

Abstract
The Kras gene is mutated to an oncogenic form in most pancreatic tumors. However, early attempts to use this molecule as a specific biomarker of the disease, or inhibit its activity as a cancer therapy, failed. This left a situation in which everyone was aware of the association between this important oncogene and pancreatic cancer, but no one knew what to do about it. Recent findings have changed this picture—many assumptions made about KRAS and its role in pancreatic cancer were found to be incorrect. Several factors have contributed to increased understanding of the activities of KRAS, including creation of genetically engineered mouse models, which have allowed for detailed analyses of pancreatic carcinogenesis in an intact animal with a competent immune system. Cancer genome sequencing projects have increased our understanding of the heterogeneity of individual tumors. We also have a better understanding of which oncogenes are important for tumor maintenance and are therefore called “drivers.” We review the advances and limitations of our knowledge about the role of Kras in development of pancreatic cancers and the important areas for future research.
Keywords: Kras, Inflammation, Pancreatic Cancer, Mouse Model
Pancreatic cancer progresses through a series of precursor lesions, the most common of which are known as pancreatic intraepithelial neoplasia (PanIN). Progression requires specific genetic changes and, at least in pancreatic tumors, each stage seems to be associated with specific mutations. Oncogenic KRAS was first associated with pancreatic cancer at least 24 years ago.1,2 At that time, Kras was shown to be mutated to an oncogenic form, most commonly KrasG12D, in most pancreatic tumors. Expression of the human oncogenic KrasG12D in the mouse pancreas duplicated, at least approximately, these precursor stages. These genetically engineered mouse models allow for the study of the earliest phases of pancreatic cancer development, as their gene expression can be manipulated. However, mouse models have inherent limitations, beyond the biologic differences between mice and humans. One limitation is that they express oncogenic KRAS in all the cells of the pancreas, unlike human pancreatic tumors. Another is the concurrent, rather than sequential, introduction of the genetic alterations associated with each stage of spontaneous tumor development. Therefore, the use of genetic mouse models needs to be balanced by other approaches, such as using human pancreatic cancer cell lines, primary human cells, and human xenograft tumors. Judicious use of all of these models provides the best picture of the initiation and progression of pancreatic cancer, and has allowed us to appreciate the roles of KRAS in these processes.
Individual vs Population of KRAS Molecules
Individual KRAS proteins function as binary molecular switches. When bound to guanosine triphosphate (GTP), they interact with signaling molecules that regulate cell activities such as proliferation, differentiation, apoptosis, and cell migration (Figure 1).3 Binding of GTP to KRAS is extremely low in the absence of interactions with guanine nucleotide exchange factors, which increase the rate of GTP loading. Many receptors for growth factors, cytokines, hormones, neurotransmitters, and other regulators are able to activate RAS, either by directly or indirectly increasing access of guanine nucleotide exchange factors.
Figure 1.

Basic KRAS biology and the positive feedback loop between KRAS and inflammation. KRAS binds either GTP or guanosine diphosphate (GDP). When occupied by GDP, KRAS does not activate downstream signaling pathways and is considered “off.” KRAS is activated by extracellular signals coming from the environment in the form of growth factors, cytokines, damage molecules (DAMPs), hormones, or other molecules. These molecules indirectly interact with guanine nucleotide exchange factors (GEFs) to replace GDP for GTP and causing KRAS to be “on.” Active KRAS will then interact with a large number of downstream signaling pathways. Some of these pathways in turn lead to generation of signals that activate KRAS through a positive feedback loop. Examples are inflammatory mediators that are activated by KRAS and in turn lead to further activation of KRAS. Normal KRAS is rapidly inactivated thanks to the effect of the GTPase-activating proteins (GAPs) that help hydrolyze GDP to GTP. In the presence of an oncogenic form of KRAS the return of KRAS to an “off” state is delayed, and a pathological response ensues that can lead to cancer.
Once a cell has responded to an incoming signal, KRAS activity is no longer needed, so GTP is hydrolyzed to guanosine diphosphate. KRAS itself has some limited GTP hydrolysis activity, which is increased by interactions with specific GTPase-activating proteins. Although individual KRAS molecules act as binary switches, the population of KRAS molecules in a cell acts more like a rheostat than a switch. Greater numbers of KRAS molecules bound to GTP lead to a greater overall signal. In other words, at the level of the cell, KRAS is neither “on” nor “off”—the number of active molecules determines the levels of the resulting signal.
Specific point mutations in KRAS (primarily those that affect KRAS–GTPase-activating protein interactions) reduce GTP hydrolysis and thereby cause KRAS to remain active.4 They are considered to be oncogenic because when they were first investigated, they were observed to transform cells in the absence of other manipulations, and cells that expressed these mutant forms of KRAS formed tumors in vivo. It has been estimated that approximately 30% of all tumors have oncogenic mutations in RAS family members, HRAS, NRAS, and KRAS5; oncogenic Kras is found in nearly every pancreatic tumor.6,7 Many studies have indicated that Kras activity increases after transfection with oncogenic forms of Kras, indicating that they are constitutively active.8–10 However, KRAS activation is a complex phenomenon; GTP binding is not sufficient to define active KRAS. Other factors, including subcellular localization, can influence its association with downstream effectors.11,12 Therefore, KRAS-GTP might not always stimulate effector signaling.13,14 This observation has important implications for cancer treatment and prevention.
An additional layer of complexity was added with the observation that Kras alone might not be sufficient to transform a cell. When oncogenic Kras was evaluated as a biomarker, numerous studies reported that healthy people have cells with oncogenic Kras in different organs, including the pancreas,15–17 colon,16 and lungs,18 at rates far exceeding the rates of cancer development. More recently, mice that express oncogenic Kras, either in the whole body or in specific organs, develop cancers from only a small fraction of the cells that contain the oncogenic Kras,19,20 It can therefore be assumed that other, genetic or epigenetic, factors are required to initiate carcinogenesis, even when a mutation in the Kras oncogene has been acquired. One key factor might be the level of KRAS activity. In fact, in genetically engineered mice that express oncogenic KRAS at physiologic levels (because a single allele is mutated, the cells presumably express equal amounts of oncogenic and nononcogenic KRAS), only a small fraction of the cells are transformed. In addition, the overall level of KRAS activity is lower than expected, whether it is because not all of the oncogenic KRAS molecules bind GTP or because, even when GTP bound, they do not always activate effectors (possibly because of inappropriate subcellular localization or negative-feedback mechanisms).11 Accordingly, expression of oncogenic Kras from its endogenous locus in mice is insufficient to activate downstream signaling pathways, such as the mitogen-activated protein kinase (MAPK) pathway.13 Supporting the hypothesis that a specific level of KRAS activation is required to initiate transformation, upstream stimuli were shown to accelerate the development of cancer in mice (Figure 2).13,21–24 These studies indicate that reaching a threshold level of KRAS activity might be essential to initiate the carcinogenesis process, and that the presence of a mutant copy of Kras is not sufficient to reach this threshold. Therefore, signals that act upstream of Kras, such as epidermal growth factor and inflammatory stimuli, might play an important role during the carcinogenesis process. In this light, it is noteworthy that many reagents shown to accelerate formation of pancreatic ductal adenocarcinomas (PDACs) in mice that express oncogenic Kras, also directly or indirectly activate KRAS. Likewise, many reagents shown to prevent or increase the time to development of PDAC in mouse models reduce RAS activity, directly or indirectly (Figure 3). A second aspect to be considered is the ability of the cells to withstand high levels of Kras activation. It is conceivable that the oncogenic stress associated with expression of oncogenic Kras might result in apoptosis or senescence,25,26 and factors that allow cells to overcome the senescence barrier, such as inflammation25 or loss of tumor suppressor genes, such as p16 or p53,27 allow the transformation process. Some controversy still persists, however, as other studies have indicated that oncogenic KRAS is sufficient to inhibit the onset of senescence and repress the expression of p16.28 However, neither the presence of inflammation nor the loss of tumor suppressor genes is sufficient, per se, to initiate pancreatic cancer in the absence of oncogenic Kras.29–31 These observations support a model in which Kras plays a unique role in the onset of PDAC.
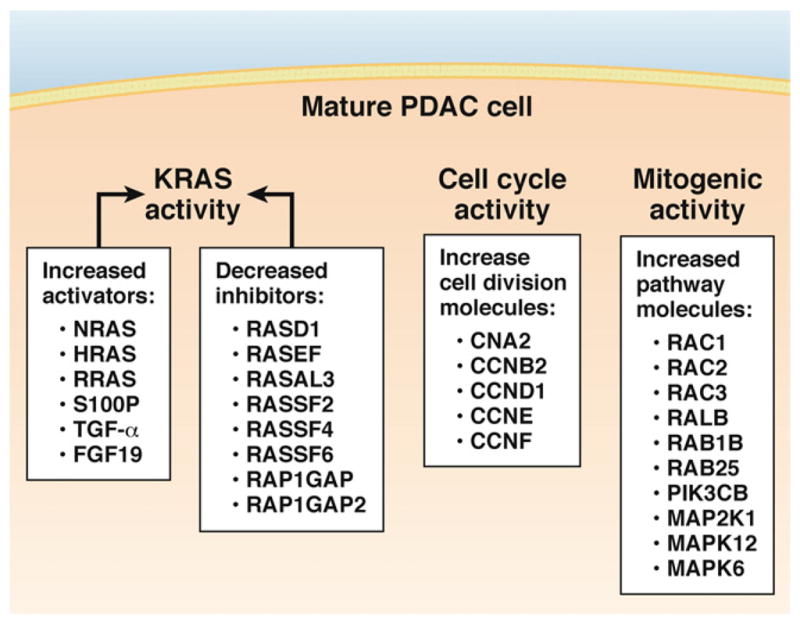
Figure 2.

Multiple changes in gene expression influence KRAS activity, the KRAS downstream pathway and cell growth occur during development of a “mature” PDAC cell. RAS activity is still affected by upstream factors, but this component might be less important in the presence of a mutant form of KRAS. The downstream effectors of KRAS are highly activated and might eventually become independent of KRAS. Finally, expression of several regulators of cell cycle are altered in cancer cells; the cyclins listed in the figure are all expressed at levels exceeding those of normal pancreatic cells.
Figure 3.
Molecules that increase KRAS activity tend to promote PDAC development, while and those that inhibit KRAS activity tend to prevent PDAC. This would be predicted by the KRAS-inflammatory feedback loop model.
Human and mouse PDAC cells have elevated KRAS activity and correspondingly high activation of KRAS effector genes. In mice that express endogenous levels of oncogenic Kras, tumor cells contain significantly higher levels of KRAS–GTP than nontumor tissues.7 In mice engineered to express elevated levels of oncogenic KRAS, approaching the elevated levels found in tumors, widespread formation of precursor lesions and rapid progression to invasive tumors are observed, in the absence of any other manipulations.13 Taken together, these results support the concept that a relatively high level of oncogenic Kras activity is necessary for cellular transformation.6 Therefore, the mechanisms that increase the levels of oncogenic Kras in cells contribute to transformation and cancer development.
What Is the Role of Oncogenic KRAS in Pancreatic Tumor Formation?
People acquire oncogenic mutations in Kras in lung, pancreas, colon, and other tissues as they age.15–18 This is a major reason that Kras was not found to be a good biomarker for cancer. So why do only a few people develop PDAC or other cancers? A major new insight has been that oncogenic KRAS is not always in the active state.14 In other words, the presence of oncogenic KRAS is not sufficient to transform cells and additional genetic or environmental factors that raise the threshold of KRAS activity and remove barriers to tumorigenesis might be required.
Oncogenic and nononcogenic forms of Kras can be activated by many factors. However, oncogenic KRAS has slower kinetics of return to its guanosine diphosphate–bound status than nononcogenic forms.9 The slower kinetics provide extra time for activated KRAS to generate a feedback loop that sustains its activity, such as activation of nuclear factor–κB, cyclo-oxygenase–2, and others. These finding apply to cancer prevention strategies, which have focused on inhibiting KRAS effectors, under the model that oncogenic KRAS is always active. However, the understanding that oncogenic KRAS needs to be activated to cause cancer means that inhibition of KRAS activation itself is a reasonable preventative strategy. This model is supported by reports that drugs that reduce cancer risk, such as nonsteroidal anti-inflammatory agents (eg, aspirin, celecoxib, etc) and various antioxidants, inhibit activation of KRAS (Figure 2). This concept also indicates that attempting to block cancer initiation downstream of KRAS will be a challenge because many effectors are activated by KRAS (Figure 1).
Kras as a Component of a Complex System
Kras is clearly important component in the pathogenesis of pancreatic cancer. Oncogenic mutations in Kras are also frequently detected in lung, colon, and other tumor types. Increased Kras activity is required for development of hepatocellular carcinoma, via alterations in GTPase-activating proteins, instead of oncogenic mutations. Increased levels of activity of KRAS effectors, or decreased levels or activity of KRAS inhibitors (or their regulatory molecules) could also lead to hepatic tumorigenesis.
Many molecular alterations are needed for PanIN lesions to progress to metastatic tumors. Hundreds of changes in gene expression occur in pancreatic cancer cells compared with normal pancreatic cells.7 Analyses of gene expression changes in human pancreatic cancer cells have identified several genes whose increased or decreased expression would affect KRAS activity (Figure 2) (C. Logsdon, unpublished observation).
KRAS is a regulator of the MAPK pathway. In fully developed PDAC, several molecules within the MAPK pathway are over- or underexpressed that are normally regulated by active KRAS (C. Logsdon, unpublished observation). Likewise, expression of cyclins and other cell cycle regulators increases in cancer cells. These alterations are likely selected for— higher levels of KRAS activity increase cell proliferation. Notwithstanding the complexity of changes that promote the activity of KRAS and its effectors in pancreatic cancer, several studies in mouse models or human cell lines suggest that at least a subset of pancreatic tumors continuously require KRAS for growth. This aspect will be discussed here in more detail.
Oncogenic Kras in Pancreatic Cancer Progression and Maintenance
There is much evidence that Kras is required for pancreatic tumorigenesis, but until recently it was assumed that nothing could be done to prevent the oncogenic effects of Kras activation, and that the only option was to slow tumor growth and progression. Researchers assumed that Kras must be important at all stages of PDAC. However, like the assumption that oncogenic Kras must be always on, this might not actually be the case. To study the role of Kras in growth and progression of PDAC, in the absence of good inhibitors, studies have relied on small hairpin RNA-mediated knockdown in cell lines, inhibitors of downstream targets of Kras, or more recently, on mouse models that allow reversible expression of oncogenic Kras.
During Early Stages of Pancreatic Cancer
Pancreatic adenocarcinoma cells have highly unstable genomes; each individual tumor is estimated to accumulate several hundred mutations.7 The progression from a normal pancreatic cell into a metastatic cancer cell requires multiple steps, each of which requires changes in gene expression. Clearly, PDAC is not the same from formation until metastasis. Early human PanIN lesions have Kras mutations, often in absence of additional genetic alterations.32,33 However, progression requires inactivation of tumor suppressors, such as p16.25,28 In humans, sporadic mutations in Kras might occur, and in most cases be cleared from the tissue by cell senescence. However, cells that escape senescence through acquisition of additional mutations might become transformed. In the pancreas of mice, these mutations can be introduced at the same time in a large number of cells.
The widely used KC (Pdx1-Cre;LSL-KrasG12D or Ptf1a-Cre; LSL-KrasG12D), KPC (most commonly Pdx1-Cre;LSL-KrasG12D;LSL-Trp53R172H or Ptf1a-Cre; LSL-KrasG12D;LSL-Trp53R172H, but also used to describe models with loss-of function or conditional inactivation of the tumor suppressor p53) and KC;Ink4a (Pdx1-Cre;LSL-KrasG12D; Ink4af/f or Ptf1a-Cre;LSL-KrasG12D;Ink4af/f) mouse models of pancreatic cancer were developed via tissue-specific, Cre-mediated expression of oncogenic Kras.29,30,34 These models recapitulate the initiation and progression of pancreatic cancer. However, because Kras expression, once induced, is irreversible, these models are not suitable for investigating whether Kras is required beyond tumorigenesis—for growth and progression. Some tumors that develop in mice have been reported to be dependent on (or addicted to) a single oncogene product; tumors that develop in mouse models of lung adenocarcinoma, breast cancer, and melanoma depend on oncogenic Kras to maintain their mass and continue growing.35–37
Researchers have recently created mice in which pancreatic expression of oncogenic Kras can be reversed, called inducible Kras* or iKras mice.38,39 These mice express oncogenic Kras under the control of a tetracycline operator; the rtTa transcription factor is expressed in a pancreas-specific manner from the Rosa26 locus. Administration of doxycycline to iKras* mice leads to expression of oncogenic Kras and withdrawal of the drug inactivates expression of the transgene. Unlike KC mice, iKras mice do not express KrasG12D from the endogenous Kras locus. The expression levels of oncogenic Kras, however, are comparable with endogenous levels, and activation of Kras in adult animals leads to formation of sporadic PanINs only with long latency and low penetrance, possibly because a threshold of KRAS activity needs to be reached before the oncogene has an effect on the tissue.
Induction of acute pancreatitis with the cholecystokinin agonist caerulein leads to rapid and widespread formation of PanINs, likely through initiation of the Kras effector loop described here. Induction of acute pancreatitis in wild-type mice leads to acinar damage, including acinar to ductal metaplasia, infiltration of immune cells, and edema; the peak level of damage occurs within 24 h of caerulein administration. Tissue repair ensues rapidly, and the pancreas resumes its normal histology within 1 week, although slightly higher proliferation of acinar cells, compared with healthy pancreata, is observed for a few weeks. In contrast, KC and iKras* pancreata fail to undergo tissue repair after caerulein administration.38,40 In these mice, acinar to ductal metaplasia progresses to form dysplastic ductal structures, surrounded by extensive fibrosis, within 1 week. Within 3 weeks, virtually all the ductal structures show characteristics of PanINs. Over time, higher-grade PanIN lesions populate the pancreas and, finally, carcinoma in situ develops.
To understand the role of Kras signaling in these changes, the effects of inactivating oncogenic Kras expression at different time points were examined. The effects of Kras inactivation were found to be time dependent (Figure 4). Inactivation in low-grade PanIN lesions results in most cells that line the dysplastic ducts to activate expression of genes of the acinar lineage, and inactivate ductal genes, in a process that is the opposite of acinar to ductal metaplasia. At the early stages of lesion formation, KRAS activity is therefore necessary for the transformation process to continue; it seems to prevent tissue repair and regulate the differentiation status of the epithelial cells, as proposed previously.40 Kras inactivation in high-grade PanIN lesions has different effects. The precancerous cells that appear require Kras for survival, and undergo apoptosis once the transgene is inactivated. These results indicate that Kras is important not only for tumor formation, but also during early stages of tumor progression.
Figure 4.

Oncogenic Kras in pancreatic cancer maintenance. Activation of oncogenic Kras in the normal pancreas leads to PanIN formation; progression to metastatic pancreatic cancer is dependent on acquisition of additional genetic alterations, such as mutation of p53. Inactivation of Kras in PanINs leads to either redifferentiation of PanIN cells to normal pancreatic lineages such as acinar cells, or to apoptosis. Inactivation of oncogenic Kras in advanced disease leads to tumor regression through apoptosis and probably additional mechanisms yet to be identified. However, a small subset of cancer cells survives Kras inactivation and persists in a dormant state; these cells can lead to tumor relapse on Kras inactivation. Other mechanisms of relapse might also exist.
A common feature of KC and iKras mice is that their lesions rarely progress to adenocarcinoma unless additional mutations are introduced. This is consistent with the observation that, in patients, pancreatic adenocarcinoma does not occur without the accumulation of multiple genetic alterations, probably over the course of decades.41 Loss, inactivation, or mutation of multiple tumor suppressors (such as Tp53 and p16) is commonly detected in human pancreatic tumors. In mice, these tumor suppressors are spontaneously lost at different rates, depending on the level of inflammation and/or Kras activity. For example, in KC mice, which express endogenous levels of oncogenic Kras, the tumor suppressor Tp53 tends to be mutated or lost at late stages of tumor development.34 In contrast, in mice engineered to express high levels of oncogenic Kras in the pancreatic cells, such as the Elastase-CreER;cLGL-KrasG12D (LGL) model, p16 is rapidly lost.42 Possibly, in presence of high levels of oncogenic Kras there is a higher selective pressure for pancreatic cells to lose p16 and therefore escape Kras-induced senescence.26 The high frequency of p16 loss might explain why LGL mice develop PDAC at a faster rate than LSL mice.
To speed cancer development in models with low Kras activity, mutant alleles of tumor suppressors can be introduced, which would resemble development of pancreatic cancer in humans. iKras mice carrying a loss-of-function allele in p53 (called iKras*p53 mice) rapidly develop pancreatic adenocarcinoma with high penetrance,38,39 and can be used to determine the effects of Kras inactivation in invasive tumors. Studies with these mice have shown that inactivation of Kras leads to rapid tumor regression through loss of tumor cell proliferation and viability.38,39 These observations have been extended to the metastases that develop with high frequency in iKras* mice, engineered to express a point mutation in p53 (p53R172H mice).43 In vivo imaging studies showed rapid regression of the primary tumor and liver metastases on Kras inactivation. Although these tumors appear to regress steadily over time, accurate in-depth characterization of the tissue after regression, as well as observations based on primary tumor cells, indicated that not all the tumor cells were eliminated on Kras inactivation. In fact, a fraction of the tumor cells appeared to persist in a dormant state and resume rapid growth on K-ras reactivation.43 It is possible that Kras inhibition is eventually be over-ridden by generation of resistant clones, which have accumulated additional mutations in the same pathways or have activated other oncogenic pathways. Lung adenocarcinoma and melanoma cells have also been shown to require oncogenic Kras.36,37
In the Tumor Microenvironment
Mice that have been genetically engineered to express oncogenic Kras have also been used to study the role of Kras on the pancreatic tumor microenvironment, which contains extensive inflammatory stroma. During the earliest stages of PanIN formation, the lesions accumulate proliferating cells of mesenchymal origin that might comprise fibroblasts and pancreatic stellate cells. PanIN formation and progression is also accompanied by the infiltration of immune cells.44 It is interesting to note that changes in the phenotype of the stellate cells occur earlier than noticeable changes in other components of the pancreas.45 Therefore, even low levels of Kras activity generate signals that influence the microenvironment.
Unlike most other solid tumors, pancreatic tumors are considered to be hypovascularized, although blood vessels are present within the tumor microenvironment. It is unclear whether the vascularity per cancer cell is lower than in other tumors or whether the abundant nonvascular extracellular matrix contributes to the low level of overall vascularity. Stellate cells produce angiogenic factors.46,47 In addition to the cellular components, the stroma comprises components of the extracellular matrix, such a collagen fibers and hyaluronic acid.48,49 Little is known about how the formation of the stroma is regulated and maintained.
Perhaps the most convincing evidence for the effect of Kras on the tumor microenvironment is found when Kras is inactivated in pancreata bearing low-grade PanINs. Under these conditions the stroma is remodeled. The activated fibroblasts that populate the stroma stop expressing markers of activation, exit the cell cycle, and are eliminated from the pancreas via an unknown mechanism. Inactivation of Kras also leads to resolution of the chronic inflammation associated with pancreatic cancer.
Kras therefore regulates production of factors that maintain an active stroma (Figure 5). These factors and their activities have not been completely identified, but appear to include sonic hedgehog, interleukin-6, and prostaglandin E,50 each of which is expressed in a Kras-dependent manner.38 Sonic Hedgehog, one of the ligands of the Hedgehog signaling pathway, is expressed by pancreatic tumor cells51,52 and functions in a paracrine manner,53 activating Hedgehog signaling in the stroma and potentially mediating its maintenance.54 The inflammatory cytokine IL-6 is overexpressed in pancreatic tumors and is important for development of PanINs in mice.55 Prostaglandin E acts directly on stellate cells through the prostaglandin E receptor 4, to stimulate production of the stroma.50 These, and likely several other factors, are generated by high levels of sustained Kras activity.
Figure 5.

Kras mediates interactions between the tumor cells and the surrounding stroma. Tumor cells expressing oncogenic Kras secrete molecules that act in a paracrine manner on surrounding components of the stroma, such as fibroblasts, innate and adaptive immune cells (black arrows). These cells in turn promote tumor growth (dashed arrows); only a small subset of the signals mediating the interaction between cancer cells and components of the stroma have been identified.
The immune cells that infiltrate the pancreas also appear to be regulated by Kras. In mouse models of pancreatic cancer, PanINs are infiltrated by immune cells, including those that suppress the immune response, such a regulatory T cells, myeloid-derived suppressor cells,44 and mast cells.56 Tumor cells secrete cytokines, such as granulocyte-macrophage colony-stimulating factor,57,58 which promote infiltration of myeloid-derived suppressor cells that inhibit anti-tumor immune responses. Kras inactivation leads to an overall reduction in the number of infiltrating immune cells. So, the inflammatory environment of pancreatic tumors also appears to be regulated by Kras, in a paracrine manner, forming part of a Kras–inflammation positive-feedback loop that requires additional study.
Tumor Metabolism
Pancreatic tumors create a hypoxic microenvironment; they are not well vascularized and contain large amounts of desmoplasia, which contribute to the low levels of oxygen.59 Cancer cells are able to adapt to the hypoxic conditions by several mechanisms, such as by up-regulating hypoxia inhibitory factor–α,60 which allows them to change their metabolic pathways. The changes in cancer cell metabolism, which have been proposed to be “hallmarks of cancer,”61 include increased glucose metabolism via the glycolytic pathway and reduced reliance on the Krebs cycle.62 The specific metabolic changes that occur in cancer cells and their regulatory mechanisms are complex and beyond the scope of this article (for a comprehensive review, see reference63), but might be targeted therapeutically.
K-ras was recently shown to regulate metabolic changes in cancer cells by controlling factors that regulate transcription of metabolic genes.39 Reagents might therefore be designed to target Kras, or its effectors, that alter pancreatic cancer metabolism, and impair the ability of the cancer cells to maintain high levels of glycolysis.64
Kras Dependency
Although Kras is known to be involved in cell transformation, pancreatic tumor formation, and early stages of tumor progression in mice, little is known about its requirement for progression of human pancreatic tumors. Mouse models were engineered to depend on Kras for quick development of tumors. Human tumors take decades to progress. Because small molecule inhibitors of Kras were not available at the time, studies to determine whether pancreatic tumor progression requires (is addicted to) KRAS activity have used RNA interference-based approaches. These studies have been conducted using pancreatic cancer cell lines (mostly derived from patients) that have been carried for years, in different laboratories. Of 7 lines analyzed, 4 were found to be Kras dependent, in that they did not undergo apoptosis when Kras levels were reduced.65
One factor associated with Kras dependency of human pancreatic cancer cells was overexpression of KRAS protein (which usually correlated with amplification of its genomic locus). The Kras gene is amplified in human and mouse pancreatic tumors.13 Another factor was increased expression of epithelial genes and functions of their products. Kras-independent lines expressed more genes that encoded mesenchymal factors, and fewer that encoded epithelial factors, than Kras-dependent cells. Expression of small hairpin RNAs against Kras slowed growth of established pancreatic xenograft tumors in mice.66
Human PDACs have been subdivided based on gene expression patterns into quasi-mesenchymal, classic, and exocrine-like (the least characterized) subtypes.67 As the nomenclature indicates, the mesenchymal subtype has mesenchymal rather than epithelial characteristics. Interestingly, in vitro studies using cell lines with the characteristics of classic and quasi-mesenchymal subtypes showed that classic PDACs depend on Kras, whereas those of the mesenchymal subtype are not. More importantly, these differences were maintained in tumor xenografts. It therefore appears that at least a subset of human pancreatic tumors depends on Kras for progression. Additional studies are necessary to elucidate the extent of Kras dependency of human pancreatic tumors. Kras dependency has not been investigated using primary human tumor xenografts, or in orthotopic tumors implanted into the pancreas.
Effectors of Kras
KRAS inhibitors might be in the pipeline,68 in fact, inhibitors of nononcogenic RAS proteins were described recently69,70; however, they have not been historically available and the current drug candidates are still probably years away from the clinic. Farnesyl transferase inhibitors, designed to prevent membrane association and thereby activation of K-ras, were found to be nonspecific and affect the activities of many other proteins. The oral farnesyl transferase inhibitor R115777 did not increase the median survival time of patients with locally advanced or metastatic pancreatic adenocarcinoma.71 It is likely that nonfarnesylated Kras can still undergo prenylation, via geranylgeranylation, to associate with the cell membrane.
An alternative approach is target effectors of Kras that are involved in tumor development. Members of several different Kras signaling pathways participate in formation and growth of pancreatic tumors in mice, such as the kinase Akt. Expression of a dominant-active form of Akt (caAkt) in the pancreas of mice caused expansion of ductal structures and activation of progenitor gene expression, but did not lead to progression of PanINs or PDAC.72 Expression of BRAFV600E (the oncogenic form of BRAF) in the pancreas of mice, but not PI3CAH1047R (the dominant-active form of phosphatidylinositol 3 kinase) led to formation of PanIN lesions. In addition, expression of BRAFV600E, but not PI3CAH1047R, in combination with a cancer-associated mutant form of p53 (TP53R270H), led to development of lethal PDACs in mice.67 However, a recent study has challenged these conclusions by showing that the PI3K pathway is sufficient and necessary to initiate pancreatic carcinogenesis; thus, the relative contribution of different Kras effectors needs further study.73 Therefore, activation of MAPK signaling, but not phosphatidylinositol 3 kinase signaling via Akt, recapitulates the effects of Kras activation in mice.
So which of these effector pathways is required for maintenance of established tumors? Inhibition of the MAPK signaling with the inhibitor PD325901 had a cytostatic effect on KPC tumors, orthotopically implanted in immune-competent, syngeneic mice. Mice given PD325901 survived longer than controls, but died soon after cessation of the drug. This finding contrasts, at least in part, with the observation that Kras inactivation in tumors affects cell survival in iKras* mice.38,39
RAF inhibitors had no effect on pancreatic cancer cell lines, and interfered with the cytostatic effects of MAP kinase kinase (MEK) inhibitors when the compounds were applied to cells in combination. However, combined inhibition of MEK and AKT had a synergistic, antiproliferative effect on a large panel of human pancreatic cancer cell lines. Similar results were obtained with the MEK and AKT inhibitors GDC0941 and AZD6244, respectively.66 In mice, the combination of these inhibitors caused regression of xenograft tumors, whereas administration of each reagent alone only slowed tumor growth. The logical next step appears to be to determine the efficacy of this treatment regimen in mice that spontaneously develop pancreatic tumors or mice with orthotopic tumors. Studies are also needed to determine the effects of these inhibitors on survival, and of discontinuing their administration.
Another, somewhat less-studied effector of pathway is the Ral guanine nucleotide exchange factor–Ral small GTPase signaling network (for review, see references74 and75), which is frequently activated in pancreatic tumors and involved in their progression.76 Inhibitors of cyclin-dependent kinase-5 have been shown to suppress KRAS–Ral signaling and block pancreatic tumor formation and progression in mice.77 The Kras-related C3 botulinum substrate 1 (Rac1) is an effector of Kras signaling from the epidermal growth factor receptor. Rac1 regulates the rearrangement of the actin cytoskeleton and cell motility (for review, see reference78). Recently, Rac1 was shown to be dispensable during development of the normal pancreas, but required for formation of pancreatic tumors in mice.79 These factors might be developed as therapeutic targets.
In development of therapeutics for pancreatic cancer, the ability of Kras to induce inflammation in surrounding tissues should also be considered. Nuclear factor–κB and signal transducer and activator of transcription 3 signaling are regulated by Kras, and can be targeted with specific inhibitors.13,80 A different way to approach Kras inhibition would be to block factors that promote KRAS activity, such as inflammatory factors or epidermal growth factor receptor signaling. However, these types of reagents might be better for blocking tumor formation than progression.
Targeting of downstream effectors of KRAS has been tested in other tumor types, such as colon and lung cancer81,82; whether the importance for specific effector pathways is different in different tumor types, or whether findings from other malignancies can be translated to pancreatic cancer will have to be addressed in the future.
A Therapeutic Target?
Experimental inhibition of Kras activity slows growth or even causes regression of pancreatic tumors in mice. Even some human pancreatic cancer cells require KRAS activity to form tumors. Therefore, strategies to inhibit KRAS directly, or inhibit its effectors, are under active investigation. But will inhibiting oncogenic KRAS be sufficient to cure patients with pancreatic cancer? In mice, inactivation of Kras leads to tumor regression, and the animals remain healthy, with no evidence of relapse, for a relatively long time.38,39,43 The mechanism of tumor regression involves apoptosis induction, and likely other components that have not been fully elucidated. However, individual tumor cells survive inactivation of Kras, presumably by maintaining a dormant state. Recent unpublished observations indicate that this dormancy can be maintained for several months (M. Collins and Marina Pasca di Magliano, unpublished results). When Kras is reactivated in cancer cells in mice, the cells begin to proliferate rapidly and the mice die within a few days.43 Whether these cells could also resume proliferation in absence of reactivation of Kras through a different mechanism is not known. Therefore, the risk of cancer relapse after withdrawal of KRAS inhibitors is likely to be high. Full tumor eradication will require identification of the mechanisms that allow a subset of tumor cells to survive Kras inactivation, and development of methods to target them.
Only a subset of human tumors remains Kras-independent over time, based on studies of human cell lines. It is important to determine whether mouse models of pancreatic cancer resemble a specific subset of human tumors. We also need to develop criteria to determine which human cell lines depend on Kras, so that individual patients can be treated appropriately.
Future Directions
The current experimental evidence indicates that a high level of sustained KRAS activity is required for pancreatic tumorigenesis. Whether the expression of an oncogenic form of Kras is sufficient to obtain these high levels of activation in tumor cells, or whether mutant RAS requires further activation by upstream signals to achieve its fully active GTP-bound state to drive cancer growth remains an unresolved issue. The observation that, at least initially, KRAS activity can be modulated by upstream signals provides a rationale to investigate new approaches to pancreatic cancer prevention. Those could conceivably include reducing factors that activate KRAS, by preventing inflammation or altering diet and lifestyle. A second focus of research might be the interaction between tumor cells and their surrounding stroma. Recent studies in mouse models indicate that KRAS activity leads to inflammation and changes in the stroma. That is not to say that inflammation and the stroma are not part of what sustains tumorigenesis; in fact, there is likely to be a positive-feedback loop that involves KRAS and the stroma and sustains tumor growth. Experiments with Kras inactivation indicate that Kras activity is not only required for pancreatic tumor formation and development but, at least in mice, the requirement for Kras is continuous. KRAS might therefore be a good therapeutic target after all.
Abbreviations used in this paper
- GTP
guanosine triphosphate
- MAPK
mitogen-activated protein kinase
- MEK
MAP kinase kinase
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
Footnotes
Conflicts of Interest
The authors disclose no conflicts.
References
- 1.Smit VT, Boot AJ, Smits AM, et al. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–7782. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 3.Spaargaren M, Bischoff JR, McCormick F. Signal transduction by Ras-like GTPases: a potential target for anticancer drugs. Gene Expr. 1995;4:345–356. [PMC free article] [PubMed] [Google Scholar]
- 4.Scheffzek K, Ahmadian MR, Kabsch W, et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2:344–358. doi: 10.1177/1947601911411084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones S, Zhang X, Parsons, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koeffler HP, McCormick F, Denny C. Molecular mechanisms of cancer. West J Med. 1991;155:505–514. [PMC free article] [PubMed] [Google Scholar]
- 9.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campbell PM, Singh A, Williams FJ, et al. Genetic and pharmacologic dissection of Ras effector utilization in oncogenesis. Methods Enzymol. 2006;407:195–217. doi: 10.1016/S0076-6879(05)07017-5. [DOI] [PubMed] [Google Scholar]
- 12.Campbell SL, Khosravi-Far R, Rossman KL, et al. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 13.Daniluk J, Liu Y, Deng D, et al. An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haojie Huang JD, Liu Yan, Chu Jun, et al. Oncogenic K-Ras requires activation for enhanced activity. Oncogene. 2013 Jan 21; doi: 10.1038/onc.2012.619. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan L, McFaul C, Howes N, et al. Molecular analysis to detect pancreatic ductal adenocarcinoma in high-risk groups. Gastroenterology. 2005;128:2124–2130. doi: 10.1053/j.gastro.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 16.Lu X, Xu T, Qian J, et al. Detecting K-ras and p53 gene mutation from stool and pancreatic juice for diagnosis of early pancreatic cancer. Chin Med J (Engl) 2002;115:1632–1636. [PubMed] [Google Scholar]
- 17.Parsons BL, Meng F. K-RAS mutation in the screening, prognosis and treatment of cancer. Biomark Med. 2009;3:757–769. doi: 10.2217/bmm.09.95. [DOI] [PubMed] [Google Scholar]
- 18.Yakubovskaya MS, Spiegelman V, Luo FC, et al. High frequency of K-ras mutations in normal appearing lung tissues and sputum of patients with lung cancer. Int J Cancer. 1995;63:810–814. doi: 10.1002/ijc.2910630611. [DOI] [PubMed] [Google Scholar]
- 19.Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 20.Guerra C, Mijimolle N, Dhawahir A, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 21.Carriere C, Young AL, Gunn JR, et al. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382:561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ardito CM, Gruner BM, Takeuchi KK, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304–317. doi: 10.1016/j.ccr.2012.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navas C, Hernandez-Porras I, Schuhmacher AJ, et al. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–330. doi: 10.1016/j.ccr.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Appleman VA, Ahronian LG, Cai J, et al. KRAS(G12D)- and BRAF(V600E)-induced transformation of murine pancreatic epithelial cells requires MEK/ERK-stimulated IGF1R signaling. Mol Cancer Res. 2012;10:1228–1239. doi: 10.1158/1541-7786.MCR-12-0340-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guerra C, Collado M, Navas C, et al. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeNicola GM, Tuveson DA. RAS in cellular transformation and senescence. Eur J Cancer. 2009;45(Suppl 1):211–216. doi: 10.1016/S0959-8049(09)70036-X. [DOI] [PubMed] [Google Scholar]
- 27.Bardeesy N, Aguirre AJ, Chu GC, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006;103:5947–5952. doi: 10.1073/pnas.0601273103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KE, Bar-Sagi D. Oncogenic KRas suppresses inflammation-associated senescence of pancreatic ductal cells. Cancer Cell. 2010;18:448–458. doi: 10.1016/j.ccr.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 31.Guerra C, Schuhmacher AJ, Canamero M, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733. e9. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong SM, Vincent A, Kanda M, et al. Genome-wide somatic copy number alterations in low-grade PanINs and IPMNs from individuals with a family history of pancreatic cancer. Clin Cancer Res. 2012;18:4303–4312. doi: 10.1158/1078-0432.CCR-12-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 35.Podsypanina K, Politi K, Beverly LJ, et al. Oncogene cooperation in tumor maintenance and tumor recurrence in mouse mammary tumors induced by Myc and mutant Kras. Proc Natl Acad Sci U S A. 2008;105:5242–5247. doi: 10.1073/pnas.0801197105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher GH, Wellen SL, Klimstra D, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–3262. doi: 10.1101/gad.947701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chin L, Tam A, Pomerantz J, et al. Essential role for oncogenic Ras in tumour maintenance. Nature. 1999;400:468–472. doi: 10.1038/22788. [DOI] [PubMed] [Google Scholar]
- 38.Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639–653. doi: 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris JP, 4th, Cano DA, Sekine S, et al. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120:508–520. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1147. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ji B, Tsou L, Wang H, et al. Ras activity levels control the development of pancreatic diseases. Gastroenterology. 2009;137:1072–1082. doi: 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collins MA, Brisset JC, Zhang Y, et al. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One. 2012;7:e49707. doi: 10.1371/journal.pone.0049707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clark CE, Hingorani SR, Mick R, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 45.Won JH, Zhang Y, Ji B, et al. Phenotypic changes in mouse pancreatic stellate cell Ca2+ signaling events following activation in culture and in a disease model of pancreatitis. Mol Biol Cell. 2011;22:421–436. doi: 10.1091/mbc.e10-10-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Z, Vonlaufen A, Phillips PA, et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177:2585–2596. doi: 10.2353/ajpath.2010.090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erkan M, Adler G, Apte MV, et al. StellaTUM: current consensus and discussion on pancreatic stellate cell research. Gut. 2012;61:172–178. doi: 10.1136/gutjnl-2011-301220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Provenzano PP, Cuevas C, Chang AE, et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Charo C, Hwang RF, Arumugam T, et al. PGE2 activation of the EP4 receptor regulates stellate cell activity in the pancreatic stromal microenvironment. Pancreas. 2013;42:467–474. doi: 10.1097/MPA.0b013e318264d0f8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thayer SP, di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berman DM, Karhadkar SS, Maitra A, et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature. 2003;425:846–851. doi: 10.1038/nature01972. [DOI] [PubMed] [Google Scholar]
- 53.Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455:406–410. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 54.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intra-epithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 56.Chang DZ, Ma Y, Ji B, et al. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2011;17:7015–7023. doi: 10.1158/1078-0432.CCR-11-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pylayeva-Gupta Y, Lee KE, Hajdu CH, et al. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hidalgo M, Maitra A. The hedgehog pathway and pancreatic cancer. N Engl J Med. 2009;361:2094–2096. doi: 10.1056/NEJMcibr0905857. [DOI] [PubMed] [Google Scholar]
- 60.Chaika NV, Gebregiworgis T, Lewallen ME, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A. 2012;109:13787–13792. doi: 10.1073/pnas.1203339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 62.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neesse A, Michl P, Frese KK, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 65.Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with ”K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hofmann I, Weiss A, Elain G, et al. K-RAS mutant pancreatic tumors show higher sensitivity to MEK than to PI3K inhibition in vivo. PLoS One. 2012;7:e44146. doi: 10.1371/journal.pone.0044146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Collisson EA, Trejo CL, Silva JM, et al. A central role for RAF→ME-K→ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov. 2012;2:685–693. doi: 10.1158/2159-8290.CD-11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cox AD, Der CJ. Ras family signaling: therapeutic targeting. Cancer Biol Ther. 2002;1:599–606. doi: 10.4161/cbt.306. [DOI] [PubMed] [Google Scholar]
- 69.Maurer T, Garrenton LS, Oh A, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109:5299–5304. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patgiri A, Yadav KK, Arora PS, et al. An orthosteric inhibitor of the Ras-Sos interaction. Nat Chem Biol. 2011;7:585–587. doi: 10.1038/nchembio.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Macdonald JS, McCoy S, Whitehead RP, et al. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest New Drugs. 2005;23:485–487. doi: 10.1007/s10637-005-2908-y. [DOI] [PubMed] [Google Scholar]
- 72.Elghazi L, Weiss AJ, Barker DJ, et al. Regulation of pancreas plasticity and malignant transformation by Akt signaling. Gastroenterology. 2009;136:1091–1103. doi: 10.1053/j.gastro.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Eser S, Reiff N, Messer M, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013 Feb 27; doi: 10.1016/j.ccr.2013.01.023. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 74.Vigil D, Cherfils J, Rossman KL, et al. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Neel NF, Martin TD, Stratford JK, et al. The RalGEF-Ral effector signaling network: the road less traveled for anti-Ras drug discovery. Genes Cancer. 2011;2:275–287. doi: 10.1177/1947601911407329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lim KH, O’Hayer K, Adam SJ, et al. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385–2394. doi: 10.1016/j.cub.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 77.Feldmann G, Mishra A, Hong SM, et al. Inhibiting the cyclin-dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ral signaling. Cancer Res. 2010;70:4460–4469. doi: 10.1158/0008-5472.CAN-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 79.Heid I, Lubeseder-Martellato C, Sipos B, et al. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology. 2011;141:719–730. doi: 10.1053/j.gastro.2011.04.043. [DOI] [PubMed] [Google Scholar]
- 80.Fukuda A, Wang SC, Morris JP, 4th, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19:441–455. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ebi H, Corcoran RB, Singh A, et al. Receptor tyrosine kinases exert dominant control over PI3K signaling in human KRAS mutant colorectal cancers. J Clin Invest. 2011;121:4311–4321. doi: 10.1172/JCI57909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engelman JA, Chen L, Tan X, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]