAKO) were used as secondary antibodies. Development was performed using CDP-Star (Roche) or ECL Advance (Amersham), respectively. For quantification, autoradiographs were scanned and analyzed by densitometry using the Quantity One® software (Bio-Rad).
Detection of GM-CSF by ELISA
Supernatants of triplicate cultures treated with medium or individual siRNA were harvested 72 h after transfection. GM-CSF was determined by ELISA (Pharmingen) according to the manufacturers instructions.
RESULTS
SiRNA targeting HuR and BRF1
We recently showed that siRNA targeting BRF1 increased mRNA stability of a GFP–ARE reporter and, concomitantly, fluorescence of transfected cells by FACS. Indeed, these siRNAs allowed distinction between cells with two wild-type BRF1 alleles from cells exhibiting a heterozygous inactivation of BRF1, solely based on the extent of the fluorescence shift (22). These data encouraged us to extend this approach to further AUBPs, arguing that if changes in GFP–ARE transcript stability are faithfully reflected in altered fluorescence intensity, this approach will enable the assessment of ARE-mRNA turnover rates in live cells without requiring the use of transcriptional inhibitors. As exemplified in Figure 1, downregulation of AUBPs stabilizing or destabilizing ARE-mRNAs will decrease or increase fluorescence intensity, respectively.
Figure 1.
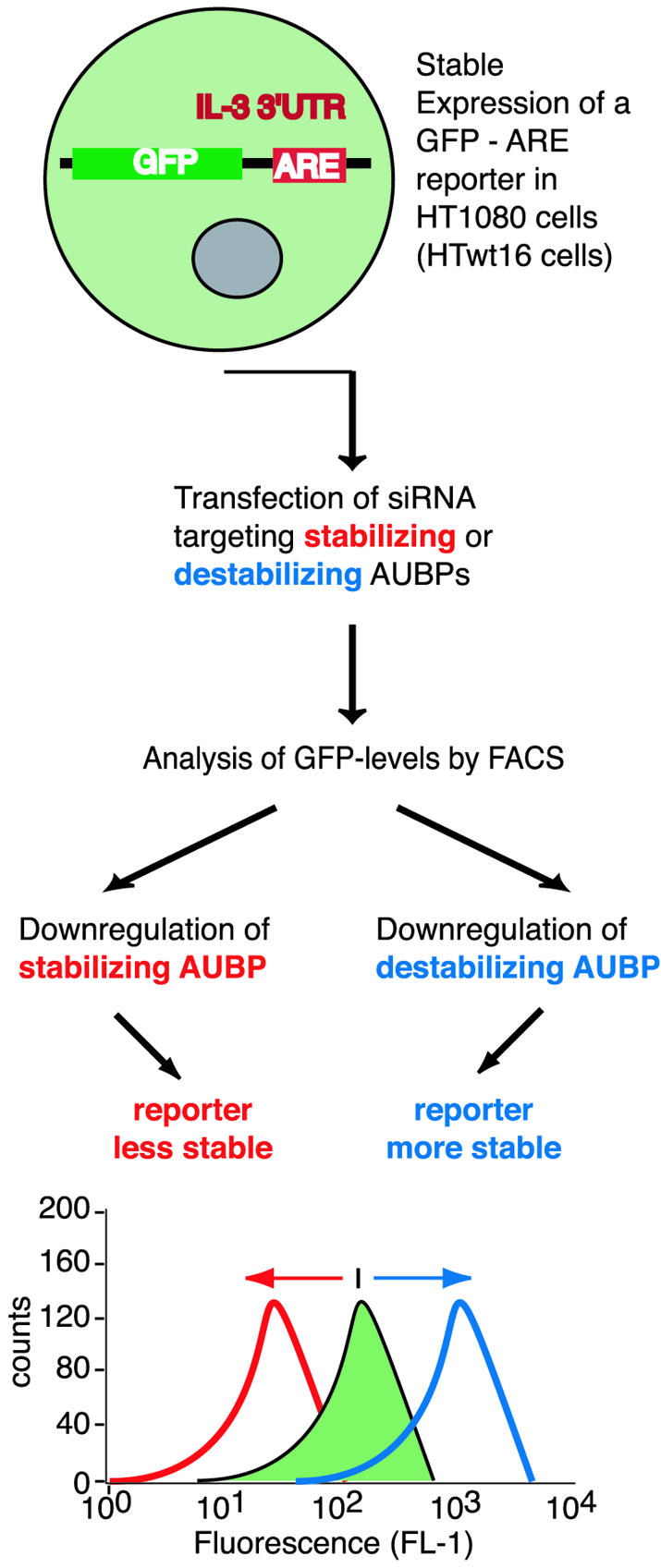
Reporter system employed to reveal effects of decreased AUBP expression on ARE-mRNA stability. HT1080 cells stably expressing a GFP–ARE reporter (HTwt16) are transfected with siRNA targeting AUBP. Decreasing the level of an AUBP that stabilizes ARE-mRNAs should result in decreased fluorescence by FACS. In contrast, downregulation of an AUBP that destabilizes ARE-mRNAs should result in increased fluorescence.
First, we investigated HuR, an AUBP known to stabilize ARE-mRNAs (15,16). A siRNA targeting HuR was designed that reduced HuR mRNA levels to 40% of medium or β-globin siRNA treated controls. By comparison, treatment of cells with BRF1 siRNA alone or in combination with β-globin or HuR siRNAs reduced BRF1 mRNA levels to 25% of controls (Fig. 2A). An additional HuR siRNA that had recently been described by others (42) reduced HuR protein levels to 60% of controls by western blot (Fig. 2B). Efficient inhibition of BRF1 by siRNA was also established in western blots using a polyclonal anti-BRF1 antibody generated in rabbits immunized with a C-terminal peptide of BRF1. Specific recognition of BRF1 by this antibody was confirmed by the absence of the signal in mutant slowC cells, which lack BRF1 expression compared to parental Htwt16 cells, which express wild-type BRF1 (22) (Fig. 2B).
Figure 2.

HuR and BRF1 siRNA have opposing effects on fluorescence and GFP–ARE mRNA stability in Htwt16 cells. (A) Northern blot analysis of HuR and BRF1 expression in Htwt16 cells treated for 72 h with control β-globin, HuR (oligo A), BRF1 siRNA or their combinations. (B) Western blots of 15 µg of cytoplasmic and 30 µg of total cell lysate to analyze cells treated with HuR (oligo B) or BRF1 siRNA. As control for the specificity of the anti-BRF1 antibody, absence of the BRF1 signal in slowC cells lacking BRF1 expression is shown in comparison to Htwt16 cells, which express wild-type BRF1. (C) FACS analysis of cells treated for 72 h. In experiment 1 (A–F) the shaded areas represent medium and the overlays siRNA-treated cells; HuR (oligo A) was employed in panels B and F. In experiment 2 (G–I) the shaded curves represent cells treated with β-globin control and the overlays represent cells treated with specific BRF1 and/or HuR (oligo B) siRNA. (D) To evaluate the effects of siRNA on GFP–ARE mRNA stability, cells were exposed to actinomycin D for the indicated time-points 72 h after transfection of medium, β-globin control, BRF1 and/or HuR (oligo A) siRNA. Values were normalized to the β-actin control. (E) Quantification of the mean ± SEM of at least three independent experiments is shown and the half-life (T1/2) of the GFP–ARE reporter for each individual treatment is indicated.
Next, we determined by FACS whether siRNA treatment affected GFP expression. The results of two independent experiments are shown in Figure 2C. For quantification, the greatest difference (D) in fluorescence between the curves obtained from cells treated with medium or siRNA was calculated using Kolmogorow–Smirnov statistics (Table 1). In experiment 1, no significant differences were observed between cells transfected with medium or a non-specific β-globin control siRNA (Fig. 2C, panel A). However, transfection of HuR siRNA decreased fluorescence (panel B) and BRF1siRNA increased fluorescence as previously reported (panel D) (22). To evaluate whether HuR siRNA might antagonize the stabilizing effect of BRF1 siRNA, cells were treated with combinations of siRNAs. Combined treatment with HuR or BRF1 and a non-specific β-globin siRNA led to the same shift in fluorescence as treatment with HuR or BRF1 siRNA alone (panel C and E). In contrast, combined treatment with BRF1 and HuR siRNA abolished the effects of the single siRNAs (panel F). We verified these findings with a second HuR siRNA (Fig. 2C, experiment 2). Again, HuR siRNA decreased fluorescence of transfected cells compared to β-globin siRNA-treated control cells (panel G) and annihilated the increase in fluorescence observed in cells treated solely with BRF1 siRNA (panels H and I).
Table 1. Treatment of Htwt16 cells with HuR siRNA decreases fluorescence, an effect that is reversed upon combined treatment with BRF1 siRNA.
| siRNA treatment | na | Fluorescenceb | DKolmogorow–Smirnowc mean ± SD | Pd |
|---|---|---|---|---|
| β-globin | 13 | no effect | 0.07 ± 0.04 | |
| HuR | 9 | decreased | 0.23 ± 0.08 | <0.05 |
| HuR + β-globin | 4 | decreased | 0.24 ± 0.23 | <0.05 |
| BRF1 | 9 | increased | 0.31 ± 0.12 | <0.05 |
| BRF1 + β-globin | 5 | increased | 0.29 ± 0.11 | <0.05 |
| HuR + BRF1 | 5 | no effect | 0.07 ± 0.03 | = 0.9 |
aNumber of experiments.
bEffect on fluorescence of medium compared to siRNA-treated cells. For statistical analysis only HuR (oligo A) was taken into consideration.
cGreatest distance (D) between curves obtained from medium- and siRNA-treated cells.
dStudent’s t-test comparing D of cells treated with control β-globin and specific siRNA.
To evaluate whether changes in fluorescence were accompanied by altered mRNA stability of the GFP–ARE reporter, actinomycin D chase experiments were performed. The results of a representative experiment are shown in Figure 2D and quantification of at least three independent experiments for each treatment is exhibited in Figure 2E. Cells treated with HuR siRNA exhibited reduced GFP–ARE mRNA levels and a slightly accelerated decay that was reflected in a decrease of the GFP–ARE half-life (t1/2 = 1.2 ± 0.2 h) compared to controls (t1/2 = 1.6 ± 0.2 h). The stabilization exerted by BRF1 siRNA (t1/2 = 3.0 ± 0.6 h) was reversed by combined treatment with HuR siRNA (t1/2 = 1.8 ± 0.4 h). In all actinomycin D chase experiments performed, we observed the decay of the GFP–ARE mRNA reporter to be fastest during the first hour of treatment. Actinomycin D has been shown to itself stabilize ARE-mRNAs (45), an effect that may be responsible for the observed impediment of decay at later time points. Together, changes in fluorescence intensity in cells treated with BRF1, HuR and combinations of these two siRNAs were qualitatively reflected in altered decay rates of the GFP–ARE reporter mRNA.
SiRNA targeting AUF1
The role of AUF1 in ARE-mediated mRNA decay is complicated by the existence of four isoforms and both destabilizing and stabilizing effects have been reported (40,41). To differentially decrease selected AUF1 isoforms we targeted siRNA to exons 1 and 3, which are common to all four isoforms, to exon 2, which is contained in p40AUF1 and p45AUF1, and to exon 7, which is present in p42AUF1 and p45AUF1 (Fig. 3A). First, to evaluate whether the selected siRNA would specifically reduce the targeted isoforms we analyzed their distribution by western blot and quantified the respective bands by densitometry (Fig. 3B). In all experiments performed, we observed a non-specific band (marked by an asterisk) at ∼55 kDa whose signal intensity correlated to the α-tubulin loading control as determined by densitometry (data not shown). The p37 and p45 isoforms were clearly distinguished in control cells (lanes 1 and 2). However, p40AUF1, which is highly abundant in these cells, resolved as a very broad band obscuring simultaneous detection of the weakly expressed p42AUF1. As expected, both siRNAs targeting exons 1 and 3 decreased expression of all four isoforms (lanes 3 and 6). Two independent oligoribonucleotide duplexes targeting exon 2 of AUF1 reduced only expression of p40AUF1, the most abundant isoform in this cell line, and p45AUF1 (lanes 4 and 5). In contrast, expression of p37AUF1 was comparable to medium or β-globin siRNA-treated controls. Of note, the low abundant p42 isoform became visible only after selective removal of p40AUF1, in cells treated with exon 2-specific siRNA. Finally, siRNA targeting exon 7 specifically reduced p45AUF1. The concomitant decrease of p42AUF1 could not be analyzed (lane 7), due to the reasons mentioned above. In conclusion, the AUF1 siRNA employed allowed selective and efficient inhibition of the targeted isoforms, with p45AUF1 being reduced to 10–27% of controls by the different siRNA.
Figure 3.

Exon-specific targeting of AUF1 allows selective reduction of specific AUF1 isoforms in Htwt16 cells. (A) Schematic drawing of the mRNAs coding for the different AUF1 isoforms and the localization of the siRNA employed. Location of the RRMs and the glutamine rich (Q) stretch are indicated. The siRNA AUF1 (exon 2/oligo A) marked by an asterisk targets 16 bp of exon 1 and 3 bp of exon 2. (B) Two independent western blots of cells treated for 72 h with AUF1 siRNA targeting exon 1 (lanes 3), exon 2 (lanes 4 and 5), exon 3 (lanes 6) and exon 7 (lanes 7). The asterisk marks a non-specific band at ∼55 kDa that was observed in all experiments performed, and whose signal intensity correlated to the α-tubulin loading control in individual samples. Quantitative analysis of experiment 2 is shown in the table.
Next, we investigated how the different AUF1 siRNA affected fluorescence of transfected cells by FACS. Figure 4A shows a representative experiment and quantification is exhibited in Table 2. Decreasing the expression of all four isoforms by targeting exon 1 or 3 did not significantly alter fluorescence compared to β-globin siRNA-treated controls (panels B and E). Downregulation of both p42AUF1 and p45AUF1 led in some experiments to a slight, not consistently observed and statistically not significant, decrease of fluorescence compared to controls (panel F and Table 2). Together, these data might indicate that AUF1 does not affect expression of the GFP–ARE reporter in this cell line. However, we observed a significant increase of fluorescence in cells exhibiting reduced p40AUF1 and p45AUF1 expression after treatment with two independent exon 2-specific oligoribonucleotide duplexes (panels C and D). To substantiate these findings we performed a titration experiment, treating cells with 240–1.92 nM of the different AUF1 siRNA employed. Whereas siRNA targeting exons 1, 3 or 7 did not increase fluorescence of transfected cells at any of the concentrations tested, both siRNA targeting exon 2 increased fluorescence, with a minimal effect still being observed at 9.6 nM (data not shown).
Figure 4.

Selective downregulation of p40AUF1/p45AUF1 increases fluorescence and stabilizes the GFP–ARE reporter in Htwt16 cells. (A) FACS analysis of cells treated for 72 h with medium or siRNA as indicated. (B) To evaluate the effects of siRNA on GFP–ARE mRNA stability, cells were exposed to actinomycin D for the indicated time-points 72 h after transfection of siRNA.
Table 2. Exon specific targeting of AUF1 reveals downregulation of p40AUF1/p45AUF1 to increase fluorescence of transfected cells.
| siRNA treatment | Isoforms targeted | na | Fluorescenceb | DKolmogorow–Smirnowc mean ± SD | Pd |
|---|---|---|---|---|---|
| β-globin | 7 | no effect | 0.09 ± 0.04 | ||
| AUF1 exon 1 | p37, p40, p42, p45 | 4 | no effect | 0.09 ± 0.05 | = 1 |
| AUF1 exon 2 (oligo A) | p40, p45 | 7 | increased | 0.46 ± 0.09 | <0.05 |
| AUF1 exon 2 (oligo B) | p40, p45 | 4 | increased | 0.29 ± 0.09 | <0.05 |
| AUF1 exon 3 | p37, p40, p42, p45 | 4 | no effect | 0.1 ± 0.08 | = 0.9 |
| AUF1 exon 7 | p42, p45 | 4 | no effect | 0.12 ± 0.03 | = 0.2 |
aNumber of experiments.
bEffect on fluorescence of medium compared to siRNA-treated cells (Fig. 4A).
cGreatest distance (D) between curves obtained from medium and siRNA-treated cells.
dStudent’s t-test comparing D of cells treated with control β-globin and specific siRNA.
Actinomycin D chase experiments (Fig. 4B) revealed that cells exhibiting an increased fluorescence due to selective reduction of p40AUF1/p45AUF1 indeed showed a slower decay rate of the GFP–ARE reporter compared to β-globin siRNA- or medium-treated controls. The half-life of the GFP–ARE reporter mRNA in cells treated with AUF1 siRNA targeting exon 2 varied between individual experiments, but a consistent increase was observed compared to controls. The reduced decay rate of the GFP–ARE mRNA in cells thus treated was reflected in an accordingly increased steady-state level of the reporter, which was, however, 15–20% higher than expected. This discrepancy between the predicted and the observed increase was within the variation of the experiment. In contrast, cells exhibiting reduced levels of all four AUF1 isoforms or selectively of p42AUF1/p45AUF1 showed decay rates of the reporter that were within the range of the controls.
In addition, NIH 3T3 cells stably expressing the GFP–ARE reporter also exhibited only an increased fluorescence if treated with AUF1 (exon 2a), but not AUF1 (exon 3) siRNA (data not shown).
Effects of HuR, BRF1 and AUF1 siRNA on endogenous GM-CSF expression
To validate these data we wished to investigate whether these siRNAs influenced expression of a physiological endogenous target gene. A recent cDNA microarray analysis had revealed that HT1080 cells lacking wild-type BRF1 (22) expressed at least six-fold higher GM-CSF mRNA and protein levels compared to cells with two intact BRF1 alleles, thus identifying GM-CSF as a BRF1 target gene (unpublished data). Therefore, HT1080 cells were treated for 72 h with the different siRNAs and secreted GM-CSF was subsequently determined by ELISA. Treatment with BRF1 siRNA increased GM-CSF production, an effect that was reduced upon simultaneous addition of HuR but not of β-globin siRNA (Fig. 5A). Extending this study to AUF1, a known regulator of GM-CSF expression (46), we found an even more powerful effect than with BRF1 siRNA. Of note, only selective reduction of p40AUF1/p45AUF1 increased GM-CSF expression, whereas reduction of all four isoforms or p42AUF1/p45AUF1 again had no effect (Fig. 5B). The increase in GM-CSF expression in cells treated with AUF1 (exon 2a) siRNA was partially reversed if cells were simultaneously treated with HuR siRNA (Fig. 5C). Therefore, not only do the relative levels of stabilizing and destabilizing AUBPs regulate expression of GM-CSF but, surprisingly, also the relative levels of individual AUF1 isoforms.
Figure 5.

Downregulation of p40AUF1/p45AUF1 increases GM-CSF secretion of transfected Htwt16 cells. Secreted GM-CSF was measured by ELISA in the culture supernatant of cells treated for 72 h with BRF1 and HuR (oligo A) siRNA (A), AUF1 siRNA (B) and AUF1 (exon 2a) and HuR (oligo A) siRNA (C).
DISCUSSION
The present study was designed to evaluate siRNA targeting AUBPs in the investigation of ARE-mRNA turnover. This seemed to be a timely objective as, with the exception of TTP (47), no AUBP knockout mice models have been reported. Results gained from loss-of-function experiments are needed to validate data previously obtained by overexpression of AUBPs. This is of particular importance because these experiments did not provide unifying conclusions (see below).
Indeed, treatment of Htwt16 cells with siRNA targeting HuR, a protein known to stabilize ARE-mRNAs (15,16) decreased fluorescence intensity and promoted decay of the GFP–ARE-mRNA reporter, thus confirming HuR to be a stabilizer of ARE-mRNAs. BRF1 siRNA induced the expected increase in fluorescence intensity and stabilized the GFP–ARE reporter, effects that were nullified in cells treated with a combination of HuR and BRF1 siRNAs. Inactive siRNAs have been claimed to compete with active siRNAs in a sequence-independent manner (48). However, non-specific effects were ruled out in our experiments, showing that cells treated with a combination of BRF1 and control β-globin siRNA exhibited the same shift in fluorescence as cells treated solely with BRF1 siRNA. Thus, we concluded that HuR and BRF1 control mRNA stability of the GFP–ARE reporter in an antagonistic fashion, presumably by competing for the ARE. Experiments revealing that BRF1 and HuR also exerted opposing effects on expression of GM-CSF, an endogenous BRF1 target gene recently identified by cDNA microarray, further validated our findings.
In contrast to BRF1 and HuR, the role of AUF1 in ARE-mRNA turnover has not been fully clarified. Conclusions, of whether this protein acts as a stabilizer or destabilizer of ARE-mRNAs, are based on indirect evidence associating AUF1 abundance with ARE-mRNA stability (36,38,39,49) and on experiments using AUF1 overexpression, which provided ambiguous results. In NIH 3T3 cells, all four isoforms stabilized ARE-mRNAs (41). Other studies, however, showed that both p37AUF1 and p40AUF1 promoted decay in several cell lines investigated, including NIH 3T3 (50). Also, in K562 cells forced expression of AUF1 antagonized stabilization of ARE-mRNAs (40). The reasons for these discrepancies are not clear. Short of performing a gene knockout experiment, exon-specific targeting of AUF1 by siRNA might provide a more physiologic approach to functionally dissect this gene. At first sight, our finding that two different siRNAs efficiently inhibiting expression of all four AUF1 isoforms did not exert an effect on cellular fluorescence or mRNA stability at any of the concentrations tested would argue that, at least in HT1080 cells, AUF1 is not involved in steady-state control of ARE-mRNA turnover. However, this possibility was rejected because the decrease of p40AUF1/p45AUF1 in cells treated with exon 2-specific siRNA strongly induced cellular fluorescence and stabilized the GFP–ARE reporter mRNA. This seems to be mainly due to reduction of the most abundant p40 isoform rather than to the decrease in p45AUF1 because downregulation of p42AUF1 together with p45AUF1 did not significantly affect fluorescence of transfected cells. We wish to stress that the AUF1 siRNA employed had the same effect on GM-CSF, a gene known to be controlled by AUF1 (46). Again, downregulation of all four isoforms had no effect, whereas selective reduction of p40AUF1 and p45AUF1 strongly stimulated GM-CSF production. Based on these results, one might draw the conclusion that p40AUF1, like BRF1, acts as a destabilizer of ARE-mRNAs. However, we would like to offer an alternative explanation.
The four AUF1 isoforms differ considerably in their ARE-binding affinities in vitro, with the rank order of p37>p42> p45>p40 (26) and the two isoforms with the highest binding affinity exert the most profound effect on ARE-mRNA stability (40,41). Thus, we propose that in HT1080 cells the highly abundant yet low-affinity p40AUF1 competes with p37AUF1 and p42AUF1for ARE-binding. After selective downregulation of p40AUF1, p37AUF1 and p42AUF1 gain access to the ARE and stabilize transcripts. This model does not exclude that post-transcriptional modifications of AUF1 might play an additional role, as the affinity of p40AUF1 for the ARE has been shown to be affected by phosphorylation (51–53). Our proposition that alterations in ARE-mRNA stability might be induced by changes in the relative rather than the absolute levels of AUF1 isoforms would explain why the uniform reduction of all four isoforms had no effect. A key factor may well be the ratio of p37AUF1 and possibly p42AUF1 to p40AUF1. This notion is supported by two previous observations. First, the decreased stability of the β-adrenergic receptor mRNA in the failing compared to normal human heart has been associated with an increased expression of both p40AUF1 and p45AUF1 (38). Second, differential expression of AUF1 isoforms accompanied the decreased half-life of GM-CSF mRNA in neonatal compared to adult PBMC (36,37). Both p37AUF1 and p40AUF1 were decreased in the latter, whereas expression of p42AUF1 was comparable in neonatal and adult PBMC. If our model is correct, it would follow that mRNA splicing might ultimately influence ARE-mRNA stability by determining the relative levels of individual AUF1 isoforms.
The fact that simultaneous targeting of HuR and p40AUF1/p45AUF1 partially cancelled the effect of solely targeting p40AUF1/p45AUF1 clearly indicates that the relative levels of the individual AUF1 isoforms can only be one determinant of ARE-mRNA stability. We propose that the quantity of all stabilizing (HuR) and destabilizing AUBPs (TTP, BRF1, BRF2), together with the relative amount of the individual AUF1 isoforms, determines the net stability of ARE- containing transcripts in a balanced fashion. Thus, silencing a single destabilizing factor will tip the balance towards increased stabilization despite the fact that other ARE-destabilizing factors may still be present. In return, decreasing the expression of any stabilizing factor will favor destabilization despite the potential presence of additional stabilizing AUBPs.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to thank Don Benjamin and Martin Schmidlin for critical reading of the manuscript. This work was supported by grant 31-57065.99 from the Swiss National Science Foundation.
REFERENCES
- 1.Ross J. (1996) Control of messenger RNA stability in higher eukaryotes. Trends Genet., 12, 171–175. [DOI] [PubMed] [Google Scholar]
- 2.Guhaniyogi J. and Brewer,G. (2001) Regulation of mRNA stability in mammalian cells. Gene, 265, 11–23. [DOI] [PubMed] [Google Scholar]
- 3.Wilusz C.J., Wormington,M. and Peltz,S.W. (2001) The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell. Biol., 2, 237–246. [DOI] [PubMed] [Google Scholar]
- 4.Shaw G. and Kamen,R. (1986) A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell, 46, 659–667. [DOI] [PubMed] [Google Scholar]
- 5.Chen C.Y. and Shyu,A.B. (1995) AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci., 20, 465–470. [DOI] [PubMed] [Google Scholar]
- 6.Shyu A.B., Belasco,J.G. and Greenberg,M.E. (1991) Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mRNA decay. Genes Dev., 5, 221–231. [DOI] [PubMed] [Google Scholar]
- 7.Chen C.Y., Gherzi,R., Ong,S.E., Chan,E.L., Raijmakers,R., Pruijn,G.J., Stoecklin,G., Moroni,C., Mann,M. and Karin,M. (2001) AU binding proteins recruit the exosome to degrade ARE-containing mRNAs. Cell, 107, 451–464. [DOI] [PubMed] [Google Scholar]
- 8.Mukherjee D., Gao,M., O’Connor,J.P., Raijmakers,R., Pruijn,G., Lutz,C.S. and Wilusz,J. (2002) The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J., 21, 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keyomarsi K. and Pardee,A.B. (1993) Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc. Natl Acad. Sci. USA, 90, 1112–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lebwohl D.E., Muise-Helmericks,R., Sepp-Lorenzino,L., Serve,S., Timaul,M., Bol,R., Borgen,P. and Rosen,N. (1994) A truncated cyclin D1 gene encodes a stable mRNA in a human breast cancer cell line. Oncogene, 9, 1925–1929. [PubMed] [Google Scholar]
- 11.Dixon D.A., Tolley,N.D., King,P.H., Nabors,L.B., McIntyre,T.M., Zimmerman,G.A. and Prescott,S.M. (2001) Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Invest., 108, 1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nair A.P., Hahn,S., Banholzer,R., Hirsch,H.H. and Moroni,C. (1994) Cyclosporin A inhibits growth of autocrine tumour cell lines by destabilizing interleukin-3 mRNA. Nature, 369, 239–242. [DOI] [PubMed] [Google Scholar]
- 13.Jacob C.O., Lee,S.K. and Strassmann,G. (1996) Mutational analysis of TNF-alpha gene reveals a regulatory role for the 3′-untranslated region in the genetic predisposition to lupus-like autoimmune disease. J. Immunol., 156, 3043–3050. [PubMed] [Google Scholar]
- 14.Kontoyiannis D., Pasparakis,M., Pizarro,T.T., Cominelli,F. and Kollias,G. (1999) Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity, 10, 387–398. [DOI] [PubMed] [Google Scholar]
- 15.Peng S.S., Chen,C.Y., Xu,N. and Shyu,A.B. (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J., 17, 3461–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan X.C. and Steitz,J.A. (1998) Overexpression of HuR, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of ARE-containing mRNAs. EMBO J., 17, 3448–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piecyk M., Wax,S., Beck,A.R., Kedersha,N., Gupta,M., Maritim,B., Chen,S., Gueydan,C., Kruys,V., Streuli,M. and Anderson,P. (2000) TIA-1 is a translational silencer that selectively regulates the expression of TNF-alpha. EMBO J., 19, 4154–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carballo E., Lai,W.S. and Blackshear,P.J. (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science, 281, 1001–1005. [DOI] [PubMed] [Google Scholar]
- 19.Lai W.S., Carballo,E., Strum,J.R., Kennington,E.A., Phillips,R.S. and Blackshear,P.J. (1999) Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol., 19, 4311–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carballo E., Lai,W.S. and Blackshear,P.J. (2000) Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood, 95, 1891–1899. [PubMed] [Google Scholar]
- 21.Stoecklin G., Ming,X.F., Looser,R. and Moroni,C. (2000) Somatic mRNA turnover mutants implicate tristetraprolin in the interleukin-3 mRNA degradation pathway. Mol. Cell. Biol., 20, 3753–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stoecklin G., Colombi,M., Raineri,I., Leuenberger,S., Mallaun,M., Schmidlin,M., Gross,B., Lu,M., Kitamura,T. and Moroni,C. (2002) Functional cloning of BRF1, a regulator of ARE-dependent mRNA turnover. EMBO J., 21, 4709–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stoecklin G., Stoeckle,P., Lu,M., Muehlemann,O. and Moroni,C. (2001) Cellular mutants define a common mRNA degradation pathway targeting cytokine AU-rich elements. RNA, 7, 1578–1588. [PMC free article] [PubMed] [Google Scholar]
- 24.Brewer G. (1991) An A + U-rich element RNA-binding factor regulates c-myc mRNA stability in vitro. Mol. Cell. Biol., 11, 2460–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang W., Wagner,B.J., Ehrenman,K., Schaefer,A.W., DeMaria,C.T., Crater,D., DeHaven,K., Long,L. and Brewer,G. (1993) Purification, characterization and cDNA cloning of an AU-rich element RNA-binding protein, AUF1. Mol. Cell. Biol., 13, 7652–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagner B.J., DeMaria,C.T., Sun,Y., Wilson,G.M. and Brewer,G. (1998) Structure and genomic organization of the human AUF1 gene: alternative pre-mRNA splicing generates four protein isoforms. Genomics, 48, 195–202. [DOI] [PubMed] [Google Scholar]
- 27.Arao Y., Kuriyama,R., Kayama,F. and Kato,S. (2000) A nuclear matrix-associated factor, SAF-B, interacts with specific isoforms of AUF1/hnRNP D. Arch. Biochem. Biophys., 380, 228–236. [DOI] [PubMed] [Google Scholar]
- 28.Sarkar B., Lu,J.L. and Schneider,R.J. (2003) Nuclear import and export functions in the different isoforms of the AUF1/hnRNP D protein family. J. Biol. Chem., 31, 31. [DOI] [PubMed] [Google Scholar]
- 29.Laroia G. and Schneider,R.J. (2002) Alternate exon insertion controls selective ubiquitination and degradation of different AUF1 protein isoforms. Nucleic Acids Res., 30, 3052–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laroia G., Cuesta,R., Brewer,G. and Schneider,R.J. (1999) Control of mRNA decay by heat shock-ubiquitin-proteasome pathway. Science, 284, 499–502. [DOI] [PubMed] [Google Scholar]
- 31.Laroia G., Sarkar,B. and Schneider,R.J. (2002) Ubiquitin-dependent mechanism regulates rapid turnover of AU-rich cytokine mRNAs. Proc. Natl Acad. Sci. USA, 99, 1842–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eversole A. and Maizels,N. (2000) In vitro properties of the conserved mammalian protein hnRNP D suggest a role in telomere maintenance. Mol. Cell. Biol., 20, 5425–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuentes-Panana E.M., Peng,R., Brewer,G., Tan,J. and Ling,P.D. (2000) Regulation of the Epstein–Barr virus C promoter by AUF1 and the cyclic AMP/protein kinase A signaling pathway. J. Virol., 74, 8166–8175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiledjian M., DeMaria,C.T., Brewer,G. and Novick,K. (1997) Identification of AUF1 (heterogeneous nuclear ribonucleoprotein D) as a component of the alpha-globin mRNA stability complex. Mol. Cell. Biol., 17, 4870–4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grosset C., Chen,C.Y., Xu,N., Sonenberg,N., Jacquemin-Sablon,H. and Shyu,A.B. (2000) A mechanism for translationally coupled mRNA turnover: interaction between the poly(A) tail and a c-fos RNA coding determinant via a protein complex. Cell, 103, 29–40. [DOI] [PubMed] [Google Scholar]
- 36.Buzby J.S., Lee,S.M., Van Winkle,P., DeMaria,C.T., Brewer,G. and Cairo,M.S. (1996) Increased granulocyte-macrophage colony-stimulating factor mRNA instability in cord versus adult mononuclear cells is translation-dependent and associated with increased levels of A + U-rich element binding factor. Blood, 88, 2889–2897. [PubMed] [Google Scholar]
- 37.Buzby J.S., Brewer,G. and Nugent,D.J. (1999) Developmental regulation of RNA transcript destabilization by A + U-rich elements is AUF1-dependent. J. Biol. Chem., 274, 33973–33978. [DOI] [PubMed] [Google Scholar]
- 38.Pende A., Tremmel,K.D., DeMaria,C.T., Blaxall,B.C., Minobe,W.A., Sherman,J.A., Bisognano,J.D., Bristow,M.R., Brewer,G. and Port,J. (1996) Regulation of the mRNA-binding protein AUF1 by activation of the beta-adrenergic receptor signal transduction pathway. J. Biol. Chem., 271, 8493–8501. [DOI] [PubMed] [Google Scholar]
- 39.Sirenko O.I., Lofquist,A.K., DeMaria,C.T., Morris,J.S., Brewer,G. and Haskill,J.S. (1997) Adhesion-dependent regulation of an A+U-rich element-binding activity associated with AUF1. Mol. Cell. Biol., 17, 3898–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Loflin P., Chen,C.Y. and Shyu,A.B. (1999) Unraveling a cytoplasmic role for hnRNP D in the in vivo mRNA destabilization directed by the AU-rich element. Genes Dev., 13, 1884–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu N., Chen,C.Y. and Shyu,A.B. (2001) Versatile role for hnRNP D isoforms in the differential regulation of cytoplasmic mRNA turnover. Mol. Cell. Biol., 21, 6960–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mazan-Mamczarz K., Galban,S., Lopez de Silanes,I., Martindale,J.L., Atasoy,U., Keene,J.D. and Gorospe,M. (2003) RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl Acad. Sci. USA, 100, 8354–8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nair A.P., Hirsch,H.H. and Moroni,C. (1992) Mast cells sensitive to v-H-ras transformation are hyperinducible for interleukin 3 expression and have lost tumor-suppressor activity. Oncogene, 7, 1963–1972. [PubMed] [Google Scholar]
- 44.Gough N.M. (1988) Rapid and quantitative preparation of cytoplasmic RNA from small numbers of cells. Anal. Biochem., 173, 93–95. [DOI] [PubMed] [Google Scholar]
- 45.Chen C.Y., Xu,N. and Shyu,A.B. (1995) mRNA decay mediated by two distinct AU-rich elements from c-fos and granulocyte-macrophage colony-stimulating factor transcripts: different deadenylation kinetics and uncoupling from translation. Mol. Cell. Biol., 15, 5777–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhattacharya S., Giordano,T., Brewer,G. and Malter,J.S. (1999) Identification of AUF-1 ligands reveals vast diversity of early response gene mRNAs. Nucleic Acids Res., 27, 1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taylor G.A., Carballo,E., Lee,D.M., Lai,W.S., Thompson,M.J., Patel,D.D., Schenkman,D.I., Gilkeson,G.S., Broxmeyer,H.E., Haynes,B.F. and Blackshear,P.J. (1996) A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity, 4, 445–454. [DOI] [PubMed] [Google Scholar]
- 48.Holen T., Amarzguioui,M., Wiiger,M.T., Babaie,E. and Prydz,H. (2002) Positional effects of short interfering RNAs targeting the human coagulation trigger Tissue Factor. Nucleic Acids Res., 30, 1757–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lapucci A., Donnini,M., Papucci,L., Witort,E., Tempestini,A., Bevilacqua,A., Nicolin,A., Brewer,G., Schiavone,N. and Capaccioli,S. (2002) AUF1 Is a bcl-2 A + U-rich element-binding protein involved in bcl-2 mRNA destabilization during apoptosis. J. Biol. Chem., 277, 16139–16146. [DOI] [PubMed] [Google Scholar]
- 50.Sarkar B., Xi,Q., He,C. and Schneider,R.J. (2003) Selective degradation of AU-rich mRNAs promoted by the p37 AUF1 protein isoform. Mol. Cell. Biol., 23, 6685–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilson G.M., Sun,Y., Lu,H. and Brewer,G. (1999) Assembly of AUF1 oligomers on U-rich RNA targets by sequential dimer association. J. Biol. Chem., 274, 33374–33381. [DOI] [PubMed] [Google Scholar]
- 52.Wilson G.M., Lu,J., Sutphen,K., Suarez,Y., Sinha,S., Brewer,B., Villanueva-Feliciano,E.C., Ysla,R.M., Charles,S. and Brewer,G. (2003) Phosphorylation of p40AUF1 regulates binding to A + U-rich mRNA-destabilizing elements and protein-induced changes in ribonucleoprotein structure. J. Biol. Chem., 278, 33039–33048. [DOI] [PubMed] [Google Scholar]
- 53.Wilson G.M., Lu,J., Sutphen,K., Sun,Y., Huynh,Y. and Brewer,G. (2003) Regulation of A + U-rich element-directed mRNA turnover involving reversible phosphorylation of AUF1. J. Biol. Chem., 278, 33029–33038. [DOI] [PubMed] [Google Scholar]