

Abstract
Objective:
To identify early cognitive and neuroimaging features of sporadic nonfluent/agrammatic variant of primary progressive aphasia (nfvPPA) caused by frontotemporal lobar degeneration (FTLD) subtypes.
Methods:
We prospectively collected clinical, neuroimaging, and neuropathologic data in 11 patients with sporadic nfvPPA with FTLD-tau (nfvPPA-tau, n = 9) or FTLD–transactive response DNA binding protein pathology of 43 kD type A (nfvPPA-TDP, n = 2). We analyzed patterns of cognitive and gray matter (GM) and white matter (WM) atrophy at presentation in the whole group and in each pathologic subtype separately. We also considered longitudinal clinical data.
Results:
At first evaluation, regardless of pathologic FTLD subtype, apraxia of speech (AOS) was the most common cognitive feature and atrophy involved the left posterior frontal lobe. Each pathologic subtype showed few distinctive features. At presentation, patients with nfvPPA-tau presented with mild to moderate AOS, mixed dysarthria with prominent hypokinetic features, clear agrammatism, and atrophy in the GM of the left posterior frontal regions and in left frontal WM. While speech and language deficits were prominent early, within 3 years of symptom onset, all patients with nfvPPA-tau developed significant extrapyramidal motor signs. At presentation, patients with nfvPPA-TDP had severe AOS, dysarthria with spastic features, mild agrammatism, and atrophy in left posterior frontal GM only. Selective mutism occurred early, when general neurologic examination only showed mild decrease in finger dexterity in the right hand.
Conclusions:
Clinical features in sporadic nfvPPA caused by FTLD subtypes relate to neurodegeneration of GM and WM in frontal motor speech and language networks. We propose that early WM atrophy in nfvPPA is suggestive of FTLD-tau pathology while early selective GM loss might be indicative of FTLD-TDP.
The nonfluent/agrammatic variant of primary progressive aphasia (nfvPPA)1,2 falls under the umbrella of clinical syndromes caused by frontotemporal lobar degeneration (FTLD) pathology.3–5 The major molecular classes of FTLD associated with nfvPPA are microtubule-associated protein tau (either 3 repeat [3R] or 4 repeat [4R])5,6 (FTLD-tau) and transactive response DNA binding protein of 43 kD (TDP-43) type A (TDP-A)7 (FTLD-TDP).8,9
Several studies have evaluated speech and language, and/or anatomical data, in pathologically confirmed cases of nfvPPA.9–13 However, studies including complete datasets of prospectively collected cognitive, neuroimaging, and pathologic data are still scarce. Furthermore, the distinctive features characterizing sporadic nfvPPA caused by different FTLD pathologic subtypes are still not established. Some authors have proposed that agrammatism might be a marker for FTLD-TDP pathology,12 whereas motor speech deficits would be more typical of FTLD-tau.14 Pathologic and neuroimaging evidence in a variety of frontotemporal dementia (FTD)-spectrum disorders suggests that greater white matter (WM) than gray matter (GM) changes might be characteristic of the FTLD-tau subtype.15–17
Here, we present a prospective cognitive and neuroimaging study of 11 patients with nfvPPA with sporadic disease and pathologically confirmed FTLD-tau (nfvPPA-tau) or FTLD–TDP-A (nfvPPA-TDP). The aim of the study was to identify antemortem clinical and neuroimaging features suggestive of each FTLD pathologic subtype in this comprehensively characterized, homogeneous clinical group.
METHODS
Subjects.
We recruited 11 subjects (7 women, mean age 68.5 ± 7.6 years) diagnosed with nfvPPA at the University of California at San Francisco (UCSF) Memory and Aging Center (MAC) (table 1). Inclusion criteria involved clinical diagnosis based on current criteria,18 the availability of speech and language and cognitive testing, an MRI scan within 6 months of first visit to the UCSF MAC, and postmortem pathologic FTLD diagnosis. Exclusion criteria included family history for dominantly inherited FTD or dementia at an early age of onset, and the presence of a known genetic mutation.
Table 1.
Demographic and cognitive data in nfvPPA groups vs controls at presentation
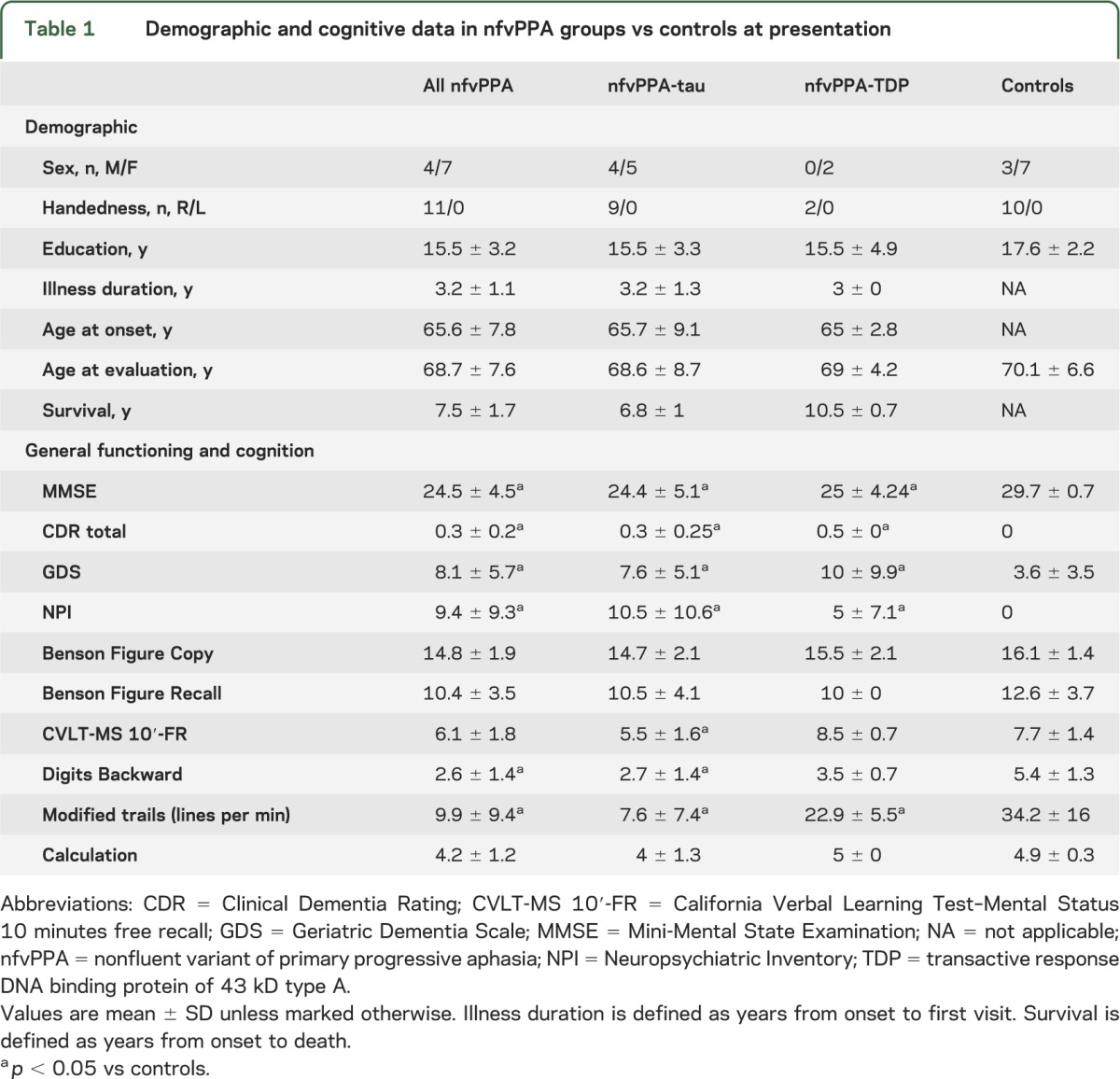
We followed patients for an average of 4.5 years and evaluated trajectories of clinical progression by considering data from longitudinal neurologic examinations.
Cognitive testing.
Cognitive functioning at presentation was assessed using the UCSF neuropsychological and speech and language batteries described elsewhere2,19 (appendix e-1 on the Neurology® Web site at www.neurology.org).
Cognitive data from 10 age-matched, right-handed healthy subjects (3 men, mean age 70.1 ± 6.6 years) were used as control for the cognitive analysis. We compared cognitive scores between subjects with nfvPPA and controls at presentation using the Kruskal-Wallis and Mann-Whitney U tests (table 1). Furthermore, each patient's scores on speech and language tests were transformed into standardized z scores (table 2).
Table 2.
Summary of language scores at first evaluation in each patient with nfvPPA
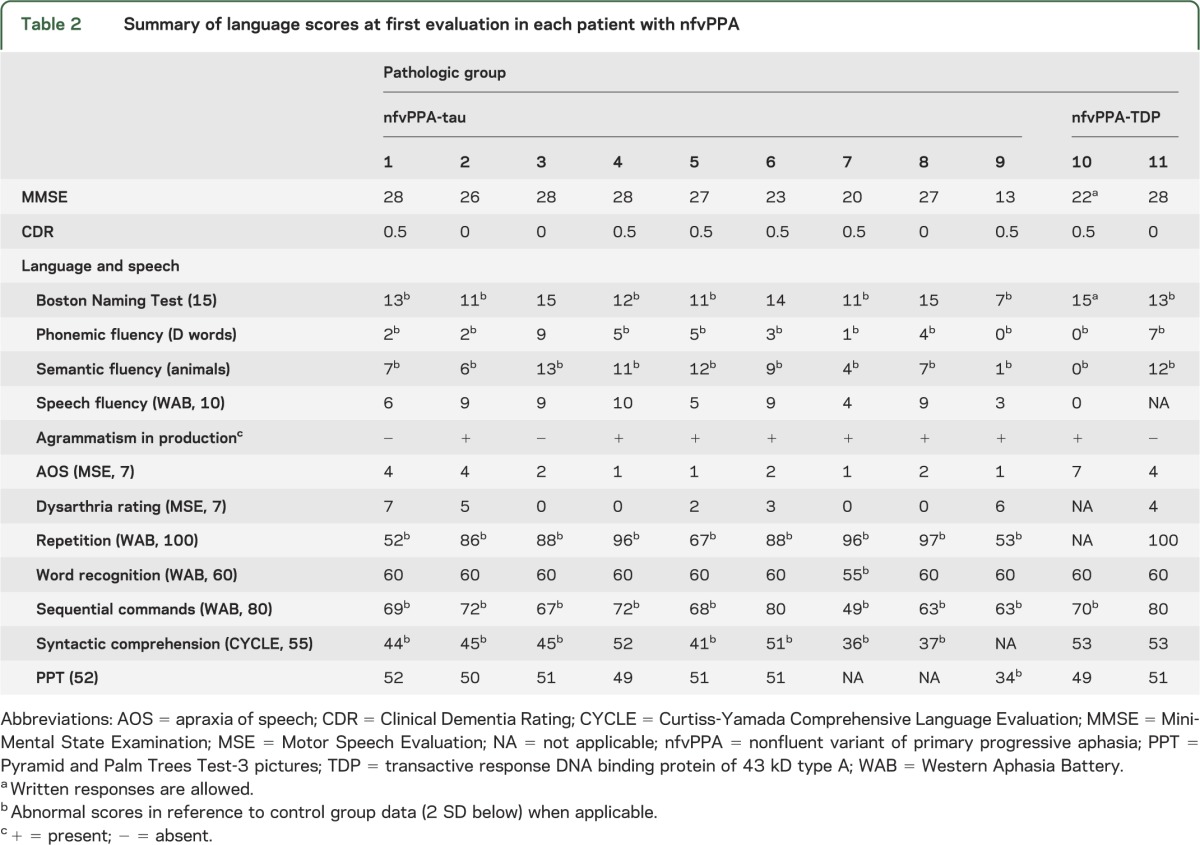
Neuropathology.
Autopsies were performed at UCSF (n = 7), University of Pennsylvania (cases 1, 3, and 5), and Vancouver General Hospital (case 7) (table 3). Pathologic diagnosis was based on consensus criteria for FTLD20 and Alzheimer disease21 following standard procedures described elsewhere.22 Nine patients with nfvPPA had FTLD-tau, and 2 had FTLD-TDP pathology. Among the FTLD-tau group, 2 had progressive supranuclear palsy (PSP) (cases 1 and 2), 6 had corticobasal degeneration (CBD) (cases 3–8), and one had an unclassifiable 4R tauopathy (case 9). Both TDP cases (cases 10 and 11) were classified as TDP-A.
Table 3.
Findings on neurologic examination at first and last visits at the UCSF MAC and language deficits in the nfvPPA cohort
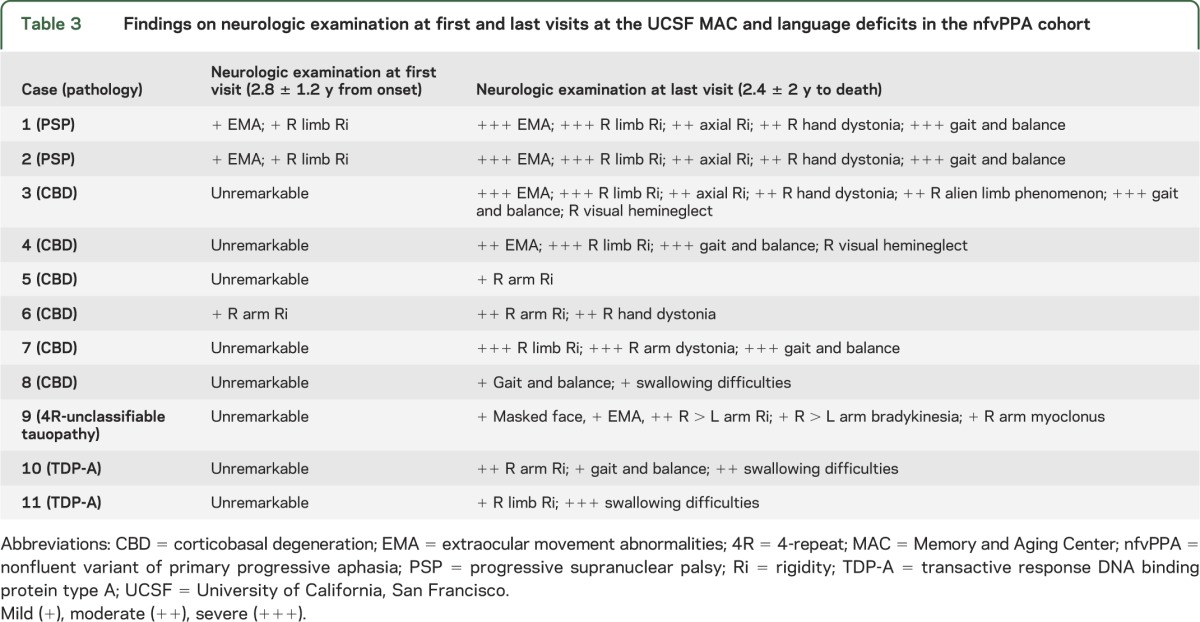
MRI acquisition and analysis.
All images were acquired on a 1.5T Siemens Magnetom VISION system (Siemens, Iselin, NJ) using a magnetization-prepared rapid gradient echo sequence.2
We used voxel-based morphometry (VBM)23 in SPM8 (Statistical Parametric Mapping; Wellcome Department of Imaging Neuroscience, London, UK) to investigate volume differences in GM and WM using standard methods.24,25 The VBM control group included 53 age- and sex-matched, right-handed healthy subjects (16 men, mean age 68.5 ± 8.6 years).
Statistical analyses first compared all 11 cases of nfvPPA with controls. We then separated the nfvPPA-tau (n = 9) from the nfvPPA-TDP (n = 2) cases and compared each pathologic subtype with controls.
We accepted a level of significance of p < 0.05, corrected for familywise error (FWE). Subsequently, VBM results were tested at p < 0.001, uncorrected, to avoid false negatives that can occur in single-subject or small-group VBM analyses.
We applied the Montreal Neurological Institute coordinate system for GM atrophy localization, and attempted attribution of WM volume loss to the main fasciculi using the JHU-MNI-ss atlas (http://cmrm.med.jhmi.edu).
Standard protocol approvals, registrations, and patient consents.
All participants gave written informed consent, and the study was approved by the Committee on Human Research at UCSF.
RESULTS
Demographic data.
All nfvPPA cases (7 women and 4 men) were right-handed with at least a high school level of education (12 years) (table 1). No significant differences were found between subjects with nfvPPA-tau and those with nfvPPA-TDP for age at onset, illness duration (years from symptom onset to first neurologic evaluation), or education.
Cognitive findings.
All nfvPPA-FTLD.
At first visit, the whole nfvPPA group showed a neuropsychological profile (table 1) consistent with previous reports using the same battery.2 Symptoms of apraxia of speech (AOS) and/or dysarthria were present in all patients with nfvPPA. Clear agrammatism in production or comprehension was detected in all nfvPPA-tau and in one of the 2 nfvPPA-TDP cases (table 3). Case 11 was the only patient who did not show clear grammatical deficits at presentation but developed some difficulties 6 months later. In this cohort, agrammatism never occurred in isolation, without motor speech impairment. Case 7 (CBD), previously described because of her artistic talent,26 was the only patient who showed mild AOS but severe agrammatism, with production limited to single words. Further details and evolution of language symptoms are discussed below.
nfvPPA-tau.
All nfvPPA-tau cases presented with varying degrees of motor speech impairment (table 2) consistent with either isolated AOS (4 CBD cases) or mixed AOS and dysarthria (2 CBD + 2 PSP + 1 unclassifiable 4R tauopathy). Speech was characterized by slowed rate, delayed initiation, prosodic insufficiency, and frequent pauses, with increased intra- and intersegment duration. Patients with nfvPPA-tau showed visible groping, especially on syllable-initial consonant clusters, and sequencing errors that were particularly evident on multiple repetitions of multisyllabic words. Motor speech evaluations revealed the presence of both sound distortions (including prolongations, particularly for consonants) and phonemic errors (including sound substitutions, i.e., “glass” for “grass” and insertions “pinknic” for “picnic”).
Dysarthria presented with mixed prominent hypokinetic (monopitch, reduced stress, monoloudness, inappropriate silence, and speech festination) and spastic features (strained, harsh vocal quality, hypernasality, bursts of loudness, low pitch, slowed rate, and imprecise articulation). Furthermore, 5 patients with nfvPPA-tau showed mild buccofacial apraxia at first visit.
At presentation, agrammatism in production was detected in 7 of 9 patients with nfvPPA-tau. The 2 patients who did not have initially detectable agrammatic errors in language production nevertheless showed impaired comprehension of syntactically complex sentences. Later in the disease course, both patients developed agrammatism in oral and written samples. All patients with nfvPPA-tau presented with various degree of impairment in comprehension of syntactically complex sentences.
Over the course of their disease, all patients with nfvPPA-tau developed buccofacial apraxia that gradually worsened over time. They remained able to execute simple mouth movements on command until very late in the disease course. Performance on single-word comprehension and semantic tasks remained relatively spared.
nfvPPA-TDP.
The 2 nfvPPA-TDP cases showed the most severe speech output deficits (table 3) and buccofacial apraxia that led to mutism without any extrapyramidal motor deficits affecting everyday life. Below we report their detailed clinical features.
One patient, case 10, was first seen at the Alzheimer's Disease Center of the Northwestern University Feinberg School of Medicine reporting 2 years of progressive language output difficulties. She was then diagnosed as having PPA with moderately severe motor speech impairment and mild agrammatism. Speech was characterized by slow rate, sequencing errors, and distortions suggestive of AOS. She also showed features of spastic dysarthria, such as weak and hoarse voice. Minor grammatical difficulties were observed on written samples. Comprehension of syntactically complex statements was also impaired. In subsequent months, speech continued to deteriorate and at her first language evaluation at the UCSF MAC (approximately 1 year later and 3 years after onset) she was functionally mute (table 3). She could only produce a few sounds and answer yes or no with great effort. Dysarthria could no longer be classified because of insufficient production, but the patient's voice was clearly weak and hypernasal. Written production was much more preserved than spoken output but few agrammatic errors were detected. Comprehension of syntactically complex sentences was impaired but lexical retrieval abilities, semantic memory, and single-word comprehension were spared. At 1-year follow-up, she was unable to produce any speech sounds, despite other cognitive and language domains being unchanged. Buccofacial apraxia had worsened to the point that the only movement she could perform on command was to open her mouth. She reported some swallowing difficulties. EMG was performed and found to be negative.
Case 11 had a comparable history. At presentation, she was 66 years old and reported a 3-year history of speech output difficulties. At her first language evaluation (table 3), she showed moderate AOS and her voice was soft and hypernasal, consistent with moderate spastic dysarthria. At that point, she did not show clear signs of agrammatism in spoken or written language, although her written picture description was short and simple. Syntactic comprehension was spared. At the 1-year follow-up visit, she was functionally mute, producing only a few sounds, which were weak and hypernasal. The speech pathologist who examined her defined her deficit as “apraxia of phonation.” At that time, mild grammatical deficits appeared in writing and in sentence comprehension; single-word comprehension, confrontation naming, and semantic memory remained relatively spared. As in case 10, buccofacial apraxia became severe and she was unable to perform even simple mouth movements or cough on command or imitation. She reported loss of sensation in the lips and tongue and mild swallowing problems. EMG results were negative. At the last evaluation, when she was aphonic, general motor deficits were mild and she was still able to dance with her husband. She became progressively unable to swallow and in the last year of disease, a feeding tube was placed.
General cognitive evaluation was nonstandard because of severe motor speech deficits, but both patients appeared relatively spared in all domains.
Neurologic examination at presentation and follow-up.
By definition, all patients with nfvPPA showed early, isolated speech or language difficulties that were the only cause of limitation of daily living activities. The review of longitudinal neurologic evaluations revealed that, during the disease course, all patients developed various degrees of motor impairment (table 3), as previously reported for FTD-spectrum disorders.27,28 However, the trajectory appeared different in the 2 nfvPPA-FTLD subtypes. At first visit, 3 nfvPPA-tau cases (cases 1, 2, and 6) showed mild extrapyramidal motor signs on neurologic examination only, with no subjective complaints or functional impact (table 3). All patients with nfvPPA-tau developed a moderate to severe extrapyramidal syndrome within a mean of 3 years from (language) symptom onset. Five subjects with nfvPPA-tau (cases 1–4 and 7) were wheelchair-bound approximately 5 years after (language) symptom onset.
The nfvPPA-TDP cases did not show extrapyramidal motor signs at first evaluation, despite having moderate to severe motor speech deficits (table 2). They developed early mutism, when general neurologic examination still only showed decreased finger dexterity in the right hand. Extrapyramidal signs remained mild for most of the disease course. At last evaluation before death (8 years from language symptom onset), case 10 showed moderate right limb rigidity and needed some assistance in walking, and case 11 was still able to walk autonomously and dance with her husband 6 years from the onset of language symptoms.
Mean survival (years from onset to death) of the entire nfvPPA group was 7.5 years. Patients with nfvPPA-TDP lived almost 3 years longer than those in the nfvPPA-tau group.
Neuroimaging.
All nfvPPA-FTLD vs controls.
VBM analysis revealed GM atrophy in patients with nfvPPA-FTLD along left motor and premotor cortices, including precentral gyrus, superior frontal gyrus, middle frontal gyrus, pars opercularis of the inferior frontal gyrus (IFG-po), supplementary motor area (SMA), dorsal anterior insula, and basal ganglia (p < 0.05, FWE).
WM atrophy was extensive within the left frontal lobe. The JHU-MNI-ss atlas placed the atrophy in the superior longitudinal fasciculus (SLF), corona radiata, and the body of the corpus callosum (p < 0.05, FWE). At an uncorrected threshold, WM atrophy was also detected in the right frontal region and left brainstem, likely the cerebral peduncle (figure e-1).
nfvPPA-tau vs controls.
Subjects with nfvPPA-tau showed GM atrophy in the left premotor cortex, comprising the precentral gyrus, IFG-po, superior frontal gyrus, middle frontal gyrus, and putamen (figure 1, table e-1). At an uncorrected threshold, the left SMA, middle cingulate cortex, dorsal anterior insula, and caudate were also involved.
Figure 1. Gray matter atrophy in nfvPPA pathologic subtypes vs controls.

Voxel-based morphometric analysis on gray matter regions in nfvPPA pathologic subtypes relative to healthy controls. Statistical maps have been thresholded at p < 0.05 for FWE (top) and at p < 0.001 uncorrected (bottom). Statistical maps have shown in the coronal (coordinates [mm]: +0, +8) and axial (coordinates [mm]: +24, +48) sections of a T1-weighted MRI template image in DARTEL space. The color bar (hot) represents the t score. DARTEL = diffeomorphic anatomical registration through exponentiated lie; FTLD = frontotemporal lobar degeneration; FWE = familywise error; nfvPPA = nonfluent variant of primary progressive aphasia; TDP = transactive response DNA binding protein of 43 kD type A.
WM atrophy was severe in subjects with nfvPPA-tau and mirrored the pattern of the entire group (figure 2), including part of the SLF, corona radiata, and the body of the corpus callosum (p < 0.05, FWE). At an uncorrected threshold, WM atrophy was also detected contralaterally and in the left brainstem.
Figure 2. White matter atrophy in nfvPPA pathologic subtypes vs controls.

Voxel-based morphometric analysis on white matter regions in nfvPPA pathologic subtypes relative to healthy controls. Statistical maps have been thresholded at p < 0.05 for FWE (top) and at p < 0.001 uncorrected (bottom). Statistical maps have shown in the coronal (coordinates [mm]: +0, +8) and sagittal (coordinates [mm]: −18, −4) sections of a T1-weighted MRI template image in DARTEL space. The color bar (winter) represents the t score. DARTEL = diffeomorphic anatomical registration through exponentiated lie; FTLD = frontotemporal lobar degeneration; FWE = familywise error; nfvPPA = nonfluent variant of primary progressive aphasia; TDP = transactive response DNA binding protein of 43 kD type A.
nfvPPA-TDP vs controls.
GM atrophy was found in the left posterior, inferior frontal area, including IFG-po and the posterior part of precentral gyrus, in the face, mouth, and pharyngeal motor representations (p < 0.05; figure 1, table e-1). At p < 0.001 uncorrected, atrophy included the posterior part of the left SMA, insula, middle cingulum, bilateral inferior parietal lobule (IPL), and inferior temporal and right supramarginal gyri.
The nfvPPA-TDP cases did not show WM atrophy at p < 0.05 FWE (figure 2). Even at an uncorrected threshold, only a small area in the left frontal region was detected, suggesting that the WM atrophy seen in the overall nfvPPA-FTLD group derived from the nfvPPA-tau cases.
DISCUSSION
We report clinical, cognitive, and neuroimaging findings in a cohort of patients with sporadic nfvPPA with autopsy-confirmed FTLD-tau or FTLD–TDP-A pathology. The aim of the study was to identify early clinical and neuroimaging nfvPPA features associated with each molecular FTLD subtype. Our data show that AOS is the most frequent manifestation of nfvPPA caused by both FTLD-tau and -TDP pathology. Distinctive features of nfvPPA-TDP cases included early mutism with severe buccofacial apraxia, spastic dysarthria, and selective inferior frontal GM atrophy with relative sparing of WM. Typical nvfPPA-tau features included extensive frontal WM damage, early mixed dysarthria with prominent hypokinetic features, and later development of significant extrapyramidal motor signs. We propose that earlier and greater WM damage in nfvPPA-tau could explain the subtle distinctions between the clinical manifestations of nfvPPA-tau and -TDP.
AOS is a disorder characterized by an impaired ability to coordinate articulatory movements. It can occur in isolation without other speech, language, or movement deficits and is associated with damage in the left inferior frontal/insular region. AOS has been previously reported as a common clinical feature in nfvPPA.2,11 Our findings suggest that it is associated with both FTLD-tau and -TDP, likely in relation to left inferior frontal atrophy, which is common to both pathologic subtypes.
Previous studies have shown significant WM damage in patients with clinically diagnosed nfvPPA.29–31 One recent diffusion tensor imaging (DTI)-MRI study revealed WM damage in an nfvPPA cohort including patients with FTLD-tau pathology or non-Alzheimer CSF biomarkers.13 We suggest that in nfvPPA-tau, early GM and WM degeneration produces a network-level dysfunction in both motor speech and language systems. GM degeneration of cortical regions and disconnection between cortical and basal ganglia motor control systems would result in AOS and hypokinetic dysarthria first32,33 and, later in the disease course, in the development of a generalized extrapyramidal motor syndrome. GM damage in more anterior portions of the IFG and disconnection between frontal and temporal language areas caused by pathology in the SLF32 could instead result in early grammatical deficits.
By contrast, at presentation, patients with nfvPPA-TDP showed atrophy only in the GM of the left inferior motor and premotor regions without significant WM involvement. The early clinical features of nfvPPA-TDP might thus be related to focal damage to these specific cortical hubs rather than to a network-level dysfunction. Both patients showed severe AOS, spastic dysarthric features, and severe buccofacial apraxia, as previously shown in patients with focal lesions of the inferior frontal region.34,35 Agrammatism was present but mild, possibly because damage to a single cortical hub within the distributed grammar network is not sufficient to cause severe deficits.36 Similarly, patients with nfvPPA-TDP did not show dysarthric features typical of subcortical damage, nor development of a prominent general extrapyramidal syndrome, even later in the disease, if not very close to death. This nfvPPA-TDP clinical picture resembles an upper motor neuron variant of amyotrophic lateral sclerosis, with which the nfvPPA variant shares common TDP-related pathology.9
Biological features of FTLD-tau and -TDP molecules support our neuroimaging findings. The widespread WM damage in patients with FTLD-tau early in the disease is consistent with the hypothesis that tau deposition might primarily affect the axon, with retrograde GM degeneration, as suggested by previous studies in animal models.37,38 Pathology could then spread in a “prion-like” manner39 along connected networks,40 in our case those related to motor and language functions. In contrast, TDP-43 is a nuclear protein7 that is redistributed from the nucleus to the cytoplasm and dendritic processes during neurodegeneration. However, despite these differences in cellular pathogenesis, caution should be used in this interpretation because GM and WM are both heavily involved in FTLD-tau and FTLD-TDP in late-stage disease, when pathologic analysis occurs.
This study has some clear limitations. First, the patient group was small, reflecting the rarity of the disease and our strict inclusion criteria. We believe that the strengths of our study are the selection of a homogeneous patient cohort and the extensive clinical characterization. Specific hypotheses based on such detailed assessment of a small cohort can eventually be tested in larger, less extensively studied patient cohorts. The second limitation is that DTI data were not available. Nevertheless, VBM proved sensitive for detecting WM atrophy in the FTLD-tau group and GM damage in both groups, consistent with recent DTI findings in a more heterogeneous clinical population. We do believe that DTI could enable even earlier detection of WM damage in FTLD.
Our results suggest that AOS and agrammatism are not distinctive features of an underlying FTLD-tau or FTLD–TDP-A pathology. Early WM damage on neuroimaging might provide a biomarker for FTLD-tau pathology in the nfvPPA syndrome and might be associated with subtle differential clinical features.
Because future treatments will be directed toward specific molecules, predicting pathology in nfvPPA is an increasingly important endeavor.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families for the time and effort they dedicated to the research. The authors thank Marsel Mesulam, Sandy Weintraub, and their colleagues at the Alzheimer's Disease Center of the Northwestern University Feinberg School of Medicine for providing data from the Northwestern clinical and neuropsychological evaluations of case 10. The authors thank Ian Mackenzie, MD (Department of Pathology, University of British Columbia, Vancouver, Canada) for assistance with one of the autopsies.
GLOSSARY
- AOS
apraxia of speech
- CBD
corticobasal degeneration
- DTI
diffusion tensor imaging
- 4R
4 repeat
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- FWE
familywise error
- GM
gray matter
- IFG-po
pars opercularis of the inferior frontal gyrus
- MAC
Memory and Aging Center
- nfvPPA
nonfluent variant of primary progressive aphasia
- PSP
progressive supranuclear palsy
- SLF
superior longitudinal fasciculus
- SMA
supplementary motor area
- TDP
transactive response DNA binding protein of 43 kD type A
- UCSF
University of California, San Francisco
- VBM
voxel-based morphometry
- WM
white matter
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Francesca Caso: study concept and design, acquisition of data, drafting/revising the manuscript for content, analysis or interpretation of data, statistical analysis. Maria Luisa Mandelli: acquisition of data, drafting/revising the manuscript for content, statistical analysis. Maya Henry: acquisition and analysis of data, drafting/revising the manuscript for content. Benno Gesierich: acquisition and analysis of data, revising the manuscript for content. Brianne M. Bettcher: acquisition and analysis of data, drafting/revising the manuscript for content. Jennifer Ogar: acquisition and analysis of data, revising the manuscript for content. Massimo Filippi: drafting/revising the manuscript for content. Giancarlo Comi and Giuseppe Magnani: revising the manuscript for content. Manu Sidhu, John Q. Trojanowski, Eric J. Huang, and Lea T. Grinberg: acquisition and analysis of data, revising the manuscript for content. Bruce L. Miller: acquisition of data, drafting/revising the manuscript for content. Nina Dronkers: acquisition and analysis of data, revising the manuscript for content. William W. Seeley: acquisition and analysis of data, drafting/revising the manuscript for content. Maria Luisa Gorno-Tempini: study concept and design, revising the manuscript for content, analysis or interpretation of data, study supervision or coordination, obtaining funding.
STUDY FUNDING
Supported by the NIH (NINDS R01 NS050915, NIA P50 AG023501, NIA P01 AG019724); State of California (DHS 04-35516); Alzheimer's Disease Research Center of California (09-11410 DHS/ADP/ARCC); Larry L. Hillblom Foundation; John Douglas French Alzheimer's Foundation; Koret Family Foundation; Consortium for Frontotemporal Dementia Research; and the McBean Family Foundation.
DISCLOSURE
Francesca Caso, Maria Luisa Mandelli, Maya Henry, Benno Gesierich, Brianne M. Bettcher, and Jennifer Ogar report no disclosures. Massimo Filippi serves on scientific advisory boards for Bayer Schering Pharma, Merck Serono, Teva Pharmaceutical Industries Ltd., Biogen Idec, and Genmab A/S; and has received speaker honoraria from the same entities stated above; and grants/grants pending from the same entities stated above, CurePSP, Fondazione Italiana Sclerosi Multipla, and the Italian Ministry of Health. Giancarlo Comi serves on scientific advisory boards for Bayer Schering Pharma, Merck Serono, Teva Pharmaceutical Industries Ltd., Sanofi-Aventis, Novartis, and Biogen Idec; and has received speaker honoraria from the entities stated above and from Serono Symposia International Foundation. Giuseppe Magnani and Manu Sidhu report no disclosures. John Q. Trojanowski may accrue revenue in the future on patents submitted by the University of Pennsylvania wherein he is coinventor and he received revenue from the sale of Avid to Eli Lilly as coinventor on imaging-related patents submitted by the University of Pennsylvania. Eric J. Huang and Lea T. Grinberg report no disclosures. Bruce L. Miller serves on scientific advisory boards for TauRx Ltd., Allon Therapeutics, Bristol-Myers Squibb, Siemens Molecular Imaging, and Lilly USA. Nina Dronkers reports no disclosures. William W. Seeley serves on scientific advisory boards for Bristol-Myers Squibb, and has received speaker honoraria from Novartis-Korea. Maria Luisa Gorno-Tempini reports no disclosures. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Mesulam MM. Slowly progressive aphasia without generalized dementia. Ann Neurol 1982;11:592–598 [DOI] [PubMed] [Google Scholar]
- 2.Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol 2004;55:335–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mesulam M, Wicklund A, Johnson N, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol 2008;63:709–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grossman M. Primary progressive aphasia: clinicopathological correlations. Nat Rev Neurol 2010;6:88–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Josephs KA, Hodges JR, Snowden JS, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122:137–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998;393:702–705 [DOI] [PubMed] [Google Scholar]
- 7.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006;314:130–133 [DOI] [PubMed] [Google Scholar]
- 8.Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 2007;114:31–38 [DOI] [PubMed] [Google Scholar]
- 9.Rohrer JD, Lashley T, Schott JM, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain 2011;134:2565–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodges JR, Davies RR, Xuereb JH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol 2004;56:399–406 [DOI] [PubMed] [Google Scholar]
- 11.Josephs KA, Duffy JR, Strand EA, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 2006;129:1385–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deramecourt V, Lebert F, Debachy B, et al. Prediction of pathology in primary progressive language and speech disorders. Neurology 2010;74:42–49 [DOI] [PubMed] [Google Scholar]
- 13.Grossman M, Powers J, Ash S, et al. Disruption of large-scale neural networks in non-fluent/agrammatic variant primary progressive aphasia associated with frontotemporal degeneration pathology. Brain Lang Epub 2012 Dec 3 [DOI] [PMC free article] [PubMed]
- 14.Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol 2008;21:688–692 [DOI] [PubMed] [Google Scholar]
- 15.Kim EJ, Rabinovici GD, Seeley WW, et al. Patterns of MRI atrophy in tau positive and ubiquitin positive frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry 2007;78:1375–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Josephs KA, Whitwell JL, Dickson DW, et al. Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging 2008;29:280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McMillan CT, Irwin DJ, Avants BB, et al. White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry 2013;84:949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogar JM, Dronkers NF, Brambati SM, Miller BL, Gorno-Tempini ML. Progressive nonfluent aphasia and its characteristic motor speech deficits. Alzheimer Dis Assoc Disord 2007;21:S23–S30 [DOI] [PubMed] [Google Scholar]
- 20.Mackenzie IR, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 2006;112:539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 1997;18:S1–S2 [PubMed] [Google Scholar]
- 22.Lee SE, Rabinovici GD, Mayo MC, et al. Clinicopathological correlations in corticobasal degeneration. Ann Neurol 2011;70:327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner J, Andersson JL, Friston KJ. Image registration using a symmetric prior—in three dimensions. Hum Brain Mapp 2000;9:212–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashburner J, Friston KJ. Unified segmentation. Neuroimage 2005;26:839–851 [DOI] [PubMed] [Google Scholar]
- 25.Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage 2007;38:95–113 [DOI] [PubMed] [Google Scholar]
- 26.Seeley WW, Matthews BR, Crawford RK, et al. Unravelling Boléro: progressive aphasia, transmodal creativity and the right posterior neocortex. Brain 2008;131:39–49 [DOI] [PubMed] [Google Scholar]
- 27.McMonagle P, Blair M, Kertesz A. Corticobasal degeneration and progressive aphasia. Neurology 2006;67:1444–1451 [DOI] [PubMed] [Google Scholar]
- 28.Kertesz A, McMonagle P, Jesso S. Extrapyramidal syndromes in frontotemporal degeneration. J Mol Neurosci 2011;45:336–342 [DOI] [PubMed] [Google Scholar]
- 29.Galantucci S, Tartaglia MC, Wilson SM, et al. White matter damage in primary progressive aphasias: a diffusion tensor tractography study. Brain 2011;134:3011–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Agosta F, Canu E, Sarro L, Comi G, Filippi M. Neuroimaging findings in frontotemporal lobar degeneration spectrum of disorders. Cortex 2012;48:389–413 [DOI] [PubMed] [Google Scholar]
- 31.Schwindt GC, Graham NL, Rochon E, et al. Whole-brain white matter disruption in semantic and nonfluent variants of primary progressive aphasia. Hum Brain Mapp 2013;34:973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilson SM, Henry ML, Besbris M, et al. Connected speech production in three variants of primary progressive aphasia. Brain 2010;133:2069–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duffy JR. Motor Speech Disorders: Substrates, Differential Diagnosis, and Management, 2nd ed St. Louis: Elsevier Mosby; 2005 [Google Scholar]
- 34.Tognola GVL. Brain lesions associated with oral apraxia in stroke patients: a clinico-neuroradiological investigation with the CT scan. Neuropsychologia 1980;18:257–272 [DOI] [PubMed] [Google Scholar]
- 35.Groswasser ZKC, Groswasser-Reider I, Solzi P. Mutism associated with buccofacial apraxia and bihemispheric lesions. Brain Lang 1988;34:157–168 [DOI] [PubMed] [Google Scholar]
- 36.Wilson SM, Dronkers NF, Ogar JM, et al. Neural correlates of syntactic processing in the nonfluent variant of primary progressive aphasia. J Neurosci 2010;30:16845–16854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin WL, Zehr C, Lewis J, Hutton M, Yen SH, Dickson DW. Progressive white matter pathology in the spinal cord of transgenic mice expressing mutant (P301L) human tau. J Neurocytol 2005;34:397–410 [DOI] [PubMed] [Google Scholar]
- 38.Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci 2001;114:1179–1187 [DOI] [PubMed] [Google Scholar]
- 39.Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild-type tau fibrils specified by templated conformation change. J Biol Chem 2009;284:3546–3551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron 2009;62:42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}