

Abstract
Chromosomal translocations are critically involved in the molecular pathogenesis of B-cell lymphomas, and highly recurrent and specific rearrangements have defined distinct molecular subtypes linked to unique clinicopathological features1,2. In contrast, several well-characterized lymphoma entities still lack disease-defining translocation events. To identify novel fusion transcripts resulting from translocations, we investigated two Hodgkin lymphoma cell lines by whole-transcriptome paired-end sequencing (RNA-seq). Here we show a highly expressed gene fusion involving the major histocompatibility complex (MHC) class II transactivator CIITA (MHC2TA) in KM-H2 cells. In a subsequent evaluation of 263 B-cell lymphomas, we also demonstrate that genomic CIITA breaks are highly recurrent in primary mediastinal B-cell lymphoma (38%) and classical Hodgkin lymphoma (cHL) (15%). Furthermore, we find that CIITA is a promiscuous partner of various in-frame gene fusions, and we report that CIITA gene alterations impact survival in primary mediastinal B-cell lymphoma (PMBCL). As functional consequences of CIITA gene fusions, we identify downregulation of surface HLA class II expression and overexpression of ligands of the receptor molecule programmed cell death 1 (CD274/PDL1 and CD273/PDL2). These receptor–ligand interactions have been shown to impact anti-tumour immune responses in several cancers3, whereas decreased MHC class II expression has been linked to reduced tumour cell immunogenicity4. Thus, our findings suggest that recurrent rearrangements of CIITA may represent a novel genetic mechanism underlying tumour–microenvironment interactions across a spectrum of lymphoid cancers.
In Hodgkin lymphoma, translocations or chromosomal breakpoints have only rarely been described, whereas in PMBCL no recurrent translocation events have been reported5. Massively parallel, paired-end sequencing of expressed transcripts (RNA-seq) provides an analytical platform suitable for genome-wide mapping of translocation breakpoints, sequence variants and quantitative expression6,7. Thus, we used this technology to detect novel gene fusions in the two Hodgkin lymphoma cell lines KM-H2 and L428, including 82.9 million paired-end reads for KM-H2, of which 71.1 million mapped to the reference human genome (86%, 3.6 gigabases), and 61.5 million paired-end reads for L428, of which 55.5 million (90%, 2.8 gigabases) mapped to the reference genome (Supplementary Fig. 1). Using a novel gene-fusion discovery method, we obtained 14 distinct fusion transcript predictions for KM-H2 and five for L428 (Supplementary Table 1). Figure 1 shows the fusion prediction with the highest read support involving CIITA and an uncharacterized gene BX648577 (FLJ27352/hypothetical LOC145788) found in KM-H2.
Figure 1. CIITA–BX648577 gene fusion observed using paired-end massively parallel whole transcriptome sequencing.
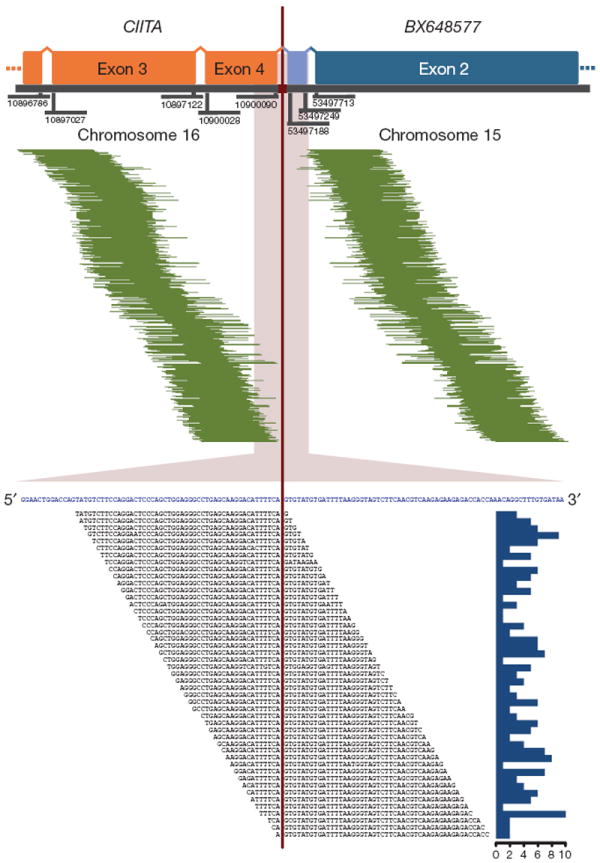
In the upper panel, 468 mate-pair sequences are shown aligning on either side of the breakpoint (pairing CIITA and BX648577). The genomic coordinates of the exon boundaries are given. In light blue, the transcribed intronic BX648577 sequence is shown as part of a transcript variant resulting from the fusion. In the lower panel, 191 split-reads are depicted lying on the breakpoint (in blue: merged reference sequence of CIITA and BX648577). The histogram on the right describes the absolute frequency of each sequence read spanning the breakpoint.
Next, we validated three fusion predictions by direct sequencing and fluorescence in situ hybridization (FISH): the first involving the genes BAT2L1 (chromosome 9q34.13) and MGMT (chromosome 10q26.3) in KM-H2 (Supplementary Fig. 2), the second involving the genes SLCO3A1 (chromosome 15q26.1) and ELMO1 (chromosome 7p14.1–14.2) in L428 (Supplementary Fig. 2), and lastly the previously mentioned CIITA–BX648577 gene fusion (Fig. 2 and Supplementary Fig. 3). In all three cases we could confirm the breakpoint sequences as predicted. Although all of the identified gene fusions are likely to contribute to the specific phenotype of the affected cells, we focused on the CIITA–BX648577 fusion transcripts given the known involvement of CIITA in B-cell immune function and the high read-support. The CIITA gene was initially studied in patients with bare lymphocyte syndrome, a rare autosomal recessive disease, in which CIITA mutations lead to loss of MHC class II expression and clinical manifestations due to an immunodeficiency phenotype8. CIITA was found to be the essential transactivator of MHC class II expression functioning in a complex of transcription factors (RFX, NFY, X2BP) that bind to class II MHC promoters9,10.
Figure 2. Molecular characterization of the gene fusion CIITA–BX648577 in Hodgkin lymphoma cell line KM-H2.
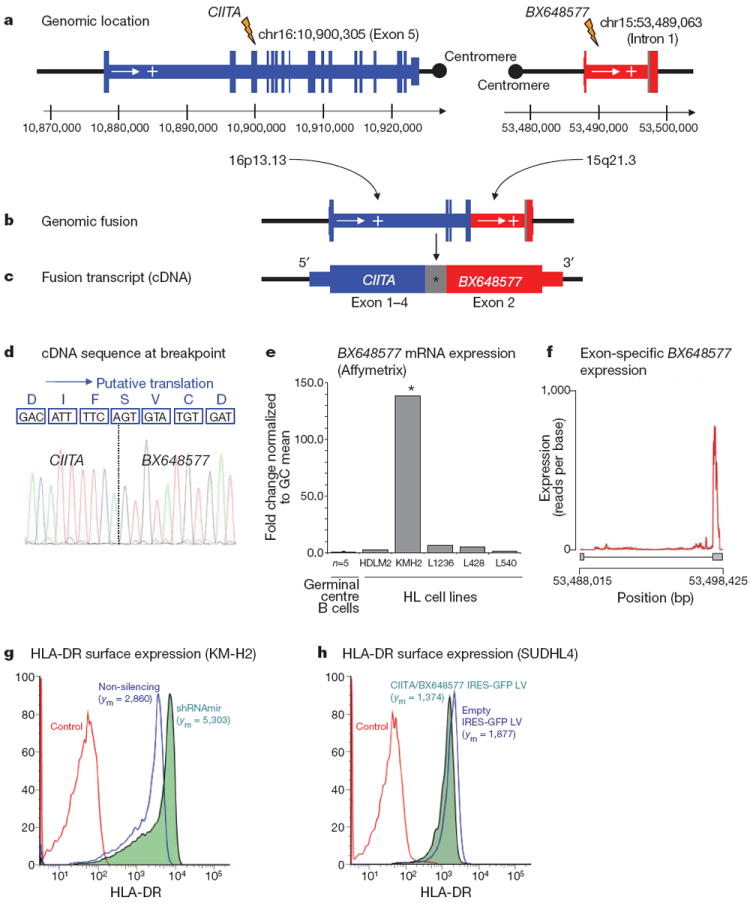
a, Genomic location: exon structure and genomic breakpoints. b, Genomic fusion: rearranged genomic location with fusion of CIITA exons 1–5 with intron 1 of BX648577. c, Fusion transcript: the longest fusion transcript with transcribed intronic BX648577 sequence (*) is shown. Shorter splice variants exist. d, Reading frame at the breakpoint and putative translation: CIITA exons 1–4 and BX648577 exon 2 original reading frames are conserved. The shorter splice variants leading to premature translational stop at the breakpoint are not shown. e, BX648577 gene expression: gene expression array data (Affymetrix HG UA133 2.0 Plus probe set ID 243309_at) showing overexpression of BX648577 in KM-H2 compared with microdissected germinal centre B cells and other Hodgkin lymphoma cell lines. f, BX648577 exon-specific expression is biased towards exon 2 as part of the CIITA–BX648577 gene fusion. g, RNA interference with the gene fusion in KM-H2 cells increases surface HLA-DR expression compared with the non-silencing control. h, Forced expression of the CIITA–BX648577 fusion decreases surface HLA-DR expression on SUDHL4 cells compared with empty vector controls. Mean fluorescence intensities (ym) are indicated.
Using PCR, the genomic breakpoint coordinates were mapped to chr15:53,489,063 and chr16:10,900,305 (NCBI Build 36.1) falling within CIITA exon 5 and BX648577 intron 1 (Fig. 2a, b). Two major transcripts of this gene fusion were identified. Sequences of the first alternative transcript aligned to exons 1–4 of CIITA (chromosome 16) and exon 2 of BX648577 (chromosome 15), whereas the second alternative transcript contained an additional sequence of 62 base pairs aligning to BX648577 intron 1, which has not been previously annotated as expressed (Fig. 2c, d). Gene expression analysis demonstrated that BX648577 was highly overexpressed in KM-H2 compared with other Hodgkin lymphoma cell lines (35.0-fold) and microdissected germinal centre B cells (138.3-fold) (Fig. 2e, f). The longer isoform has an open reading frame consistent with a predicted 255 amino-acid protein containing the amino (N)-terminal MHC class II transactivator domains 1–4, an amino-acid sequence originating from the novel BX648577 exon and the carboxy (C) terminus of the hypothetical BX648577 protein (Fig. 2d and Supplementary Information).
We next performed immunofluorescence to investigate the cellular localization of the fusion protein. We found a strong perinuclear cytoplasmic staining pattern in KM-H2 compared with primary peripheral blood B cells in which we observed a predominantly nuclear localization of wild-type CIITA (Supplementary Fig. 4). This staining pattern is in agreement with loss of a C-terminal nuclear localization sequence11 of CIITA in the chimaeric protein.
Using high-resolution single nucleotide polymorphism analysis, we found that the rearrangement involving chromosomes 15 and 16 was associated with relative genomic imbalances at the breakpoints (Supplementary Fig. 5). Multicolour-FISH in representative metaphases of KM-H2 and interphase-FISH showed complex chromosomal rearrangements with various marker chromosomes and subclonal variation as shown by others for this cell line12 (Supplementary Fig. 6). Locus-specific FISH using Fosmid probes for CIITA and BX648577 confirmed co-localization of the CIITA and BX648577 gene loci on two derivative chromosomes 16, both harbouring insertions of chromosome 15 material. These complex rearrangements are likely to place the fusion gene in a different positional context that might add to the high promoter activity of CIITA driving expression of the fusion transcripts in this cell line.
To investigate the functional consequence of the CIITA–BX648577 gene fusion in KM-H2 cells, we generated stable KM-H2 fusion transcript knockdowns by RNA interference. Quantitative reverse transcriptase PCR (RT–PCR) showed reduction of fusion transcript levels by 85%, and western blotting demonstrated a marked reduction in CIITA–BX648577 fusion protein compared with non-silencing controls (Supplementary Fig. 7). As CIITA is an essential transactivator of MHC class II expression9 and deletion mutants have been described that inhibit wild-type CIITA function in vitro13,14, we studied transcriptional changes of short hairpin RNA (shRNA)-transduced cells using gene expression microarrays (Supplementary Table 2). Most strikingly, overrepresentation analysis identified genes of the ‘antigen presentation pathway’ significantly enriched among the upregulated genes (P = 0.03), and flow cytometry confirmed increased surface expression of HLA-DR in KM-H2 cells with fusion transcript knockdown (Fig. 2g). We also transduced diffuse large B-cell lymphoma (DLBCL) cell line SUDHL4 with a lentiviral vector containing the full-length coding complementary DNA (cDNA) of the CIITA–BX648577 fusion (second alternative transcript), which led to decreased surface HLA-DR expression (Fig. 2h). These data demonstrate that the CIITA–BX648577 fusion suppresses expression of HLA class II genes in KM-H2 and SUDHL4. Diminished expression of HLA class II in B-cell lymphomas has been linked to reduced immunogenicity, escape from immunosurveillance and inferior survival15,16.
To determine if breakpoints within CIITA are recurrent in cases of primary Hodgkin lymphoma, we used FISH on tissue microarrays and found eight out of 55 cases (15%) rearranged (Supplementary Table 3). Representative FISH images of the break-apart assay are shown in Fig. 3, demonstrating involvement of all cells of the malignant cell compartment (Hodgkin Reed–Sternberg cells that carry the CD30 antigen). In four of the eight cases we detected an unbalanced rearrangement. Because PMBCL shares clinical and biological features with cHL, we also studied PMBCL and found 29 (38%) of 77 cases positive for break-apart of the CIITA locus. Only four out of 131 studied cases of DLBCL (3%) showed break-apart of the CIITA locus. Hence, the occurrence of CIITA rearrangements in PMBCL and cHL was significantly higher than in DLBCL (P < 0.0001).
Figure 3. FISH on tissue microarrays showing recurrent CIITA break-apart in cHL and PMBCL.
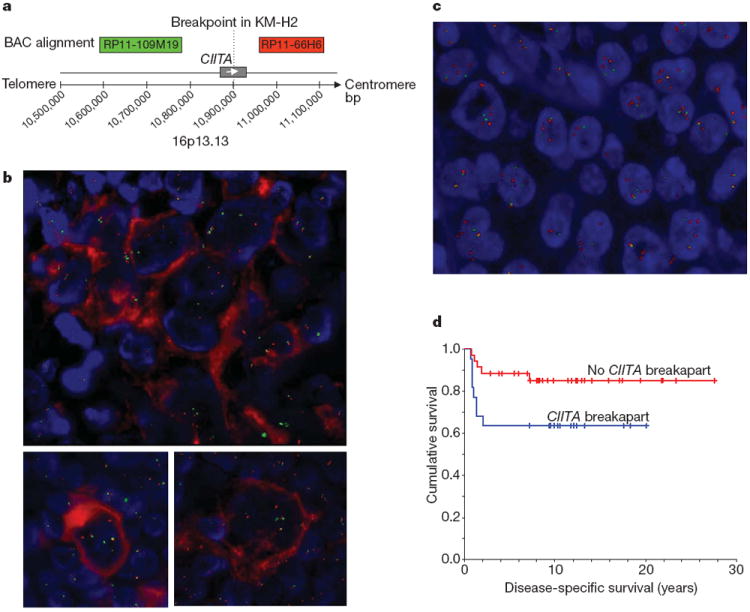
Representative images are shown. a, Design of the break-apart assay using bacterial artificial chromosome probes RP11-109M19-SpG (green signals) plus RP11-66H6-SpO (red signals). b, Combined immunofluorescence for CD30 (red staining) and FISH shows CIITA rearrangement in Hodgkin Reed–Sternberg cells that carry the CD30 antigen. Upper image: Hodgkin Reed–Sternberg cell-rich area with CIITA break-apart, lower images: individual Hodgkin Reed–Sternberg cells with break-apart. c, PMBCL case with CIITA break-apart in almost all cells represented in the section. Signal constellation indicates CIITA polyploidy and rearrangement of multiple alleles. d, Disease-specific survival of 57 patients with PMBCL treated with multi-agent chemotherapy (with or without radiation) according to CIITA rearrangement status. The presence of a CIITA rearrangement significantly correlated with shorter disease-specific survival (P = 0.044)
We also studied the correlation of CIITA translocation with clinical outcome in 57 patients with PMBCL who were treated with curative intent using multi-agent chemotherapy with or without radiotherapy17. The clinical characteristics of this cohort are summarized in Supplementary Table 4. Presence of a CIITA rearrangement significantly correlated with a shorter disease-specific survival (10-year disease-specific survival 63.6% compared with 85.0%, P = 0.044) (Fig. 3d). Multivariate testing using a Cox regression model including the International Prognostic Index18 score and the individual clinical prognostic factors showed that the presence of CIITA rearrangement had independent prognostic significance (P = 0.013) for decreased disease-specific survival together with elevated LDH serum levels (P = 0.001) (Supplementary Table 5).
To determine fusion partners involved in CIITA rearrangements, we used 3′ rapid amplification of cDNA ends (RACE) in eight cases of PMBCL that were positive for CIITA translocation by FISH. In total, we identified five new translocation partners (CD274, CD273, RALGDS, RUNDC2A and C16ORF75) (Supplementary Table 6). Precise mapping revealed that the corresponding genomic breakpoints in CIITA all fell within a 1.6-kilobase (kb)-spanning breakpoint cluster region in intron 1, indicating a ‘hot spot’ area for chromosomal breaks (Supplementary Fig. 8). In the case of CD274 (PDL1), CD273 (PDL2) and RUNDC2A, the fusion transcripts merged exon 1 of CIITA with exon 2 of the respective fusion partners, resulting in open reading frames (Supplementary Information). Of note, in both cases with CIITA–CD273 fusions, mutations to the CIITA start codon were detected resulting in translational reading frames beginning at the original CD273 start site (Supplementary Information). Overexpression of CD274 and CD273 closely linked to copy number gain has been previously described in PMBCL and Hodgkin lymphoma19-21, and expression of CD274 has been shown to correlate with poor prognosis in other cancers22. Furthermore, re-analysis of gene expression profiling data confirmed overexpression of both genes in over 50% of cases of PMBCL (Fig. 4a)23. Here, we could show by quantitative RT–PCR that in all three cases CD274 and CD273 were highly overexpressed (fold changes 451, 4,115 and 1,729, respectively) as part of the respective CIITA fusions compared with CD77+ centroblasts (Fig. 4b). Importantly, expression levels markedly exceeded the levels of those in cases of PMBCL without translocations. Overall, these results indicate that substitution of active CIITA promoters24 can lead to overexpression of partner genes in a B-cell context.
Figure 4. CD274/CD273 expression on PMBCL cells inhibits T-cell activation.
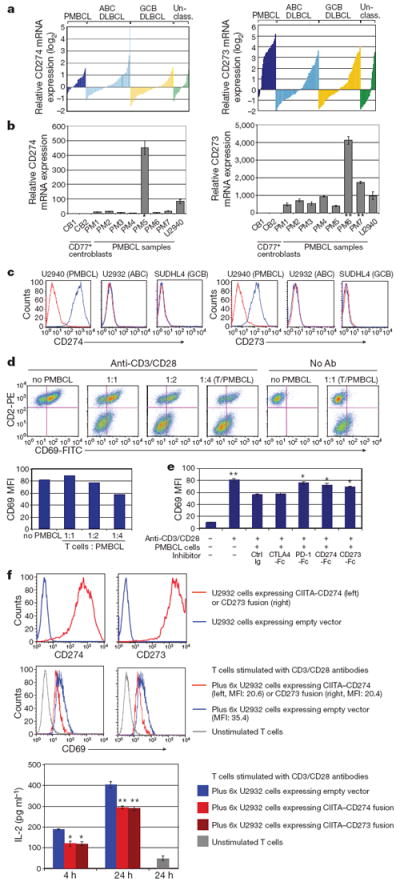
a, mRNA overexpression of CD274 and CD273 in molecular subtypes of DLBCL including PMBCL. Normalized relative log2 ratios are shown (Affymetrix gene expression profiling)23. Probeset intensities of CD274 and CD273 correlate with each other (Pearson coefficient 0.541). b, Fold change of CD274 and CD273 mRNA expression in PMBCL compared with germinal centre B cells including cases with CIITA–CD274 (*), CIITA–CD273 (**) fusions and cell line U2940. c, Flow cytometric analysis of CD274/CD273 expression. Expression levels of CD274 or CD273 (blue histogram) and isotype controls (red histogram) are shown. d, Inhibition of T-cell activation by PMBCL cells. Dot plots and bar graph show that CD69 expression on Jurkat T cells (CD2 positive) is increasingly reduced by co-incubation with increasing numbers of U2940 cells. e, T-cell inhibition is PD1 dependent. CD274-Fc, CD273-Fc, PD1-Fc, CTLA4-Fc or control Ig were added to the co-cultures, respectively, in which Jurkat T cells were mixed with U2940 cells. Mean fluorescence intensities (MFI) are shown. Each bar is the mean of triplicate cultures, the error bars indicating the standard deviation (Student’s t-test, *P < 0.05, **P < 0.01, compared with control Ig). f, Forced expression of CIITA–CD274 and CIITA–CD273 fusion transcripts in U2932. The top panel shows the surface protein expression in transduced cells compared with empty vector control cells. The middle and bottom panels show significantly reduced CD69 expression on Jurkat T cells and IL-2 levels in the supernatant after admixture of CIITA–CD274 and −CD273 fusion-expressing U2932 cells. Each bar is the mean of triplicate cultures, the error bars indicating the standard deviation (Student’s t-test, *P < 0.05, **P < 0.01, compared with the empty vector control).
Next, we sought to study the functional consequences of CD274 and CD273 wild-type overexpression. We selected cell line U2940 (ref. 25) (consistent with PMBCL features) expressing high levels of both CD274 and CD273 messenger RNA (mRNA) and membrane protein (Fig. 4b, c). To study the effects of CD274 and CD273 surface expression on T-cell activation in particular, we analysed a co-culture system in which Jurkat T cells were co-incubated with U2940 cells. First, we confirmed that PD-1 expression was induced in Jurkat T cells upon T-cell receptor stimulation (Supplementary Fig. 9a). After 4 h of co-culture, Jurkat T-cell activation, measured by CD69 expression, was markedly decreased with increasing admixture of U2940 cells (Fig. 4d). In contrast, inhibition of T-cell activation was not observed when U2932 and SUDHL4 cells were co-incubated that did not express CD274 or CD273 on their surface (Supplementary Fig. 9b). We could also show that inhibition of T-cell activation by U2940 cells was reversible by incubation with inhibitors against PD-1, CD274 and CD273, but not against CTLA4 (Fig. 4e). Furthermore, overexpression of PD-1 in Jurkat cells further augmented T-cell inhibition, indicating dependence on the PD-1 pathway (Supplementary Fig. 9c).
We also investigated the effects of forced expression of the CIITA–CD274 and CIITA–CD273 gene fusions in U2932 cells, which do not express wild-type PD-1 ligands on their surface. Ectopic expression of the full-length coding cDNA of the fusions in U2932 markedly increased surface expression of the respective proteins in transduced cells (Fig. 4f) and, similar to U2940, decreased CD69 and interleukin-2 (IL-2) expression of PD-1-overexpressing Jurkat T cells compared with empty vector controls. In summary, these data show that both wild-type PD-1 ligand expression and the identified CIITA–CD274/CD273 gene fusions negatively regulate Jurkat T-cell activation.
Taken together, we have discovered breakpoints in the master regulator of MHC class II expression CIITA that are novel and highly recurrent in PMBCL. Moreover, the occurrence of CIITA breaks in cHL further substantiates the relatedness of cHL and PMBCL19,26. We observed CIITA breaks in only 3% of cases of DLBCL, contrasting with 38% in PMBCL, which highlights the relative specificity of this genetic event. In PMBCL, CIITA breaks were significantly correlated with outcome, demonstrating the potential clinical importance of these rearrangements, a finding that will require validation in additional cohorts of patients. Consequences of CIITA rearrangements appear to be diverse as evidenced by multiple fusion partners, and concomitant chromosomal imbalances as described previously in PMBCL. However, functional study of the most frequent specific gene fusions suggests that escape from immunosurveillance through various mechanisms might play an important role in the pathogenesis of these lymphomas. Furthermore, our data raise the possibility that deletion of tumour suppressor genes27, overexpression of oncogenes resulting from gene fusion, and CIITA loss of function might be concurrent consequences of a single genetic event.
METHODS
Materials and patient samples
The Hodgkin lymphoma cell lines KM-H2 and L428 were selected for whole transcriptome paired-end sequencing (RNA-seq) analysis. Both cell lines originated from patients with cHL (Epstein Barr virus negative mixed cellularity and nodular sclerosis, respectively). Additionally, cHL cell lines L540, L1236, HDLM2 and microdissected germinal centre cells were used for comparison of gene expression levels. All cell lines were obtained from the German Collection of Microorganisms and Cell Cultures and cultures were grown following standard recommendations (http://www.dsmz.de). DNA and RNA were extracted using Allprep extraction kits (Qiagen).
For validation in primary tissue samples, we studied paraffin material from 263 patients using FISH. All cases were identified through the Lymphoid Cancer Database of the British Columbia Cancer Agency including 131 patients with DLBCL, 77 with PMBCL and 55 with cHL. Patients with PMBCL were treated with CHOP and CHOP-like chemotherapy as previously described17. Additionally, we studied eight cases of PMBCL using RACE. All cases included in this study were independently reviewed by two haematopathologists (R.D.G. and P.F.) before analysis. Ethical approval for this study was obtained from the University of British Columbia – British Columbia Cancer Agency Research Ethics Board. Previously published gene expression profiling data of 203 biopsy samples with defined molecular subtypes of DLBCL, including PMBCL, were re-analysed23.
For functional in vitro studies, we used cell line U2940, derived from a DLBCL with features consistent with PMBCL25, U2932 (DLBCL, activated B-cell phenotype)31 and SUDHL4 (DLBCL, germinal centre B-cell phenotype)32. Jurkat T cells were used for co-culture experiments measuring T-cell activation.
Whole transcriptome paired-end sequencing (RNA-seq)
RNA-seq was performed as previously described28,33. In brief, double-stranded cDNA was synthesized from polyadenylated RNA and sheared. The 190–210 base-pair fraction was isolated and amplified with ten cycles of PCR using the Illumina Genome Analyser paired-end library protocol (Illumina). The resulting libraries were then sequenced on an Illumina Genome Analyser II.
Short read sequences obtained from the Genome Analyser II were mapped to the reference human genome (NBCI build 36.1, hg18, http://genome.ucsc.edu/cgi-bin/hgGateway), and spliced (cDNA) and unspliced gene sequences (Ensembl version 54) were aligned using the Bowtie algorithm34. Fusion transcript discovery was performed using deFuse (http://compbio.bccrc.ca). Briefly, deFuse identifies fusion transcripts by clustering discordantly aligning paired-end reads as potential evidence of reads that span a fusion breakpoint. Clusters of discordantly aligning reads are then used to inform a targeted search for reads split by fusion breakpoints. The results produced by deFuse were further filtered to reduce the number of false positives: (1) predictions had to be supported by at least eight reads spanning a fusion breakpoint and five reads split by a fusion breakpoint; (2) fusions between adjacent genes were removed unless implied in genomic inversion or eversion; (3) predictions involving ribosomal proteins or small nuclear ribosomal proteins were removed; (4) the results were filtered against fusion predictions from 41 ovarian cancer libraries in an attempt to remove systematic technical artefacts. After these series of filters were applied, a candidate list of fusions was carried forward for further analysis.
Gene expression levels were determined according to the total coverage of a gene, which was defined as the sum of the coverage of each non-redundant exonic nucleotide normalized by total mapped nucleotides.
Gene expression profiling of Hodgkin lymphoma cell lines
RNA-seq data were compared with gene expression data of Hodgkin lymphoma cell lines (KM-H2, L428, L540, L1236 and HDLM2) and microdissected germinal centre cells (n = 5) derived from Affymetrix HG 2.0 Plus gene expression arrays using two-cycle labelling reactions according to the standard protocol. For comparison of KM-H2 knockdown cultures (see below) we used freely available software (DCHIP, http://biosun1.harvard.edu/complab/dchip). Overrepresentation and pathway analysis was performed using Ingenuity Pathway Analysis (Ingenuity Systems).
High-resolution single nucleotide polymorphism analysis
Genome-wide copy number analysis was performed using Human SNP 6.0 arrays (Affymetrix) following the standard protocol and the manufacturer’s instructions. Hybridized arrays were washed, stained and scanned using Affymetrix Fluidics Station 450 and an Affymetrix GeneChip Scanner. The Affymetrix SNP 6.0 data were processed using the R-based aroma.affymetrix package.35 Total copy number was estimated according to the CRMA procedure, calibrating for allelic cross talk, nucleotide-position probe sequence effects, robust probe summarization and correction for fragment length effects. CRMA-normalized data were then compared with a reference derived from the HapMap 270 set of arrays downloaded from the Affymetrix website. Breakpoints were determined by a modified version of a hidden Markov model described before36. Software (including visualization routines) is available as part of the CNA-HMMer package available at http://compbio.bccrc.ca.
Genomic and RT–PCR and FISH
The validation of fusion transcripts was performed using both genomic and RT–PCR with forward and reverse primer combinations designed within the margins of the paired-end read sequences detected by RNA-seq (Supplementary Table 7). Expressed fusion transcript variants were cloned into pCR 2.1-TOPO vectors for amplification in chemically competent Escherichia coli (TOPO TA cloning kit, Invitrogen). Consensus sequences were determined from at least five separate colonies. For further validation by FISH, fusion probes were used to confirm gene fusions in interphase and metaphase nuclei of KM-H2 and L428. For probe design see Supplementary Fig. 10a–c. For detailed cytogenetic characterization of KM-H2, we used multicolour-FISH using 24Xcyte multi-colour probes (MetaSystems) as previously described37 and locus-specific FISH using Fosmid probes available from the Children’s Hospital & Research Center of Oakland for the gene loci of CIITA (WI2-1388I10, G248P83694E5) and BX648577 (WI2-2329H16, G248P88223D8).
Lentiviral transductions
RNA interference with the CIITA–BX648577 fusion transcript was performed using lentiviral transduction of a vector (pGIPZ-sh.FLJ27352, clone V2LHS_212659, Thermo Scientific Open Biosystems) expressing an shRNA (TGCTGTTGACAGTGAGCGCGACAGCCACCTCACTATCAAATAGTGAAGCCACAGATGTATTTGATAGTGAGGTGGCTGTCTTGCCTACTGCCTCGGA) that, after processing to mature siRNA, interferes with BX648577 exon 2 sequence as part of the CIITA–BX648577 fusion transcript. A vector with a non-interfering shRNA insert (non-silencing control) was used for comparison. In brief, the shRNAmir constructs were co-transfected along with the helper and packaging plasmids (pHDM-G, pHDM-Hgpm2, pHDM-tat1b and pRC/CMV-rev1b) into 293T cells by calcium-phosphate-mediated transfection (CalPhos Mammalian Transfection kit (Clontech)). Supernatant was collected 48 h and 72 h after transfection and concentrated by ultracentrifugation for 2 h at 60,000g, at 4 °C. The lentiviral particles were resuspended in IMDM and then used for transduction of KM-H2 cells. KM-H2 parental cells were transduced by adding 8 μg ml−1 polybrene, with both pGIPZ-sh.FLJ27352 and non-silencing control virus, and green fluorescent protein (GFP)-positive cells were sorted on a BD FACSAria cell sorter (BD Biosciences) 3 days after transduction and cultured using standard recommendations (http://www.dsmz.de). Purity (GFP positivity) was greater 93% for all experiments. Efficiency of RNA interference was evaluated by measuring residual expression of the fusion transcript by quantitative RT–PCR (see Supplementary Table 7 for primer sequences). Samples were run in triplicate on an iQ5 real-time PCR instrument (BioRad) under standard conditions using SYBR Green (Applied Biosystems) for detection. Measurements were quantified using the ΔΔCT method (Pfaffl) and expressed relative to the expression in parental (wild type) KM-H2 cells.
For forced expression of the CIITA–BX648577 gene fusion, we PCR-amplified the full-length cDNA from KM-H2 cells, and cloned it into the pGEM-T easy TA cloning vector (Promega). The fusion gene was then transferred into the pCAD-IRES-GFP lentiviral vector29. An empty vector was used as a negative control. For lentivirus production, we co-transfected lentiviral vectors and helper plasmids into 293T cells, harvested the supernatant and concentrated the lentiviral particles by ultracentrifugation (L7-65, Beckman) at 60,000g, 2 h, 4 °C. The resultant concentrated lentiviral suspension was then used to transduce SUDHL4 target cells. Three to five days after transduction, we sorted GFP-positive cells, which were used for subsequent experiments.
Western blotting and immunofluorescence
Semi-quantitative measurement of fusion protein expression was performed by semi-dry western blotting using a mouse monoclonalanti-CIITA primary antibody (7-1H, Santa Cruz Biotechnology) directed against the N terminus of CIITA, and a goat anti-mouse IgG-HRP secondary antibody (Santa Cruz). An anti-β-actin goat polyclonal antibody (C-11) followed by a donkey anti-goat IgG-HRP (both Santa Cruz) served as a control. RNA extraction of cell cultures and gene expression profiling using Affymetrix GeneChip HG 133 2.0 plus arrays was performed as previously described38.
KM-H2 cells and primary human B cells (CD20+) were attached to poly-lysine-coated slides. To fix the cells, we incubated them with 4% PFA for 10 min at room temperature and treated with 0.15% Triton X in PBS for 2 min at room temperature. After blocking, cells were incubated with an anti-CIITA antibody (clone 7-1H, Santa Cruz Biotechnology) overnight at 4 °C. After washing, cells were incubated with Alexa Fluor 594 rabbit anti-mouse IgG (Invitrogen) for 45 min at 37 °C. After stringent washing, cells were counterstained with DAPI and covered. Slides were analysed on a laser confocal microscope (Leica SP5 AOBS).
FISH on tissue microarrays
Tissue microarrays were constructed using archival, formalin-fixed, paraffin-embedded diagnostic biopsy specimens and 0.6-mm duplicate cores for DLBCL and PMBCL cases, and 1.5-mm cores for cHL, respectively. Optimal areas of the biopsies were chosen containing frequent large B or Hodgkin Reed–Sternberg cells, respectively. FISH was performed as previously described39 using in-house bacterial artificial chromosome break-apart and fusion probes for the loci of CIITA and BX648577 (Supplementary Fig. 10a). For fluorescence immunophenotyping and interphase cytogenetics as a tool for the investigation of neoplasms (FICTION) experiments, we used a primary monoclonal mouse CD30 antibodies (Ber-H2, Dako) and Alexa Fluor 594, goat anti-mouse (IgG, Invitrogen, Molecular Probes), following a standard protocol as previously described40. Commercially available Vysis LSI BCL6 Dual Colour, Break Apart Rearrangement probes (Abbott Molecular) were used in selected samples. All cases were independently scored by C.S. and S.B.N. For DLBCL and PMBCL, cases were recorded as rearranged for a certain locus if break-apart occurred in more than 5% of nuclei in three different high-power fields. All other signal constellations were regarded as negative. cHL cases were scored positive if at least ten cells with a signal split were detected in typically large cells distributed across the biopsy core. Unbalanced rearrangements were recorded if unequal numbers of red or green signals were seen in most nuclei.
RACE and quantitative RT–PCR
We used 3′ RACE to identify aberrant transcript variants of CIITA in selected PMBCL samples. Total RNA was extracted from eight cases with PMBCL with fresh-frozen lymph node material available after mechanical homogenization using Allprep extraction kits (Qiagen). cDNA was prepared from 150 ng to 1 μg of total RNA using the SMARTer RACE cDNA Amplification Kit (Clontech Laboratories) following the manufacturer’s protocol. 3′ RACE PCR amplification of the cDNA was performed using gene-specific primers for CIITA exon 1 and Universal Primer Mix. Aberrant lengths products were gel-purified and sequenced directly or were cloned into pCR 2.1-TOPO vectors (TOPO TA cloning kit, Invitrogen) and fully sequenced. Chimaeric mRNAs were confirmed by RT–PCR of randomly primed cDNA of the respective cases using gene-specific primers.
Quantitative RT-PCT was performed to measure expression levels of CD274 and CD273 mRNA using an Applied Biosystems 7900HT real-time PCR system. Primers were designed for SYBR Green (Applied Biosystems) detection of both wild-type and fusion transcripts for each gene (Supplementary Table 7). RNA was extracted from cell line U2940 and seven PMBCL lymph node samples (cell disaggregates) using Allprep extraction kits (Qiagen). PMBCL cases included one with previously identified CIITA–CD274 fusion and two with CIITA–CD273 fusions. As expression controls, magnetically enriched CD77+ cells were used (benign tonsillar cell disaggregates) (Miltenyi Biotec) as previously described41.
Flow cytometric analysis and ELISA
U2940, U2932 and SUDHL4 cell pellets were washed in 1×PBS with 1% FCS buffer and incubated with antibodies against CD69, CD2, CD274, CD273 and PD-1 (all from BD Pharmingen). Jurkat T cells were stimulated with anti-CD3 and anti-CD28 antibodies (BD Pharmingen). Recombinant human PD-1-Fc, CD274-Fc, CD273-Fc, CTLA4-Fc and control IgG (R&D Systems) were used for specific inhibition of surface molecules. KM-H2 and SUDHL4 cells were incubated with PE-Cy5-conjugated anti-human HLA-DR antibody (Beckman Coulter), and analysed on a BD FACSAria II instrument (BD Biosciences). IL-2 levels were measured using the Human IL-2 ELISA Kit (R&D Systems, number D2050) according to the manufacturer’s instructions. Plates were read at 405 nm using an ELISA reader (Molecular Devices).
Retroviral transduction
PD-1 cDNA (Invitrogen) was subcloned into an pBMN-IRES-EGFP vector, which was modified from the original Moloney LTR vector pBMN-IRES-Lyt2 (provided by the G. Nolan laboratory, Stanford University) by replacing Lyt2 with EGFP.
Lipofectamine 2000 (Invitrogen) was used to transfect 293T producer cells with a plasmid mixture for gag and pol, a mutant ecotropic env and each particular retrovirus. After 2 days, supernatant was passed through a 0.45-μm filter, mixed with polybrene (8 μg ml−1) and used for centrifugal transduction of target cells expressing the ecotropic retroviral receptor. In some instances a second infection was performed, using fresh supernatant collected 3 days after transfection of producer cells.
U2932 cells were first transduced with a feline endogenous virus expressing the ecotropic retroviral receptor. Ecotropic receptor-expressing cells were then transduced with a retrovirus expressing the bacterial tetracycline repressor using blasiticidin as the selectable marker. Single-cell clones were screened for tetracycline repressor expression. An inducible retroviral vector was used to drive expression of CIITA–CD274 or CIITA–CD273 fusions (for full-length sequences see Supplementary Information) under the control of a cytomegalovirus promoter containing tetracycline repressor binding sites as described previously30. A mixture of CIITA–CD274- or CIITA–CD273-containing plasmid DNA, the mutant ecotropic envelope-expressing plasmid pHIT/EA6×3* and gag-pol expressing plasmid pHIT60 was used to transfect 293T cells using the Lipofectamine 2000 reagent (Invitrogen). Two days after transfection, retrovirus supernatants were collected to infect the engineered U2932 cells in the presence of 8 mg ml−1 polybrene (Sigma) in a single spin infection, and puromycin was used to select for stable integrants over 6 days. Fusion gene expression was induced by doxycycline (20 ng ml−1) for 2 days before flow cytometry analysis and Jurkat T-cell co-culture experiments. For flow cytometric analysis of intracellular cleaved poly(ADP-ribose) polymerase (PARP) and active caspase 3 in U2932, cells were fixed in PBS containing 2% paraformaldehyde, then permeabilized and co-stained with anti-PARP (BD Pharmingen, clone F21-852) and anti-caspase-3 (BD Pharmingen number 51-68655X) antibodies diluted in FACS buffer containing 0.25% saponin (Sigma).
Statistical analysis
Group comparisons were performed using Student’s t-tests and χ2 analysis. For time-to-event analyses, we used disease-specific survival as the primary endpoint. Disease-specific survival was defined as the time from initial diagnosis to lymphoma- or treatment-related death. Time to event analyses using the Kaplan–Meier method was performed using SPSS version 11.0.0. Multivariate analysis was performed using a Cox regression model (forward stepwise) with the input variables of age, tumour stage, serum lactate dehydrogenase concentration, World Health Organization performance status, number of extranodal sites and presence of CIITA rearrangement.
Supplementary Material
Acknowledgments
This work is supported by a postdoctoral fellowship of the Cancer Research Society (Steven E. Drabin Fellowship) to C.S., the Michael Smith Foundation for Health Research to C.S. and S.P.S., the Lymphoma Research Foundation to C.S. and the Canadian Breast Cancer Foundation to S.P.S. Operational funds were available through the Canadian Institutes of Health Research, grant number 178536 to R.D.G. R.D.G., J.M.C., M.A.M. and D.E.H. are also supported by the Terry Fox Foundation (number 019001). This work was in part supported by an infrastructure grant of Genome Canada/Genome BC. U.S. is the recipient of a Howard Temin Award of the National Institutes of Health/National Cancer Institute (R00CA131503), a new investigator award of the Leukemia Research Foundation, and is the Diane and Arthur B. Belfer Faculty Scholar in Cancer Research of the Albert Einstein College of Medicine. We thank G. Simkin, C. Polumbo and T. Vogler for technical support. L.R. is the recipient of a CJ Martin Fellowship from the National Health and Medical Research Council of Australia.
Footnotes
Author Contributions C.S. designed the research, performed FISH, PCR and direct sequencing, interpreted results and wrote the paper. S.P.S. designed the research, analysed the transcriptome data and wrote the paper. B.W.W. performed PCR and interpreted results. M.K., U.C.O. and L.R. performed in vitro functional analyses. P.F. reviewed pathology and constructed the tissue microarrays. N.A.J. performed single nucleotide polymorphism analyses. Y.Z. performed library construction and RNA-seq. A.T. performed nucleic acid extraction and quantitative RT-PCR. B.M. and S.B.N. performed FISH. A.M., M.S., G.L. and S.J.J. analysed the transcriptome data. A.D., A.B., L.R. and D.E.H. interpreted results. J.M.C. and K.J.S. provided clinical data. D.G.H. designed the research. L.M.S. and U.S. designed the research and interpreted results. M.A.A. designed the research. R.D.G. designed the research, constructed the tissue microarrays, interpreted results and wrote the paper.
Array data are deposited in NCBI Gene Expression Omnibus under accession number GSE25990.
The authors declare no competing financial interests.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Dyer MJ. The pathogenetic role of oncogenes deregulated by chromosomal translocation in B-cell malignancies. Int J Hematol. 2003;77:315–320. doi: 10.1007/BF02982637. [DOI] [PubMed] [Google Scholar]
- 2.Kerckaert JP, et al. LAZ3, a novel zinc-finger encoding gene, is disrupted by recurring chromosome 3q27 translocations in human lymphomas. Nature Genet. 1993;5:66–70. doi: 10.1038/ng0993-66. [DOI] [PubMed] [Google Scholar]
- 3.Blank C, Gajewski TF, Mackensen A. Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T cells as a mechanism of immune evasion: implications for tumor immunotherapy. Cancer Immunol Immunother. 2005;54:307–314. doi: 10.1007/s00262-004-0593-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rimsza LM, et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103:4251–4258. doi: 10.1182/blood-2003-07-2365. [DOI] [PubMed] [Google Scholar]
- 5.Martin-Subero JI, et al. Chromosomal breakpoints affecting immunoglobulin loci are recurrent in Hodgkin and Reed-Sternberg cells of classical Hodgkin lymphoma. Cancer Res. 2006;66:10332–10338. doi: 10.1158/0008-5472.CAN-06-1992. [DOI] [PubMed] [Google Scholar]
- 6.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 7.Palanisamy N, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nature Med. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reith W, Mach B. The bare lymphocyte syndrome and the regulation of MHC expression. Annu Rev Immunol. 2001;19:331–373. doi: 10.1146/annurev.immunol.19.1.331. [DOI] [PubMed] [Google Scholar]
- 9.Silacci P, Mottet A, Steimle V, Reith W, Mach B. Developmental extinction of major histocompatibility complex class II gene expression in plasmocytes is mediated by silencing of the transactivator gene CIITA. J Exp Med. 1994;180:1329–1336. doi: 10.1084/jem.180.4.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masternak K, et al. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14:1156–1166. [PMC free article] [PubMed] [Google Scholar]
- 11.Cressman DE, Chin KC, Taxman DJ, Ting JP. A defect in the nuclear translocation of CIITA causes a form of type II bare lymphocyte syndrome. Immunity. 1999;10:163–171. doi: 10.1016/s1074-7613(00)80017-5. [DOI] [PubMed] [Google Scholar]
- 12.Joos S, et al. Hodgkin’s lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including REL and JAK2. Int J Cancer. 2003;103:489–495. doi: 10.1002/ijc.10845. [DOI] [PubMed] [Google Scholar]
- 13.Chin KC, Li G, Ting JP. Activation and transdominant suppression of MHC class II and HLA-DMB promoters by a series of C-terminal class II transactivator deletion mutants. J Immunol. 1997;159:2789–2794. [PubMed] [Google Scholar]
- 14.Zhou H, Su HS, Zhang X, Douhan J, III, Glimcher LH. CIITA-dependent and -independent class II MHC expression revealed by a dominant negative mutant. J Immunol. 1997;158:4741–4749. [PubMed] [Google Scholar]
- 15.Roberts RA, et al. Loss of major histocompatibility class II gene and protein expression in primary mediastinal large B-cell lymphoma is highly coordinated and related to poor patient survival. Blood. 2006;108:311–318. doi: 10.1182/blood-2005-11-4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diepstra A, et al. HLA class II expression by Hodgkin Reed-Sternberg cells is an independent prognostic factor in classical Hodgkin’s lymphoma. J Clin Oncol. 2007;25:3101–3108. doi: 10.1200/JCO.2006.10.0917. [DOI] [PubMed] [Google Scholar]
- 17.Savage KJ, et al. Favorable outcome of primary mediastinal large B-cell lymphoma in a single institution: the British Columbia experience. Ann Oncol. 2006;17:123–130. doi: 10.1093/annonc/mdj030. [DOI] [PubMed] [Google Scholar]
- 18.The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin’s lymphoma. N Engl J Med. 1993;329:987–994. doi: 10.1056/NEJM199309303291402. [DOI] [PubMed] [Google Scholar]
- 19.Rosenwald A, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–862. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamamoto R, et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood. 2008;111:3220–3224. doi: 10.1182/blood-2007-05-085159. [DOI] [PubMed] [Google Scholar]
- 21.Green MR, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–3277. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson RH, et al. Costimulatory B7–H1 in renal cell carcinoma patients: Indicator of tumor aggressiveness and potential therapeutic target. Proc Natl Acad Sci USA. 2004;101:17174–17179. doi: 10.1073/pnas.0406351101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lenz G, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. 2008;105:13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lennon AM, et al. Isolation of a B-cell-specific promoter for the human class II transactivator. Immunogenetics. 1997;45:266–273. doi: 10.1007/s002510050202. [DOI] [PubMed] [Google Scholar]
- 25.Sambade C, et al. U-2940, a human B-cell line derived from a diffuse large cell lymphoma sequential to Hodgkin lymphoma. Int J Cancer. 2006;118:555–563. doi: 10.1002/ijc.21417. [DOI] [PubMed] [Google Scholar]
- 26.Savage KJ, et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood. 2003;102:3871–3879. doi: 10.1182/blood-2003-06-1841. [DOI] [PubMed] [Google Scholar]
- 27.Melzner I, et al. Biallelic deletion within 16p13.13 including SOCS-1 in Karpas1106P mediastinal B-cell lymphoma line is associated with delayed degradation of JAK2 protein. Int J Cancer. 2006;118:1941–1944. doi: 10.1002/ijc.21485. [DOI] [PubMed] [Google Scholar]
- 28.Morin R, et al. Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques. 2008;45:81–94. doi: 10.2144/000112900. [DOI] [PubMed] [Google Scholar]
- 29.Steidl U, et al. A distal single nucleotide polymorphism alters long-range regulation of the PU.1 gene in acute myeloid leukemia. J Clin Invest. 2007;117:2611–2620. doi: 10.1172/JCI30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ngo VN, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 31.Amini RM, et al. A novel B-cell line (U-2932) established from a patient with diffuse large B-cell lymphoma following Hodgkin lymphoma. Leuk Lymphoma. 2002;43:2179–2189. doi: 10.1080/1042819021000032917. [DOI] [PubMed] [Google Scholar]
- 32.Epstein AL, et al. Biology of the human malignant lymphomas. IV. Functional characterization of ten diffuse histiocytic lymphoma cell lines. Cancer. 1978;42:2379–2391. doi: 10.1002/1097-0142(197811)42:5<2379::aid-cncr2820420539>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 33.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 34.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bengtsson H, Wirapati P, Speed TP. A single-array preprocessing method for estimating full-resolution raw copy numbers from all Affymetrix genotyping arrays including Genome Wide SNP 5 & 6. Bioinformatics. 2009;25:2149–2156. doi: 10.1093/bioinformatics/btp371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shah SP, et al. Integrating copy number polymorphisms into array CGH analysis using a robust HMM. Bioinformatics. 2006;22:e431–e439. doi: 10.1093/bioinformatics/btl238. [DOI] [PubMed] [Google Scholar]
- 37.Lestou VS, et al. Multicolour fluorescence in situ hybridization analysis of t(14;18)-positive follicular lymphoma and correlation with gene expression data and clinical outcome. Br J Haematol. 2003;122:745–759. doi: 10.1046/j.1365-2141.2003.04502.x. [DOI] [PubMed] [Google Scholar]
- 38.Steidl C, et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N Engl J Med. 2010;362:875–885. doi: 10.1056/NEJMoa0905680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shustik J, et al. Correlations between BCL6 rearrangement and outcome in patients with diffuse large B-cell lymphoma treated with CHOP or R-CHOP. Haematologica. 2010;95:96–101. doi: 10.3324/haematol.2009.007203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martinez-Ramirez A, et al. Simultaneous detection of the immunophenotypic markers and genetic aberrations on routinely processed paraffin sections of lymphoma samples by means of the FICTION technique. Leukemia. 2004;18:348–353. doi: 10.1038/sj.leu.2403230. [DOI] [PubMed] [Google Scholar]
- 41.Klein U, et al. Transcriptional analysis of the B cell germinal center reaction. Proc Natl Acad Sci USA. 2003;100:2639–2644. doi: 10.1073/pnas.0437996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.