


Abstract
We showed previously that rad50 and mre11 genes of thermophilic archaea are organized in an operon-like structure with a third gene (nurA) encoding a 5′ to 3′ exonuclease. Here, we show that the rad50, mre11 and nurA genes from the hyperthermo philic archaeon Sulfolobus acidocaldarius are co-transcribed with a fourth gene encoding a DNA helicase. This enzyme (HerA) is the prototype of a new class of DNA helicases able to utilize either 3′ or 5′ single-stranded DNA extensions for loading and subsequent DNA duplex unwinding. To our knowledge, DNA helicases capable of translocating along the DNA in both directions have not been identified previously. Sequence analysis of HerA shows that it is a member of the TrwB, FtsK and VirB4/VirD4 families of the PilT class NTPases. HerA homologs are found in all thermophilic archaeal species and, in all cases except one, the rad50, mre11, nurA and herA genes are grouped together. These results suggest that the archaeal Rad50–Mre11 complex might act in association with a 5′ to 3′ exonuclease (NurA) and a bipolar DNA helicase (HerA) indicating a probable involvement in the initiation step of homologous recombination.
INTRODUCTION
Rad50/SbcC and Mre11/SbcD are ubiquitous proteins involved in many DNA metabolic pathways that maintain genome integrity. In Eucarya, the Rad50 and Mre11 proteins, in association with a third protein partner (Xrs2 in yeast, Nbs1 in vertebrates) play a key role in homologous recombination, non-homologous end-joining, telomere maintenance, cell’s response checkpoint to double-stranded breaks (DSBs) and DSB formation during meiosis (reviewed in 1). In Bacteria, these proteins, called SbcC and SbcD, are involved in homologous recombination (at least when the major initiation complex of the pathway, RecBCD, is not functional), and in the elimination of palindromic sequences during DNA replication (2). In Archaea, the Rad50 and Mre11 homologs are also expected to have a key function in DNA metabolism pathway(s), but presently, no genetic evidence is available. In particular, these proteins could play a major role at the initiation step of homologous recombination. Indeed, as in the case of Eucarya, no homologs to the two major bacterial protein complexes involved in such a process, RecBCD and RecFOR, are found in Archaea (3,4). During the initiation of homologous recombination, DNA ends have to be processed in 3′ overhangs which are necessary for the loading and the activity of recombinases (5). This step is well understood in Bacteria and is mostly performed by the RecBCD complex. This complex exhibits helicase, endonuclease and both 3′ to 5′ and 5′ to 3′ exonuclease activities. After recognition of cis-acting ‘chi’ sites on the chromosome, the 5′ to 3′ exonuclease activity is up-regulated leading to the generation of long 3′ DNA tails (6). Concerning the Rad50/SbcC and Mre11/SbcD pathway, these proteins have been shown in all kingdoms, to form a complex that exhibits enzymatic activities inherent to the SbcD/Mre11 phosphoesterase, i.e. single-stranded endonuclease and 3′ to 5′ double-stranded exonuclease activities, as well as a mechanical function relevant to the Rad50/SbcC protein (7–11). Structural and microscopy studies suggest that the role of the Rad50/SbcC protein, which is a member of the structural maintenance of chromosomes (SMC) protein family, could be to maintain DNA molecules in close contact (12,13). In Eucarya, the third protein partner Xrs2/Nbs1, for which no homolog is found in Bacteria and Archaea, seems to regulate (at least in humans) the endonuclease activity associated with the Rad50–Mre11 complex in an ATP-dependent fashion, and this regulation leads to the cleavage of 3′ DNA tails (14). The activities associated with the Rad50/SbcC–Mre11/SbcD complex do not lead to an understanding of how this complex acts in the processing of DNA ends in 3′ overhangs during homologous recombination. Thus, several workers suggested that other protein partners, such as a 5′ to 3′ nuclease and/or a DNA helicase, could be associated with the complex in order to process DNA ends (1,14). In agreement with this hypothesis, we recently showed that the rad50 and mre11 genes from most thermophilic archaea are linked to a gene, nurA, encoding a 5′ to 3′ exonuclease that might be involved in such a process and is the prototype of a novel nuclease family (15).
In the present report, we show that a fourth, previously uncharacterized, gene is co-transcribed with rad50, mre11 and nurA genes in the thermophilic archaeon Sulfolobus acidocaldarius and that the four genes cluster is present in the genomes of most of the thermophilic archaeal species. Computer-aided sequence analysis indicates that the protein encoded by the uncharacterized gene is related to the PilT/VirD4 class of NTPases. Characterization of the S.acidocaldarius protein shows that it is a previously undetected type of DNA helicase, which has the striking property to load on either a 3′ or a 5′ DNA tail for subsequent DNA unwinding (we called this enzyme HerA for ‘helicase repair of Archaea’, to correspond to NurA, ‘nuclease repair of Archaea’).
These results strongly suggest that the Rad50, Mre11, NurA and HerA proteins from thermophilic archaea could act in DNA ends processing at the initiation step of homologous recombination.
MATERIALS AND METHODS
Genomic context and sequences analyses
Genomic context analyses were performed using the GENOMAPPER program developed by Y. Zivanovic in the IGM, Orsay. The non-redundant protein sequence database (NCBI, NIH, Bethesda, MD) was searched using the PSI-BLAST program (16) and multiple sequence alignments were constructed using the T-Coffee program (17), with manual corrections.
Nucleic acids, enzymes and other reagents
[γ-32P]ATP was obtained from Amersham Pharmacia Biotech and T4 polynucleotide kinase from Promega. Single-stranded M13mp19 DNA and PBR322 DNA were from Invitrogen. The pET-30Ek/lic vector was from Novagen and oligonucleotides were synthesized by MWG biotech.
RT–PCR analyses
Sulfolobus acidocaldarius DSM 639 was grown as previously described (18) until OD600 0.5. Cells were washed with 200 mM Tris–HCl pH 8, resuspended in 50 mM EDTA, and SDS and sodium acetate were added to a final concentration of 1% and 50 mM, respectively. Total RNA was purified by hot acid–phenol extraction and submitted to a DNAse, Rnase free, treatment. One microgram of total RNA was used to perform each reverse transcription (RT) assay as described by the manufacturer (thermoscript RT–PCR System, Invitrogen) using either a nurA-specific primer (5′-TTAATAAAATTG GCTAGGC-3′) or a rad50-specific primer (5′-TTATC TATCATAACTTGAC-3′). PCRs were performed on RT products with the specific primers indicated in the legend of Figure 3. Reaction products were analyzed by electrophoresis on 0.7% agarose gel and ethidium bromide staining.
Figure 3.

Co-transcription of S.acidocaldarius herA, mre11, rad50 and nurA genes. (a) Schematic representation of RT–PCR products obtained using either a nurA- or a rad50-specific primer for the RT reaction. (b and c) Analyses of RT–PCR products. (b) Lane 1, DNA ladder; lane 2, control PCR using 5′mre11 and 3′nurA primers on S.acidocaldarius total RNA; lanes 3–6, PCR products obtained with nurA reverse products and the following primers: lane 3, 5′mre11 and 3′nurA primers; lane 4, rad50 primers; lane 5, mre11 primers; lane 6, nurA primers. (c) Lane 1, DNA ladder; lanes 2–5, PCR products obtained with rad50 reverse products and the following primers: lane 2, 5′herA and 3′rad50 primers; lane 3, herA primers; lane 4, mre11 primers; lane 5, rad50 primers; lane 6, control PCR using 5′herA and 3′rad50 primers on S.acidocaldarius total RNA.
Cloning of the S.acidocaldarius herA gene
Sulfolobus acidocaldarius genomic DNA was prepared as described previously (19) and the herA gene was amplified by PCR using the Pfu polymerase (Promega) and specific herA primers. The 3′ primer was designed from the sequencing of the S.acidocaldarius rad50–mre11 operon (15) and the 5′ primer was determined from the S.acidocaldarius genome sequencing project communicated by R. Garret’s laboratory. The herA gene was inserted into pET-30 Ek/LIC Vector as described by the manufacturer. Site-directed mutagenesis of the lysine 153 was performed using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene) and the mutagenic oligonucleotide, 5′-GGCAGCAACAGGGTCAGGTGCATC AAATAC-3′ (the introduced mutated codon GCA instead of AAA is underlined). The sequencing of the wild-type and the mutated herA genes inserted into pET-30 vector was performed using the fmol DNA cycle sequencing system from Promega.
Expression and purification of the recombinant protein
The HerA protein was overproduced in BL21(DE3) Rosetta Escherichia coli cells (Novagen). When the culture reached an OD600 of 0.5, gene expression was induced by the addition of 1 mM β-d-thiogalactoside. After a further 3 h of growth, the cells were harvested, resuspended in buffer A [20 mM HEPES pH 7.5, 1 M NaCl, 0.03% (v/v) Tween-20, 1 mM PMSF, 5 mM β-mercaptoethanol, 1 µg/ml pepstatin, 1 µg/ml leupeptin], and disrupted by sonication. After a 30 min centrifugation at 12 000 g, the soluble fraction was mixed with 1 ml of Ni-NTA resin (Qiagen) and incubated for 2 h at 4°C with gentle shaking. The resin was then packed, and the column was washed with 10 volumes of buffer A followed by a 5 volume wash with 20 mM imidazole in buffer B (buffer A containing 100 mM NaCl). Proteins were eluted with 200 mM imidazole in buffer B. Imidazole was removed with a PD-10 column (Bio-Rad) in buffer B and the fraction was loaded onto a 1 ml Source 30S column (Amersham). The column was washed with 10 volumes of buffer B and a 30 ml linear salt gradient from 100 mM to 1 M NaCl was applied. HerA protein was eluted at 480 mM NaCl and dialyzed against buffer B. Protein concentration was determined by the Bradford (Bio-Rad) method with bovine serum albumin as the standard. Proteins were stored at –80°C. The HerA K153A protein was overproduced and purified using the same protocol.
Preparation of substrates for helicase assays
Four oligonucleotides were synthesized and used for the preparation of the helicase substrates. Oligonucleotides were purified by extraction from a 10% denaturing-polyacrylamide gel (7 M urea) and labeled in a 50 µl reaction mixture containing 10 pmol of oligonucleotide, 1× polynucleotide kinase buffer, 50 µCi of [γ-32P]ATP and 10 units of T4 kinase for 30 min at 37°C. Labeled oligonucleotides were separated from free nucleotides with a G-25 spin column (Amersham).
The substrate used in the standard DNA helicase reaction was prepared by annealing the 5′ end-labeled 48mer oligonucleotide (5′-CACGACGTTGTAAAACGACGGCCA GTGAATTCGAGCTCGGTACCCGGG-3′) to M13mp19 single-stranded DNA. The single-stranded overhang substrates were prepared by annealing either the 30mer labeled oligonucleotide (5′-GGTCAGTGCTGCAACATTTTGCTG CCGGTC-3′) for the 5′ overhang substrate, or the 30mer labeled (5′-GCCCCTAGGAGATCTCAGCTGGACGTC CGT-3′) for the 3′ overhang substrate, to the 79mer oligonucleotide (5′-ACGGACGTCCAGCTGAGATCTCC TAGGGGCCCATGGCTCGAGCTTAAGTGACCGGCAG CAAAATGTTGCAGCACTGACC-3′). The blunt DNA substrate was prepared by annealing the labeled 30mer oligonucleotide used for the 3′ overhang substrate to the 30mer complementary oligonucleotide (5′-ACGGACGTCC AGCTGAGATCTCCTAGGGGC-3′). For each substrate, annealing was performed as followed: 3 pmol of labeled oligonucleotide were mixed with 1.5 pmol of non-labeled oligonucleotide or ss M13mp19 in a 30 µl reaction mixture containing 100 mM NaCl, 20 mM HEPES, pH 7.5 and 15 mM MgCl2. Reactions were placed in a heat block at 100°C and then slowly cooled to room temperature. The annealed substrate was separated from free oligonuleotides with microSpin S-400 columns (Pharmacia Biotech).
DNA helicase assays
DNA helicase activity was performed in reaction mixtures (20 µl) containing 20 mM HEPES pH 7.5, 50 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol, 200 µg of bovine serum albumin, 2.5 mM ATP, 10 fmol of helicase substrate and the indicated amounts of HerA protein for 30 min at 70°C (Fig. 5b and c). Time courses of helicase activity (Fig. 5d and e) were performed in the presence of 400 nM of HerA in order to obtain a linear rate of unwinding for the time range used (in Fig. 5e, each point represents the average of triplicate determinations and the determination coefficient R2 is ∼0.97). Reactions were stopped by the addition of 5 µl of a 5× stop solution (final concentrations: 50 mM EDTA, 0.5% SDS, 30% glycerol, 0.3% bromophenol blue, 0.3% xylene cyanol) and then run on a 10% polyacrylamide TBE gel at a constant voltage (100 V) and exposed to a PhosphorImager screen (Molecular Dynamics).
Figure 5.

ATPase and helicase activities associated with HerA protein. (a) Time course of HerA ATPase activity: ATP hydrolysis was followed at 70°C for the indicated times in the absence of DNA (triangles) or in the presence of single-stranded DNA (squares) or double-stranded DNA (circles). (b) Helicase activity recovered with a primed circular single-stranded DNA substrate and 20–300 nM wild-type HerA protein or 300 nM HerAK153A protein in 30 min at 70°C. (c) Helicase activity recovered after a 30 min incubation at 70°C using 5′ overhang, 3′ overhang or blunt linear DNA substrates in the presence of 100–500 nM HerA protein. (d and e) Time course of HerA helicase activity. HerA (400 nM) was incubated at 70°C with 5′ overhang (triangles) or 3′ overhang (circles) DNA substrates for the time points shown. Squares correspond to the quantification of the control experiment [data not shown in (d)] performed without enzyme. In (b) and (c), control lanes correspond to the DNA substrate incubated for 30 min at 70°C without enzyme and in (b), (c) and (d), boiled lanes correspond to the heat-denatured substrate.
ATPase assay
The time course of the ATPase activity was performed in 120 µl reaction mixtures containing 20 mM HEPES pH 7.5, 50 mM NaCl, 10 mM MgCl2, 100 µM ATP, 9 nM [γ-32P]ATP, 1 mM dithiothreitol, 200 µg of bovine serum albumin and 40 nM HerA protein in the absence or presence of double-stranded or single-stranded DNA (1 nM pBR322 or 1 nM φX174) at 70°C. At the indicated times, aliquots of 20 µl were chilled on ice and 2 µl of 500 mM EDTA was added to stop the reaction. One microliter of each aliquot was spotted onto a polyethyleneimine-cellulose thin-layer plate, and ATP and Pi were separated by chromatography in 1 M formic acid/0.5 M LiCl. The extent of ATP hydrolysis was quantitated by PhosphorImager analyses. The amount of HerA protein was selected in order to obtain a linear rate of hydrolysis for the time range used. The initial rate of ATP hydrolysis was evaluated with determination coefficients (R2) of 0.98 and 0.96 for single-stranded DNA and double-stranded DNA, respectively.
RESULTS
An uncharacterized gene is grouped with mre11, rad50 and nurA genes in most thermophilic archaeal species
We previously isolated an archaeal gene, nurA, which is linked to the rad50 and mre11 genes in most thermophilic archaeal species. The rad50, mre11 and nurA genes cluster with a fourth gene of ∼1500 nt in all thermophilic archaeal genomes available, with the only exception of Methanococcus jannaschii; the gene order in this cluster is variable (Fig. 1). In all species, this gene encodes a highly conserved protein of ∼55 kDa (30–40% identity between phylogenetically distant archaea). Multiple alignment of amino acid sequences shows that the N-terminal and C-terminal thirds are most highly conserved (Fig. 2). The N-terminal portion of the protein contains the Walker A motif and the C-terminal portion contains the B motif (Fig. 2). These motifs are hallmarks of P-loop NTPases (20,21), which leads to the prediction that proteins of this family have NTPase activity, as noticed previously during the annotation of the COG database (COG0433) (22). Members of the HerA family are present in all archaea, with at least one and often two or three members per archaeal genome. They are also found in several bacterial lineages, but are not detectable in any of the eukaryotic genomes sequenced so far. None of these genes has been presently functionally characterized. In Methanobacterium, the herA gene is apparently split into two distinct genes (MTH541 and MTH542). The MTH542 protein contains the N-terminal half of the predicted NTPase. The MTH541 protein contains the C-terminal half of the NTPase module fused to the Mre11 nuclease domain within the same polypeptide. This gene organization suggests that the MTH541 and MTH542 proteins form a complex that reconstitutes a functional NTPase domain and also that Mre11 nuclease might interact physically with the C-terminal domain of HerA in all archaea.
Figure 1.

Genomic organization of the herA, mre11, rad50 and nurA genes from thermophilic archaea. Sulfolobales designed S.acidocaldarius, S.tokodaii and S.solfataricus P2, and Pyrococcales designed P.abyssi, P.horikoshii OT3 and P.furiosus.
Figure 2.


Multiple alignment of the HerA protein family with selected proteins of the FtsK, TrwB/VirB4 and VirD4 ATPase families. Highly conserved residues are shown with bold type. The positions of the first and last residues of the aligned segments in the respective proteins are indicated by numbers. Poorly conserved regions not included in the alignment are denoted by the number of amino acid residues in parentheses. The secondary structure assignments are from the crystal structure of TrwB (PDB code 1E9R): H indicates α-helix and E indicates extended conformation (β-strand). The sequences are arranged in the following groups (designated to the right of the alignment): I, HerA proteins encoded within conserved archaeal operons; II, the remaining archaeal HerA proteins; III, bacterial HerA proteins; IV, bacteriophage ATPase; V, FtsK family ATPases; VI, TrwB/VirB4 family ATPases; VII, VirD4 family ATPases. The HerA family is represented in its entirety; for the other families, only selected sequences are shown. The sequences are denoted with the gene name followed by abbreviated species name, and the GI numbers. The S.acidocaldarius sequence has been submitted to the DDBJ/EMBL/GenBank databases under accession no. CAE51870. Species abbreviations are as follows: Archaea: Af, Archaeoglobus fulgidus; Ape, Aeropyrum pernix; Hsp, Halobacterium sp.; Mac, Methanosarcina acetivorans; Mj, Methanocaldococcus jannaschii; Mka, Methanopyrus kandleri; Mma, Methanosarcina mazei; Mta, Methanothermobacter thermoautotrophicus; Pab, Pyrococcus abyssi; Paer, Pyrobaculum aerophilum; Sac, Sulfolobus acidocaldarius; Sso, Sulfolobus solfataricus; Tac, Thermoplasma acidophilum; Tvo, Thermoplasma volcanium. Bacteria: Aae, Aquifex aeolicus; Atu, Agrobacterium tumefaciens; Bjap, Bradirhizobium japonicum; Bme, Brucella melitensis; Bper, Bordetella pertussis; Cau, Chloroflexus aurantiacus; Cje, Campylobacter jejuni; Ct, Chlamydia trachomatis; Ec, Escherichia coli; Fnu, Fusobacterium nucleatum; Hp, Helicobacter pylori; Lpne, Legionella pneumophila; Mtu, Mycobacterium tuberculosis; Npu, Nostoc punctiforme; Pae, Pseudomonas aeruginosa; Pput, Pseudomonas putida; Rme, Ralstonia metallidurans; Rsph, Rhodobacter sphaeroides; Rp, Rickettsia prowazekii; Sau, Staphylococcus aureus; Sme, Sinorhizobium meliloti; Ssp, Synechocystis sp.; Sty, Salmonella typhi; Tel, Thermosynechococcus elongatus BP-1; Tp, Treponema pallidum; Wol, Wolinella succinogenes.
The S.acidocaldarius herA, mre11, rad50 and nurA genes are part of the same transcription unit
In order to determine if the four genes are co-transcribed, we analyzed their expression in S.acidocaldarius by RT–PCR assays. In a first step, total RNA from S.acidocaldarius was purified and subjected to RT reactions. In order to limit the length of the expected products and to optimize reaction yields, we performed two reactions using either a nurA-specific primer or a rad50-specific primer (Fig. 3a). In a second step, the products of each RT reaction were subjected to PCR analyses. Using nurA reverse products and primers in 5′ of the mre11 gene and in 3′ of the nurA gene, a molecule of ∼5000 bp, the length of which corresponds to the added size of rad50, mre11 and nurA genes, was amplified (Fig 3b, lane 3). Indeed, this species contains the rad50, mre11 and nurA genes, as shown by independent amplification of each of these three genes (Fig. 3b, lanes 4–6). Using rad50 reverse products and primers in 5′ of the herA gene and in 3′ of the rad50 gene, we amplified a 5500 bp molecule (Fig. 3c, lane 2), and showed that it contains the herA, rad50 and mre11 genes (Fig. 3c, lanes 3–5). In each case, PCR control performed on S.acidocaldarius total RNA gave no amplification product, demonstrating the absence of any contaminating DNA. These results demonstrate that the herA, mre11, rad50 and nurA genes are part of the same operon and might be involved in the same metabolic pathway.
Purification and characterization of the HerA protein
In order to characterize the HerA protein, the S.acidocaldarius gene was cloned into pET-30 expression vector and the recombinant protein was overproduced in E.coli as a N-terminal His6-tag protein. Following a two-step purification procedure (see Materials and Methods), the protein migrates as a single band on SDS–polyacrylamide gels with an apparent molecular mass of 60 kDa, consistent with the predicted molecular mass of the His6-tag protein (Fig. 4). The identity of the protein was further confirmed by western blotting analyses using specific antibodies raised against internal peptides of the protein (data not shown). In a first experiment, we tested the purified protein for ATPase activity in either the presence or absence of DNA substrates. The enzyme is associated with an ATPase activity (0.3 mol of ATP hydrolyzed per mole of HerA per second) which is slightly stimulated by DNA with a weak preference for double-stranded DNA (Fig. 5a). The genomic context of this DNA-dependent ATPase suggested that this protein might act as a DNA helicase associated with Mre11, Rad50 and NurA proteins. Thus, we tested the ability of the protein to unwind a labeled oligonucleotide annealed to a circular single-stranded DNA. As shown in Figure 5b, the protein is able to displace the oligonucleotide in the presence of ATP and the percentage of the oligonucleotide released is dependent on the protein/DNA ratio. Quantification of the reaction products indicate that >80% of the oligonucleotide is displaced using a protein/DNA ratio of 200, indicating that the efficiency of the HerA protein is similar to that of other helicases, such as the E.coli RecD helicase (23). Using site-directed mutagenesis, we introduced a point mutation in the Walker A motif of HerA, replacing the conserved lysine, which is essential for ATP-binding and hydrolysis in P-loop NTPases (20) with alanine (K153A). The K153A HerA protein was then overproduced in E.coli and purified using the same procedure as for the wild-type protein. As shown in Figure 5b, the purified K153A HerA protein does not exhibit any detectable helicase activity (neither ATPase activity, data not shown). This result unambiguously demonstrates that the helicase activity is not due to any contaminating protein and is intrinsic to the HerA protein. Next, we tested different DNA templates in order to determine the polarity of its translocation along DNA. A 79mer oligonucleotide was synthesized and annealed to two different 5′ radiolabeled oligonucleotides: the first one corresponds to a 30mer oligonucleotide complementary to the 3′ part of the 79mer that leads to a DNA substrate with a 49 nt 5′ overhang, and the second one corresponds to a 30mer oligonucleotide complementary to the 5′ part of the 79mer leading to a DNA substrate with a 49 nt 3′ overhang. For the two substrates, we used equal length complementary oligonucleotides with the same GC content in order to get equivalent stability at 70°C. We also verified the position of each hybridized oligonucleotide either by restriction enzyme digestions in the case of the 3′ overhang substrate or by a primer extension assay in the case of the 5′ overhang substrate (data not shown). As shown in Figure 5c, the HerA protein is able to unwind the two DNA templates and quantification of the reaction products indicates that the amount of displaced oligonucleotide is in the same range for the two DNA substrates whatever the protein/DNA ratio used. This suggested that either the helicase is able to utilize a blunt DNA end for loading and subsequent DNA unwinding or the HerA helicase is capable of utilizing both 3′ and 5′ DNA tails (the former possibility is incompatible with the results obtained with the precedent helicase assay for which no blunt end was available). Thus, we tested the helicase activity using a blunt DNA duplex. As shown in Figure 5c, no detectable helicase activity was recovered on this substrate even at a protein/DNA ratio of 500, indicating that HerA needs single-stranded DNA tails to initiate the helicase reaction. Time courses of DNA unwinding were performed using the 3′ and 5′ DNA tail substrates in the presence of 400 nM HerA (Fig. 5d, each experiment was performed in triplicate). Quantification of reaction products showed that the initial velocity of DNA unwinding is equivalent for both substrates (0.75 fmol of released oligonucleotide per minute, Fig. 5e) demonstrating that HerA utilizes 3′ and 5′ DNA tail substrates for loading and subsequent DNA unwinding with the same efficiency.
Figure 4.
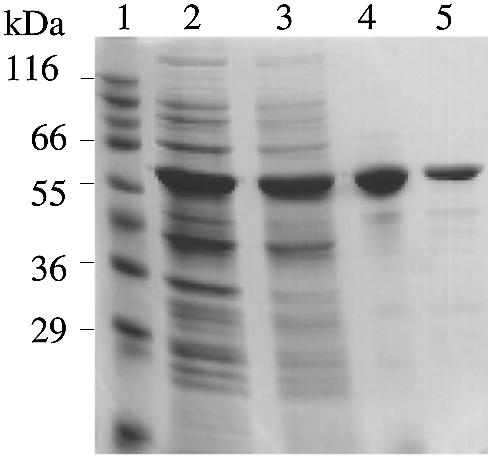
Purification of recombinant HerA protein. Purification steps were analyzed by SDS–PAGE and Coomassie blue staining. Lane 1, molecular weight standards; lane 2, overproducing extracts; lane 3, soluble fraction of overproducing extracts; lane 4, Ni2+-NTA agarose pool; lane 5, Source 30S pool.
The HerA protein family is related to the TraD/VirD4- and FtsK-like ATPases of the PilT/VirD4 class
The HerA helicase does not contain the classical helicase motifs which are seen in helicases superfamily 1/2 or helicases belonging to the AAA+ superclass. However, careful examination of the sequence shows that the P-loop NTPase domain of the HerA proteins has an additional strand after the Walker B strand that ends in a polar residue. These features suggest that the HerA family belongs to the additional strand conserved glutamate (ASCE) division of the P-loop NTPase fold (24), which includes the AAA+ class, the ABC class, the RecA/F1/F0 class, the superfamily 1/2 helicases, and the PilT/VirD4 class. The arginine of the conserved motif downstream of the Walker B motif (Fig. 2) is predicted to function similar to the arginine finger of the AAA+ ATPAses (25–28). The conserved glutamine, which is located downstream of this arginine, might act as a sensor of the nucleotide hydrolysis, analogous to the polar residue in the sensor-1 motif of the AAA+ ATPases (25,26), motif III of SFI helicases (29), and the [ST][AG][ST] motif of the SFII helicases (30).
A search of the NR database with the PSI-BLAST program using a position-specific score matrix (PSSM) of the HerA family recovered the bacterial FtsK and the TraD/VirD4 proteins of the PilT/VirD4 class (E-values in the range of 10–7–10–12) in the second iteration. Reciprocal searches with the PSSMs of the FtsK and TraD/VirD4 families recovered the HerA family members as the best hits, to the exclusion of all other NTPases. Even when the search was conducted after masking the Walker A motif, which is strongly conserved in all P-loop NTPases, members of each of the above families recovered each other as the best hits. Examination of the multiple alignment of these NTPase families revealed a characteristic motif (typically, R-x2-s-x2-hhh-x2-Q; where ‘s’ is a small residue and ‘h’ is a hydrophobic residue, Fig. 2), which encompasses the helix and the strand located immediately downstream of the Walker B motif. Thus, the HerA family is specifically related to the TraD/VirD4- and FtsK-like ATPases of the PilT/VirD4 class. Various members of the PilT/VirD4 class are involved in diverse DNA transport-related processes: TrwB proteins encoded by numerous conjugative plasmids of proteobacteria are involved in cell-to-cell DNA translocation during conjugation (31), VirD4 proteins are involved in the transfer of bacterial plasmids of nodule- and tumor-forming α-proteobacteria into plant cells (32) and FtsK proteins are involved in the pumping of DNA into the daughter cells during bacterial cell division (33).
DISCUSSION
In all organisms studied so far, initiation of homologous recombination requires the processing of DNA ends in 3′ overhangs, which are required for recombinase loading and subsequent DNA strand invasion. This process is highly complex and involves many interactive and regulated activities. Whereas this process has been extensively characterized in the case of the bacterial RecBCD pathway (6), the Rad50/Mre11 (SbcC/SbcD) pathway, which is the major recombination initiation mechanism in Eucarya and, possibly, in Archaea, is poorly understood.
In this study, we report the characterization of a new gene encoding a novel type of DNA helicase HerA that might be involved in such a process in hyperthermophilic archaea. Overproduction and characterization of the S.acidocaldarius HerA protein show that it is indeed associated with a DNA helicase activity using a primed single-stranded circular DNA as substrate and protein/DNA ratios in the range of those used for other DNA helicases (34). The enzyme exhibits ATPase activity that is slightly stimulated by either circular single- or double-stranded DNA with a weak preference for double-stranded DNA. Stimulation of the ATPase activity associated with DNA helicases by circular double-stranded DNA was previously reported in the case of helicases either involved in DNA replication or recombination (35,36). We demonstrate unambiguously that the helicase activity is intrinsic to the HerA protein, since a single mutation in a conserved amino acid that is involved in nucleotide binding in most P-loop ATPase (the conserved lysine of the Walker A motif) completely abolished both the ATPase and the helicase activities. Moreover, we tested the polarity of the helicase using commonly used substrates for such assays, i.e. a blunt ends linear substrate and linear substrates containing a blunt end and either a 3′ or a 5′ long single-stranded DNA tail. Our results show that HerA is unable to act on substrates that contain only blunt ends but has the striking property to utilize with the same efficiency 3′ and 5′ overhangs for loading and subsequent DNA unwinding.
Amino acid sequence analysis showed that this DNA helicase does not belong to helicases superfamilies 1 or 2 (37) or to the AAA+ class of NTPases, which includes several other helicases (25). Instead, HerA is a member of a distinct family of predicted NTPases related to the TrwB, FtsK and VirB4/VirD4 families of the PilT class of NTPases involved in various transport processes, including DNA pumping during bacterial cell division. The crystal structure of the TrwB protein has been determined and it has been shown that it forms a hexameric ring structure with a central channel through which single-stranded DNA passes during conjugation (38). It seems likely that HerA adopts a similar hexameric structure, a feature typical of many helicases (39).
The most surprising finding of this work is that a DNA helicase is capable of loading on either a 3′ or a 5′ single-stranded DNA tail and translocating along and unwinding DNA in two opposite directions. To our knowledge, helicases with such properties have not been described so far; in particular, hexameric helicases that form a ring encircling the DNA substrate translocate and unwind the DNA unidirectionally (39). These results lead to two alternative hypotheses: either the same form of the HerA protein has the intrinsic property to bind DNA with either polarity and to translocate in either direction, or the protein is able to adopt two distinct conformations, each one translocating and unwinding unidirectionally. Structural and microscopy studies are needed to elucidate such intriguing properties.
In most cases, DNA helicases function in association with other protein partners as part of a complex machine. We show in the present paper that HerA homologs are found in all hyperthermophilic archaea and that in most cases, herA gene forms a cluster with the mre11, rad50 and nurA genes. Furthermore, we demonstrate that the four genes are part of the same transcription unit in the hyperthermophilic archaeon S.acidocaldarius indicating that they are probably involved in the same pathway. The archaeal Rad50–Mre11 complex exhibits single-stranded endonuclease and double-stranded 3′ to 5′ exonuclease activities similar to their eucaryal and bacterial counterparts (12). We previously showed that NurA is associated with a single-stranded endonuclease activity and a single-stranded and double-stranded exonuclease activity. This exonuclease activity degrades DNA from the 5′ ends to the 3′ ends, the opposite direction of the exonuclease associated with the Mre11 protein, and the products of the exonuclease reaction are small oligonucleotides (15), as reported for the exonuclease activities associated with the RecBCD complex (40). Here, we show that the HerA protein has a DNA helicase activity that can act on DNA substrates in the two opposite directions. Recently, Dillingham et al. (41) and Taylor and Smith (23), showed that the RecBCD complex exhibits a bipolar DNA helicase activity. In addition to the previously described 3′ to 5′ helicase activity associated with the RecB subunit, these authors showed that the RecD subunit is also associated with a helicase activity acting in the opposite direction. As stated by Dillingham et al., this finding is, at first glance, surprising; however, taking into account the antiparallelism of the DNA duplex and the nature of helicase reactions, the two RecBCD helicases can be considered as two single-stranded DNA motors that bind to opposite strands of DNA breaks and translocate in the opposite polarity but in the same direction relative to the DNA duplex, increasing the processivity of the entire complex. The bipolar helicase HerA fits this model except that both helicase activities reside in the same protein. Considering the genomic context of the herA, mre11, rad50 and nurA genes and the functional analogy with the RecBCD complex that constitutes two DNA helicases of opposite polarity, a single-stranded endonuclease, a 3′ to 5′exonuclease, and a 5′ to 3′ exonuclease, an attractive hypothesis is that archaeal NurA and HerA proteins might act together with the Rad50–Mre11 complex in the processing of DNA ends at the initiation step of homologous recombination. In such a case, the single-stranded endonuclease as well as the exonuclease activities associated with the Mre11 and the NurA proteins should be regulated in order to process DNA ends in 3′ single-stranded DNA tails. This regulation may be performed by external factors like cis-acting sequences on archaeal chromosomes and/or intrinsinc factors that might be a regulatory role of the Rad50 protein and/or might be the result of protein–protein interactions. The characterization of the concerted action of the four proteins will provide the keys of this complex mechanism.
Acknowledgments
ACKNOWLEDGEMENTS
We thank R. Garrett for the communication of S.acidocaldarius herA sequence and F. Matsunaga for helpful discussions. This work was supported by Electricité de France and the Association pour la recherche sur le cancer (ARC).
NOTE ADDED IN PROOF
During the reviewing process of this manuscript, Manzan et al. (EMBO Rep., 2004, 5, 54–59) reported an archael ATPase which gene is grouped together with rad50, mrell and nurA genes. This ATPase called MlaA corresponds to the bipolar HerA helicase that we characterized in the present paper.
DDBJ/EMBL/GenBank accession no. CAE51870
REFERENCES
- 1.Haber J.E. (1998) The many interfaces of Mre11. Cell, 95, 583–586. [DOI] [PubMed] [Google Scholar]
- 2.Connelly J.C. and Leach,D.R. (1996) The sbcC and sbcD genes of Escherichia coli encode a nuclease involved in palindrome inviability and genetic recombination. Genes Cells, 1, 285–291. [DOI] [PubMed] [Google Scholar]
- 3.Aravind L., Walker,D.R. and Koonin,E.V. (1999) Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res., 27, 1223–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White M.F. (2003) Archaeal DNA repair: paradigms and puzzles. Biochem. Soc. Trans., 31, 690–693. [DOI] [PubMed] [Google Scholar]
- 5.Seitz E.M., Haseltine,C.A. and Kowalczykowski,S.C. (2001) DNA recombination and repair in the archaea. Adv. Appl. Microbiol., 50, 101–169. [DOI] [PubMed] [Google Scholar]
- 6.Kowalczykowski S.C. (2000) Initiation of genetic recombination and recombination-dependent replication. Trends Biochem. Sci., 25, 156–165. [DOI] [PubMed] [Google Scholar]
- 7.Furuse M., Nagase,Y., Tsubouchi,H., Murakami-Murofushi,K., Shibata,T. and Ohta,K. (1998) Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J., 17, 6412–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Usui T., Ohta,T., Oshiumi,H., Tomizawa,J., Ogawa,H. and Ogawa,T. (1998) Complex formation and functional versatility of Mre11 of budding yeast in recombination. Cell, 95, 705–716. [DOI] [PubMed] [Google Scholar]
- 9.Trujillo K.M., Yuan,S.S., Lee,E.Y. and Sung,P. (1998) Nuclease activities in a complex of human recombination and DNA repair factors Rad50, Mre11 and p95. J. Biol. Chem., 273, 21447–21450. [DOI] [PubMed] [Google Scholar]
- 10.Hopfner K.P., Karcher,A., Shin,D., Fairley,C., Tainer,J.A., Carney,J.P. (2000) Mre11 and Rad50 from Pyrococcus furiosus: cloning and biochemical characterization reveal an evolutionarily conserved multiprotein machine. J. Bacteriol., 182, 6036–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharples G.J. and Leach,D.R. (1995) Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol. Microbiol., 17, 1215–1217. [DOI] [PubMed] [Google Scholar]
- 12.Hopfner K.P., Putnam,C.D. and Tainer,J.A. (2002) DNA double-strand break repair from head to tail. Curr. Opin. Struct. Biol., 12, 115–122. [DOI] [PubMed] [Google Scholar]
- 13.van Noort J., van Der Heijden,T., de Jager,M., Wyman,C., Kanaar,R. and Dekker,C. (2003) The coiled-coil of the human Rad50 DNA repair protein contains specific segments of increased flexibility. Proc. Natl Acad. Sci. USA, 100, 7581–7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paull T.T. and Gellert,M. (1999) Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev., 13, 1276–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Constantinesco F., Forterre,P. and Elie,C. (2002) NurA, a novel 5′-3′ nuclease gene linked to rad50 and mre11 homologs of thermophilic Archaea. EMBO Rep., 3, 537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Notredame C., Higgins,D.G. and Heringa,J. (2000) T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol., 302, 205–217. [DOI] [PubMed] [Google Scholar]
- 18.Lopez-Garcia P. and Forterre,P. (1999) Control of DNA topology during thermal stress in hyperthermophilic archaea: DNA topoisomerase levels, activities and induced thermotolerance during heat and cold shock in Sulfolobus. Mol. Microbiol., 33, 766–777. [DOI] [PubMed] [Google Scholar]
- 19.Constantinesco F., Benachenhou,N., Motorin,Y. and Grosjean,H. (1998) The tRNA(guanine-26,N2-N2) methyltransferase (Trm1) from the hyperthermophilic archaeon Pyrococcus furiosus: cloning, sequencing of the gene and its expression in Escherichia coli. Nucleic Acids Res., 26, 3753–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker J.E., Saraste,M., Runswick,M.J. and Gay,N.J. (1982) Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J., 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saraste M., Sibbald,P.R. and Wittinghofer,A. (1990) The P-loop—a common motif in ATP- and GTP-binding proteins. Trends Biochem. Sci., 15, 430–434. [DOI] [PubMed] [Google Scholar]
- 22.Tatusov R.L., Natale,D.A., Garkavtsev,I.V., Tatusova,T.A., Shankavaram,U.T., Rao,B.S., Kiryutin,B., Galperin,M.Y., Fedorova,N.D. and Koonin,E.V. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res., 29, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor A.F. and Smith,G.R. (2003) RecBCD enzyme is a DNA helicase with fast and slow motors of opposite polarity. Nature, 423, 889–893. [DOI] [PubMed] [Google Scholar]
- 24.Leipe D.D., Koonin,E.V. and Aravind,L. (2003) Evolution and classification of P-loop kinases and related proteins. J. Mol. Biol., 333, 781–815. [DOI] [PubMed] [Google Scholar]
- 25.Neuwald A.F., Aravind,L., Spouge,J.L. and Koonin,E.V. (1999) AAA+: A class of chaperone-like ATPases associated with the assembly, operation and disassembly of protein complexes. Genome Res., 9, 27–43. [PubMed] [Google Scholar]
- 26.Neuwald A.F. (1999) The hexamerization domain of N-ethylmaleimide-sensitive factor: structural clues to chaperone function. Structure Fold. Des., 7, R19–R23. [DOI] [PubMed] [Google Scholar]
- 27.Putnam C.D., Clancy,S.B., Tsuruta,H., Gonzalez,S., Wetmur,J.G. and Tainer,J.A. (2001) Structure and mechanism of the RuvB Holliday junction branch migration motor. J. Mol. Biol., 311, 297–310. [DOI] [PubMed] [Google Scholar]
- 28.James J.A., Escalante,C.R., Yoon-Robarts,M., Edwards,T.A., Linden,R.M., Aggarwal,A.K. (2003) Crystal structure of the SF3 helicase from adeno-associated virus type 2. Structure (Camb.), 11, 1025–1035. [DOI] [PubMed] [Google Scholar]
- 29.Dillingham M.S., Soultanas,P. and Wigley,D.B. (1999) Site-directed mutagenesis of motif III in PcrA helicase reveals a role in coupling ATP hydrolysis to strand separation. Nucleic Acids Res., 27, 3310–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson E.R. and McKay,D.B. (1999) Crystallographic structure of the amino terminal domain of yeast initiation factor 4A, a representative DEAD-box RNA helicase. RNA, 5, 1526–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cabezon E., Sastre,J.I. and de la Cruz,F. (1997) Genetic evidence of a coupling role for the TraG protein family in bacterial conjugation. Mol. Gen. Genet., 254, 400–406. [DOI] [PubMed] [Google Scholar]
- 32.Hamilton C.M., Lee,H., Li,P.L., Cook,D.M., Piper,K.R., von Bodman,S.B., Lanka,E., Ream,W. and Farrand,S.K. (2000) TraG from RP4 and TraG and VirD4 from Ti plasmids confer relaxosome specificity to the conjugal transfer system of pTiC58. J. Bacteriol., 182, 1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aussel L., Barre,F.X., Aroyo,M., Stasiak,A., Stasiak,A.Z. and Sherratt,D. (2002) FtsK Is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell, 108, 195–205. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan D.L., Davey,M.J. and O’Donnell,M. (2003) Mcm4,6,7 uses a ‘pump in ring’ mechanism to unwind DNA by steric exclusion and actively translocate along a duplex. J. Biol. Chem., 278, 49171–4182. [DOI] [PubMed] [Google Scholar]
- 35.Marrione P.E. and Cox,M.M (1995) RuvB protein-mediated ATP hydrolysis: functional asymmetry in the RuvB hexamer. Biochemistry, 34, 9809–9818. [DOI] [PubMed] [Google Scholar]
- 36.Shechter D.F., Ying,C.Y. and Gautier,J. (2000) The intrinsic DNA helicase activity of Methanobacterium thermoautotrophicum DH minichromosome maintenance protein. J. Biol. Chem., 275, 15049–15059. [DOI] [PubMed] [Google Scholar]
- 37.Gorbalenya A.E. and Koonin,E.V. (1993) Helicase-amino acid sequence comparisons and structure–function relationships. Curr. Opin. Struct. Biol., 3, 419–429. [Google Scholar]
- 38.Gomis-Ruth F.X., Moncalian,G., Perez-Luque,R., Gonzalez,A., Cabezon,E., de la Cruz,F. and Coll,M. (2001) The bacterial conjugation protein TrwB resembles ring helicases and F1-ATPase. Nature, 409, 637–641. [DOI] [PubMed] [Google Scholar]
- 39.Patel S.S. and Picha,K.M. (2000) Structure and function of hexameric helicases. Annu Rev. Biochem., 69, 651–697. [DOI] [PubMed] [Google Scholar]
- 40.MacKay V. and Linn,S. (1974) The mechanism of degradation of duplex deoxyribonucleic acid by the recBC enzyme of Escherichia coli K-12. J. Biol. Chem., 249, 4286–4294. [PubMed] [Google Scholar]
- 41.Dillingham M.S., Spies,M. and Kowalczykowski,S.C. (2003) RecBCD enzyme is a bipolar DNA helicase. Nature, 423, 893–897. [DOI] [PubMed] [Google Scholar]