


Abstract
hDIS3 is a mainly nuclear, catalytic subunit of the human exosome complex, containing exonucleolytic (RNB) and endonucleolytic (PIN) active domains. Mutations in hDIS3 have been found in ∼10% of patients with multiple myeloma (MM). Here, we show that these mutations interfere with hDIS3 exonucleolytic activity. Yeast harboring corresponding mutations in DIS3 show growth inhibition and changes in nuclear RNA metabolism typical for exosome dysfunction. Construction of a conditional DIS3 knockout in the chicken DT40 cell line revealed that DIS3 is essential for cell survival, indicating that its function cannot be replaced by other exosome-associated nucleases: hDIS3L and hRRP6. Moreover, HEK293-derived cells, in which depletion of endogenous wild-type hDIS3 was complemented with exogenously expressed MM hDIS3 mutants, proliferate at a slower rate and exhibit aberrant RNA metabolism. Importantly, MM mutations are synthetically lethal with the hDIS3 PIN domain catalytic mutation both in yeast and human cells. Since mutations in PIN domain alone have little effect on cell physiology, our results predict the hDIS3 PIN domain as a potential drug target for MM patients with hDIS3 mutations. It is an interesting example of intramolecular synthetic lethality with putative therapeutic potential in humans.
INTRODUCTION
Multiple myeloma (MM) is a lethal neoplastic disease accounting for 10–15% of hematologic malignances and 20% of deaths related to cancer of the blood and bone marrow (1). MM originates from terminally differentiated antibody-producing B cells known as plasma cells (1).
The genetic background of MM is not completely understood. Hypermutations occurring at the time of immunoglobulin receptor affinity maturation and class switching are involved in MM pathogenesis, leading to chromosomal abnormalities such as translocations, hyperdiploidy, hypodiploidy, monosomy or partial deletion of chromosome 13 (1–3). A recent whole-genome sequencing of 38 MM patients provided a global view on the somatic mutations associated with this cancer (4). Unexpectedly, hDIS3 gene was mutated in ∼10% of MM patients (4). Importantly, these mutations were either homo- or hemizygotic. A high frequency of hDIS3 gene mutations in MM patients was recently confirmed in another high-throughput study (5). Interestingly, hDIS3 gene mutations were also found in global screens of other cancers, such as medulloblastoma and acute myeloid leukemia (6,7). Additionally, hDIS3 was identified in transcriptomic analyses as one of the genes, whose expression differentiates superficial spreading melanoma from nodular melanoma (8). Furthermore, hDIS3 overexpression was earlier observed in human colorectal cancer and in a mouse model of this cancer, where elevated levels of respective mRNA and protein positively correlated with the incidence of metastasis (9,10). Expression profiling revealed that hDIS3 is among several genes whose loss-of-function significantly reduces viability of colorectal carcinoma cell lines (11). Increased levels of hDIS3 mRNA have been also recently proposed as one of the characteristics of the epithelial ovarian cancer (12). All examples presented above strongly suggest the existence of possible molecular link between hDIS3 functions and development of different cancers [reviewed in (13)]. More specifically, it appears likely that exonucleolytic activity of hDIS3 protein—the major catalytic subunit of the exosome—might be somehow involved in this association.
hDIS3 is a catalytic subunit of the RNA exosome, which plays a crucial role in RNA processing and decay. The exosome complex has an evolutionarily conserved structure encompassing a 9-subunit ring devoid of any catalytic activity (14,15). The associated ribonucleases responsible for the enzymatic activity of the exosome belong to two different families: Dis3 proteins, similar to bacterial RNases II/R, and Rrp6 proteins, members of the RNase D family (16). In yeast, single genes code for Dis3 and Rrp6 proteins. Dis3 is the only essential catalytic subunit, present both in the nucleus and cytoplasm, while Rrp6 is restricted to the nucleus and responsible for only a subset of nuclear exosome functions (17). Dis3 is a multidomain protein with two different catalytic activities: a 3′–5′ exonucleolytic activity via the RNase II/R (RNB) domain and an endonucleolytic activity via the PilT N-terminal (PIN) domain at the N-terminus (16,18–20). The Dis3 exonuclease active site is located near the bottom of the central channel of the 9-subunit ring through which substrates are delivered (21–25). Both activities cooperate with each other, but the exonucleolytic activity is more important for cell physiology, whereas mutations abolishing the endonucleolytic activity alone have no detectable growth phenotype (18–20,24).
The human genome encodes three Dis3 homologues, of which only hDIS3 and hDIS3L were found to associate with the exosome (26,27). Notably, both of them are processive 3′–5′ hydrolytic exonucleases, whereas only hDIS3 has also retained endonuclease activity in its PIN domain. In vivo localization studies and analyses of substrate specificities revealed that hDIS3L is restricted to the cytoplasmic exosome, whereas hDIS3 is mainly a nucleoplasmic protein, with a small fraction present in the cytoplasm (26,27). Additionally, human RRP6 is mainly nuclear and significantly enriched in the nucleoli, with a minor fraction in the cytoplasm (26). Thus, human RNA exosomes, although based on the same structural scaffold as their Saccharomyces cerevisiae counterparts, exist as functionally and compositionally distinct variants in different areas of the nucleus and in the cytoplasm (28).
The third human Dis3 homologue—hDIS3L2—does not interact with the exosome core, due to the lack of PIN domain and it has been recently demonstrated by several independent studies that it is responsible for an alternative 3′–5′ RNA decay pathway in the cytoplasm (29–31). An important role of hDIS3L2 in the maintenance of proper cellular metabolism is supported by the fact that mutations in its gene were found to be associated with Perlman syndrome of overgrowth and predisposition to Wilm’s tumor development (13,32).
The exosome plays a fundamental role in most RNA metabolic pathways (16,33,34). These pathways include turnover of normal mRNAs and AU-rich element-regulated decay of unstable mRNAs (35,36); nuclear processing of stable RNA classes (small nuclear RNA (snRNA), small nucleolar RNA (snoRNA), rRNA, tRNA) (17,37); degradation of unstable transcripts arising from intergenic regions (38–40); and complete degradation of mRNA after endonucleolytic cleavage initiated by small interfering RNA (siRNA) in RNA interference (41). Furthermore, the exosome plays a crucial role in the quality control, as it is a primary enzyme degrading unwanted molecules in the nucleus and the cytoplasm, including incorrectly processed pre-mRNAs, rRNAs and tRNAs as well as translationally incompetent mRNAs (17,37,42–47). Apart from its well-documented role in RNA metabolism, a recent study has implicated the exosome in the activation-induced immunoglobulin heavy-chain class switch recombination and immunoglobulin variable region somatic hypermutation in human B lymphocytes (48). It was also suggested that the exosome is essential for activation-induced cytidine deaminase (AID) recruitment to chromatin (48), but the mechanistic details of this process remain elusive.
Here, we explored the consequences of hDIS3 gene mutations found in MM patients using biochemical, genetic and functional analyses, and examined possible synthetic genetic interactions between hDIS3 exonucleolytic activity and other catalytic activities of the nuclear exosome.
MATERIALS AND METHODS
Cell culture and generation of stable cell lines
Human HEK293 Flp-In T-REx (Invitrogen) cells were cultured as monolayers in Dulbecco’s modified Eagle’s medium (D-MEM, Gibco) supplemented with 10% tetracycline-free fetal bovine serum (TET System Approved Fetal Bovine Serum (FBS); Clontech) and antibiotics (penicillin-streptomycin; Sigma-Aldrich) at 37°C in a 5% CO2 humidified atmosphere.
The stable inducible HEK293 Flp-In T-REx cell lines were obtained in this study using highly pure DNA midipreps of pMM7-pMM14 plasmid constructs (Supplementary Table S3; see Supplementary Materials and Methods for details on construction of the plasmids) and the Flp-In™ T-REx™ system (Invitrogen), according to the protocol of the manufacturer. Established cell lines were grown in the same medium as above, supplemented with hygromycin B (100 μg/ml) and blasticidin (10–15 μg/ml) (both from Invitrogen). Transfections were performed with Lipofectamine2000 (Invitrogen). Expression of exogenous genes was induced by addition of doxycycline to the culture medium at a final concentration of 100 ng/ml.
The chicken DT40 Cre1 cell line was obtained from the laboratory of Jean-Marie Buerstedde (Institute for Molecular Radiobiology, Neuherberg-Munich, Germany). DT40 cells were cultured in D-MEM supplemented with 10% FBS (Invitrogen), 1% chicken serum (Sigma-Aldrich), 0.1 mM 2-mercaptoethanol (Sigma-Aldrich), 100 U/ml of penicillin and 0.1 mg/ml of streptomycin (Sigma-Aldrich) at 37°C in a 5% CO2 humidified atmosphere. Cell densities were kept between 0.2 and 2 million per 1 ml.
Stable integration of pMM15 and pMM16 constructs (Supplementary Table S3; see Supplementary Materials and Methods for details on construction of the plasmids) into the DT40 DIS3 locus with the aim of generating conditional knockouts was performed as follows. DT40 Cre1 cells (107) were centrifuged, resuspended in 0.8 ml of growth medium and incubated on ice for 10 min with 40 μg of the respective plasmid linearized with NotI restriction enzyme. The cells were then electroporated in ice-chilled 0.4 cm cuvettes (Biorad) using the following parameters: 700 V, 25 μF. The cells were transferred into 10 ml of medium and cultured for 24 h in small flasks. Ten milliliters of medium with the proper selection antibiotic was added (puromycin or blasticidin S, both from Invivogen; final concentrations of 0.5 μg/ml and 20 μg/ml, respectively), and cells were transferred to 96-well plates (200 μl each well). After 7–14 days, single colonies were counted under the microscope and resuspended in 1.5 ml of fresh medium containing selection antibiotics. Surviving cells were collected for further analysis. The stable heterozygotic puromycin-resistant DT40 Cre1 cell line (dis3ΔPuroR/DIS3) was generated following one round of transfection with the pMM15 construct. To obtain the stable homozygotic puromycin- and blastidicin S-resistant DT40 Cre1 cell line (dis3ΔPuroR/dis3ΔBsrR), a second round of electroporation was performed using the pMM16 construct.
The conditional DIS3 knockout in DT40 Cre1-derived cell lines obtained above was achieved through Cre-mediated LoxP excision of the hDIS3 expression cassette induced by 4-hydroxy-tamoxifen. Briefly, cells (106) were cultured for 48 h in presence of 10 μM 4-hydroxy-tamoxifen (Sigma-Aldrich). Cells were counted and subcloned in a set of dilutions to obtain single colonies on 96-well plates. After 5–7 days, colonies were counted under the microscope, and cells were resuspended and split into two media: with or without selection antibiotic(s). Colonies that did not survive exposure to antibiotic(s) were treated as cultures with complete LoxP excision.
Cell growth analyses
Stable inducible human HEK293 Flp-In T-REx cell lines were grown as described above in 100 mm plates until reaching 90% confluence. In the 48-h induction mode, 2 × 105 cells were plated directly in six-well plates in medium either lacking or containing doxycycline, and grown for another 48 h. In the 48-h + 48-h induction mode, the cells were first replated in new 100 mm plates in medium either lacking or containing doxycycline at 40% confluence. After 48 h, 2 × 105 cells were plated in six-well plates in the respective medium and grown for another 48 h. Cell growth and eGFP fluorescence were analyzed using an Olympus IX81 microscope.
Metabolic activity assays of the cell lines were performed as follows. Cells (5 × 103) were plated in triplicate (for each cell line, condition and time of measurement) in 96-well plates. Cells were either maintained in medium lacking doxycycline or subjected to treatment with the inducer. At various time points following induction (24, 72 or 120 h), 10 μl of AlamarBlue® (Invitrogen) was added to the cultures. Amounts of the reduced reagent were quantified after 120 min using the DTX880 Multimode Detector (Beckman Coulter).
Western blotting
Protein samples from yeast cultures and human or chicken cell lines were prepared according to standard protocols, run on 10–12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and immobilized on Protran nitrocellulose membranes (Whatman) by electrotransfer using Trans-Blot® SD Semi-Dry Transfer Cell (Bio-Rad). Following transfer, membranes were stained with Ponceau S Red (Sigma-Aldrich; 0.1% in 3% acetic acid), blocked in 5% nonfat milk in Tris-buffered saline (TBS) containing 0.05% Tween-20 (TBST), and incubated in the same solution with primary antibodies. The following primary antibodies were used for analyses: mouse monoclonal anti-eGFP (B2) (Santa Cruz Biotechnology; sc-9996) (1:1000), rabbit polyclonal anti-FLAG (Sigma-Aldrich; F-7425) (1:3000), rabbit polyclonal anti-hDIS3 (Sigma-Aldrich; HPA039281, lot: R37348) (1:1500), rabbit polyclonal anti-hDIS3L (Sigma-Aldrich; HPA041805, lot: R38591) (1:500), rabbit polyclonal anti-hRRP6 (Sigma-Aldrich; P4124) (1:3000), rabbit polyclonal anti-hDIS3L2 (31) (1:2000) and goat polyclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (V-18) (Santa Cruz Biotechnology; sc-20357) (1:2000). Membranes were then washed with TBST and incubated with appropriate secondary antibody [goat anti-mouse, goat anti-rabbit (Calbiochem; 401215, 401393, respectively) or rabbit anti-goat (Sigma-Aldrich; A5420)] conjugated with horseradish peroxidase. Protein A tag was detected directly using rabbit Peroxidase–Anti-Peroxidase Soluble Complex antibody (Sigma-Aldrich; P1291) (1:3000). Blots were developed in a Curix 60 machine (AGFA) using the Immun-StarTM WesternCTM Kit (Bio-Rad) and CL-XPosureTM Films (Thermo Scientific).
Heterologous expression and purification of hDIS3 proteins
For purification of the panel of hDIS3 variants, the Escherichia coli BL21-CodonPlus-RIL strain (Stratagene) was first transformed with appropriate plasmids (pHEX1, pHEX8 or pMM1-pMM6). Transformants were grown at 18°C for 48 h in 1 l of Auto Induction Media Super Broth Base Including Trace Elements (Formedium) supplemented with 2% glycerol, kanamycin (30 μg/ml) and chloramphenicol (34 μg/ml), following inoculation from a pre-culture, grown in standard Luria-Broth (LB) medium containing both antibiotics. Recombinant proteins were purified as described previously (26,49), with minor modifications.
Exoribonuclease assays
In vitro enzymatic assays of hDIS3 exoribonucleolytic activity were performed in 20 μl reaction volumes containing 10 mM Tris–HCl, pH = 8.0, 75 mM NaCl, 1 mM 2-mercaptoethanol and 100 μM MgCl2. Protein concentration was 0.1 μM, while substrate concentration was 0.2 or 2 μM for duplex or unstructured RNA molecules, respectively. Details on the preparation of radiolabeled substrates can be found in the Supplementary Materials and Methods.
For gel-based analyses, reactions were performed at 37°C for the indicated times and terminated by adding 20 μl of formamide loading dye (90% formamide, 20 mM EDTA, 0.03% bromophenol blue, 0.03% xylene cyanol in 1 × Tris-borate-EDTA (TBE)). Reaction products were resolved in denaturing 20% polyacrylamide, 8 M urea, 1× TBE gels. In the case of thin layer chromatography (TLC)-based experiments, samples were collected by mixing aliquots of the reaction mixtures with 1 μl of 0.5 M EDTA, and subsequently analyzed by running them on polyethylenimine (PEI)-cellulose plates (Schleicher & Schuell) in 0.5 M LiCl/1 M formic acid using nonradioactive uridine monophosphate (UMP) as a marker. For both types of analyses, reaction products were visualized using a FUJI PhosphorImager.
Southern hybridization
Approximately 10 μg of genomic DNA isolated from DT40-derived cell lines was digested overnight with 40 U of either NdeI or BsaI restriction enzyme (both from ThermoScientific) in a total volume of 100 μl, according to the manufacturer’s instructions. Digested DNA was precipitated according to standard laboratory procedures and loaded onto a 0.6% agarose gel stained with ethidium bromide. Electrophoresis was performed in 1×Tris-acetate-EDTA (TAE) buffer at 8°C. Following DNA separation, gel was sequentially submerged for 30 min with gentle agitation in (i) depurination solution (0.25 M HCl), (ii) denaturation solution (1.5 M NaCl, 0.5 M NaOH) and (iii) neutralization buffer (1.5 M NaCl, 0.5 M Tris–HCl, pH 7.5). The gel was briefly rinsed with H2O between different steps of incubation described above. Finally, DNA was transferred overnight onto a Hybond N+ membrane (GE Healthcare), according to the manufacturer’s instructions, and fixed by ultraviolet cross-linking.
The polymerase chain reaction (PCR) product spanning a 480-bp fragment of the chicken DIS3 right arm was amplified from the pMM15 construct with the DIS3_RF-DIS3rfinR primer pair (see Supplementary Table S2 for sequences). The amplification product was gel-purified and used as a probe for hybridization. Approximately 25 ng of PCR product was labeled with α-32P[dATP] using the DECAprime™ II Kit (Ambion) according to the manufacturer’s instructions. After denaturation and snap-cooling, labeled probe was purified using Micro Bio-Spin Column P-30 Tris (Biorad). Hybridization was performed at 68°C overnight in PerfectHyb Plus hybridization buffer (Sigma-Aldrich). The membrane was washed twice with 2×saline-sodium citrate (SSC), 0.1% SDS and once in 0.5×SSC, 0.1% SDS at 68°C for 20 min, and eventually exposed to a PhosphorImager screen (FujiFilm), which was scanned following exposure using a FLA 7000 scanner (FujiFilm).
RNA isolation and northern-blot analysis
RNA was isolated from yeast cultures and human cell lines using the standard hot acidic phenol procedure or TRI Reagent (Sigma-Aldrich), respectively. Total RNA (5–10 μg) was fractionated by electrophoresis either in denaturing 6% polyacrylamide-urea gel followed by electroblotting onto a Hybond N+ membrane in 0.5× TBE at 4°C, or in a 1% formaldehyde-agarose gel, followed by RNA immobilization on the same type of membrane by overnight capillary transfer in 20× SSC (3 M NaCl, 0.3 M sodium citrate). RNA was fixed on membranes by ultraviolet cross-linking. Hybridizations were performed in PerfectHyb Plus hybridization buffer. The blots were handled according to standard procedures and probed at 42°C (5′-labeled oligonucleotide probes) or 63°C (DNA fragments labeled by random priming). Between successive hybridizations, probes were stripped off the membranes at 65°C using boiling 0.1% SDS.
For detection of yeast 5′-external transcribed spacer (ETS) and NEL025 cryptic unstable transcript (CUT), PCR probes labeled by random priming with α-32P[dATP] and DecaLabel DNA Labeling Kit (Thermo Scientific) as described previously (18) were used. Randomly primed PCR products, obtained with the primer pairs GAPDH_F-GAPDH_R and 7SL_F-7SL-R (Supplementary Table S2), were used as probes for detection of GAPDH mRNA and 7SL RNA, respectively. Randomly primed probe, generated as previously described (26), was used for detection of the hDIS3 transcript. Randomly primed DNA probe specific to histone H2A mRNA was generated by EcoRI/HindIII-mediated excision of sequence corresponding to its full-length CDS (GenBank ID: AY131971.1), cloned between respective sites of pUC19; the construct was kindly provided by Prof. Zbigniew Dominski. For all other transcripts, 32P-labeled oligonucleotides (listed in Supplementary Table S2) were used as probes. After hybridization, membranes were washed with 2× SSC, 0.1% SDS and results were visualized using phosphorimaging as described in the previous section. Quantification was performed using Multi Gauge v. 3.0 software (FujiFilm).
Reverse transcription and quantitative PCR
Total RNA (10 μg) extracted from stable human cell lines was treated with 6 U of TURBOTM DNase (Ambion) in the presence of RiboLockTM RNase Inhibitor (Thermo Scientific), according to the manufacturer’s instructions. Following phenol:chloroform extraction and precipitation of RNA with isopropanol, 2 μg of DNase-treated RNA was reverse transcribed using a mixture of 50 pmol of an oligo(dT) primer and 250 ng of random hexamers (Invitrogen) and Superscript IIITM reverse transcriptase (Invitrogen), according to the manufacturer’s instructions, in a final volume of 20 µl. A portion (1/100) of the cDNA reaction was mixed with Platinum® Quantitative PCR SuperMix-UDG (Invitrogen), 2.5 pmol of each primer (Supplementary Table S2) and 0.3 μg of bovine serum albumin in a final volume of 10 µl and analyzed by real-time PCR in a Roche LightCycler® 480 system using an annealing temperature of 58°C. Negative controls lacking reverse transcriptase showed a negligible background. Analyses were performed in triplicate. All data were normalized to GAPDH mRNA.
RESULTS
hDIS3 proteins with MM-associated mutations display various defects in the degradation of different RNA substrates in vitro
The majority of mutations found in MM affect conserved residues in the hDIS3 RNB domain, suggesting their impact on the exonucleolytic activity of the enzyme [(4); Figure 1A]. In comparison, no cancer-associated mutations have been found in the PIN domain, entirely responsible for the endoribonucleolytic activity of hDIS3 (Figure 1A).
Figure 1.

Substitutions of conserved amino acids in the hDIS3 RNB domain decrease enzymatic activity. (A) Schematic view of domain organization of human and yeast DIS3 proteins. Amino acids substituted in MM are indicated in light gray and their positions with respect to hDIS3 domains are marked with arrows. An asterisk indicates position of D487, which was previously shown to be critical for exoribonucleolytic activity of hDIS3. Corresponding amino acids in S. cerevisiae Dis3p are marked with italics. (B) SDS-PAGE analysis of hDIS3 protein variants purified using two-step affinity chromatography, followed by gel filtration. Position of hDIS3 protein is marked with a solid arrow. Common contaminations with bacterial DnaK chaperone and hDIS3 proteolytic degradation products are indicated with an asterisk and open arrows, respectively. The PageRulerTM Prestained Protein Ladder (Fermentas) molecular marker is in the leftmost lane. (C) TLC analysis of the RNA degradation efficiency for recombinant hDIS3 proteins, shown in (B). Internally radiolabeled RNA, synthesized by in vitro transcription in the presence of [α-32P]UTP, was incubated with equal amounts of hDIS3 variants or in the absence of protein. Aliquots were collected at indicated time points and spotted onto a PEI-cellulose TLC plate, which was developed in the direction shown with the vertical gray arrow; positions of substrate and product (UMP) are indicated with solid and open arrows, respectively. S477R, G766R and R780K mutations significantly inhibit ribonucleolytic degradation.
To analyze the impact of hDIS3 mutations identified in MM patients by Chapman et al. (4) on the exonucleolytic activity of the protein, recombinant versions of hDIS3 [wild-type (WT) or bearing different mutations detected in MM] were purified from E. coli. We successfully obtained hDIS3S477R, hDIS3V504G, hDIS3G766R, hDIS3R780K and hDIS3I845V proteins (Figure 1B), but were unable to purify hDIS3A524P, which was insoluble irrespective of expression conditions. In parallel, we purified two control hDIS3 variants: hDIS3WT, which was previously shown to display robust exoribonucleolytic activity, and hDIS3D487N, a catalytically inactive mutant (exo−) with one of the aspartic acid residues coordinating magnesium in the active site of the RNB domain substituted with its amide [(26); see Supplementary Figure S6 therein, where hDIS3D487N is referred to as hDIS3RNB MUT]. All recombinant proteins were obtained with similar efficiency and comparable degree of purity; the only significant contaminant was bacterial DnaK chaperone, which was slightly increased for hDIS3S477R, likely due to its lower solubility (Figure 1B).
Next, we performed a biochemical experiment using the purified hDIS3 variants and in vitro transcribed RNA (∼400-nt-long RNA fragment encompassing Arabidopsis thaliana Lsm1 open reading frame) uniformly labeled with UTP. Time-course–based analysis of RNA degradation by TLC allowed us to tentatively assess the influence of individual mutations on exoribonucleolytic activity. While the degradative potential was almost unchanged in hDIS3V504G and hDIS3I845V compared with hDIS3WT, the activity was severely reduced for hDIS3S477R, hDIS3G766R and hDIS3R780K (Figure 1C). The phenotype of hDIS3R780K closely resembled that of the catalytic RNB mutant in the active site (exo−), hDIS3D487N (Figure 1C).
To more qualitatively assess defects in RNA degradation caused by hDIS3 mutations, we performed assays on various 5′-end labeled oligoribonucleotides: (i) a single-stranded RNA, composed of 17-mer generic sequence, followed by 14 nt-long oligo(A) tail [5′P*-ss17-(A)14]; (ii) a 44-mer, lacking oligo(A) extension (5′P*-ss44) and (iii) a structured substrate containing a 17-nt duplex followed by a 14-nt-long oligo(A) tail [5′P*-ds17-(A)14]. Decay products were analyzed on denaturing polyacrylamide gels (Figure 2). In the hDIS3I845V and hDIS3V504G mutants, patterns of final degradation products were the same as for hDIS3WT for all substrates (Figure 2A–C). The rate of degradation was, however, slightly slower for the hDIS3V504G variant compared with hDIS3WT and hDIS3I845V. In contrast, the remaining mutations had a visible impact on hDIS3 function. R780K led to virtually complete inhibition of exonucleolytic degradation of both single-stranded substrates, similar to the catalytic D487N mutation (Figure 2A and B), which confirmed the TLC results shown in the Figure 1B. hDIS3S477R and hDIS3G766R variants displayed somewhat milder defects on the 5′P*-ss17-(A)14, in regard to changes in the ratio of final degradation products compared with hDIS3WT: for the former variant, the level of 4-nt-long decay products was reduced, while with the latter, the level was increased at the expense of decreased amounts of 5-nt-long degradation products (Figure 2A). Moreover, the hDIS3G766R mutant lost processivity and became distributive in the presence of 5′P*-ss44 (Figure 2B). Importantly, in the case of partially double-stranded 5′P*-ds17-(A)14 substrate, while hDIS3S477R, hDIS3G766R and hDIS3R780K seemed to digest a single-stranded extension, they all stalled on encountering secondary structures in the substrate (this effect was most pronounced with the hDIS3R780K mutant), even though degradation of the partial duplex was not completely abolished, as was the case of hDIS3D487N catalytic mutant (Figure 2C).
Figure 2.


hDIS3 mutations result in changes of the degradation pattern of unstructured RNA substrates and inability to degrade structured substrates. (A) 5′-labeled ss17-(A)14 substrate was incubated in a buffer containing 100 μM magnesium with equal amounts of hDIS3 variants or in the absence of added protein. Reactions were terminated at the indicated time points, followed by denaturing PAGE and phosphorimaging. Positions of substrate and 3–5 nt degradation products are marked with solid and open arrows, respectively. R780K mutation abolished the activity, similar to D487N, while S477R and G766R substitutions led to a change in ratio of 5-nt/4-nt products. (B) Experiment was performed as in (A), but using 5′-labeled ss44 oligonucleotide. S477R and R780K mutations markedly reduced hDIS3 exoribonucleolytic activity. G766R mutation resulted in the loss of processivity. (C) Experiment was performed as in (A), but using 5′-labeled RNA substrate forming a partial duplex ds17-(A)14. hDIS3S477R, hDIS3G766R and hDIS3R780K proteins were not able to continue degradation of the substrate on encountering double-stranded region.
Together, these results strongly indicate that the S477R, G766R and R780K mutations in hDIS3 cause significant aberrations of hDIS3 exoribonucleolytic activity, while the V504G and I845V have subtle, if any, effects.
Mutations in yeast DIS3 gene corresponding to those found in MM patients cause cell growth defects and molecular phenotypes strongly suggesting impaired exosome function
Since S. cerevisiae is the most widely used model for studying Dis3 protein function, we started our functional analyses of hDIS3 MM mutations by constructing yeast strains expressing endogenous Dis3 with introduced equivalent amino acid changes (with the exception of hDIS3I845V, as this amino acid is not conserved in yeast). We successfully produced four mutants: dis3-G833R (corresponding to G766R in humans), dis3-R847K (R780K), dis3-V568G (V504G) and dis3-A588P (A524P), but not dis3-S541R (S477R).
We first performed growth tests with the yeast strains. Both dis3-G833R and dis3-R847K displayed a strong growth retardation phenotype compared with the WT control, even at physiological temperature (Figure 3A). The growth defect of dis3-G833R mutant was even stronger than that observed for the dis3-D551N (D487N in humans) catalytic mutant (exo−). On the other hand, while the two other mutant strains, dis3-V568G and dis3-A588P, grew normally at 30°C, they appeared to be extremely thermosensitive, as their growth was virtually completely abolished at the elevated temperature (Figure 3A). The observed phenotypes were not due to the lack of expression of the DIS3 mutants, as confirmed by western blotting against the protein A tag that was introduced at the 3′-end of all inserts during generation of the strains (Supplementary Figure S1A; see Supplementary Materials and Methods for details on strain construction). Moreover, successful purification of the other exosome subunits using protein A-tagged Dis3 variants as baits ruled out the possibility that these phenotypes may result from protein instability or inability to assemble into the complex (Supplementary Figure S1B). Therefore, the results of growth tests of yeast DIS3 mutants provided the first in vivo support that the amino acids altered in MM patients might be important for exosome function. Moreover, consistent with the previous in vitro analyses, the V568 substitution exhibited milder defects than G833 or R847 (Figure 3A).
Figure 3.

Mutations in yeast DIS3 in positions analogous to those found in MM cause growth defects, molecular phenotypes and display synthetic genetic interaction with catalytic mutation in the Dis3 PIN domain. (A) Serial dilutions of indicated yeast strains were spotted on YPD plates and incubated at 25, 30 or 37°C for 60 h. V568G and A524P mutations resulted in thermosensitivity, while G833R and R847K substitutions led to growth inhibition at all tested temperatures. The two latter DIS3 point mutations seemed to give a slight synergistic effect in rrp6Δ background. (B) Diploid heterozygotic strains DIS3 WT/dis3-D171N G833R-pA (top) and DIS3 WT/dis3-D171N R847K-pA (bottom), combining PIN domain endo− mutation with RNB mutations analogous to those found in MM were sporulated and subjected to tetrad dissection. Only two spores from each tetrad were viable, demonstrating that MM-associated RNB domain mutations are synthetically lethal with D171N amino acid change, abolishing endonucleolytic activity of Dis3 PIN domain. (C) Total RNA isolated from the strains in (A) was subjected to northern blot analysis using probes specific to typical exosome substrates. DIS3 mutations (G833R and R847K in particular) caused accumulation of 5′-ETS, NEL025 CUT and 7S precursor of 5.8S rRNA synthesis (as well as their degradation intermediates).
Previous studies of Dis3exo− mutant, which harbors a mutation in the RNB domain that abolishes exonucleolytic activity, showed that simultaneous inactivation of either Rrp6 exonuclease or Dis3 PIN domain endonuclease activity led to synthetic lethality (15,18–20). Therefore, since G833R and R847K DIS3 mutations resulted in the strongest growth phenotypes, we used these mutants to check for any synthetic genetic interactions with RRP6 deletion or the D171N point mutation in Dis3 PIN domain [previously shown to abolish endonucleolytic activity of the protein, hereinafter referred to as Dis3endo−; see (18)]. Importantly both G833R and R847K DIS3 mutations were synthetically lethal with the Dis3endo− mutation (Figure 3B), as in the case of Dis3exo− mutation. In contrast, only a slight synergistic growth inhibition was observed on combination of G833R and R847K DIS3 mutations with rrp6Δ (Figure 3A).
We next tested the molecular effects of DIS3 gene mutations in yeast by northern blot analyses on RNA isolated from mutant and control strains using probes for typical exosome substrates: the 5′-ETS by-product of ribosomal RNA processing, NEL025 CUT and 7S precursor in the 5.8S rRNA maturation pathway. All DIS3 mutations (the D551N substitution in the active site and four mutations corresponding to those found in MM patients) caused comparable accumulation of full-length 5′-ETS and 7S rRNA (without affecting the levels of mature 5.8 S rRNA in the latter case) compared with the WT strain (Figure 3C). On the other hand, on examining levels of 7S and 5′-ETS degradation intermediates, as well as NEL025 CUT, we could divide DIS3 mutations into two classes: one class that led to more significant accumulation of all above-mentioned species (D551N, G766R and R780K), and a second class with less pronounced effects (V568G and A588) (Figure 3C). The results are consistent with hDIS3 activity assays and yeast growth analysis (Figures 2 and 3A). Combining DIS3 G766R or R780K mutation with deletion of RRP6 did not seem to remarkably enhance the analyzed molecular phenotypes (except perhaps additional accumulation of NEL025 CUT in rrp6Δ dis3-G766R in comparison with dis3-G766R single mutant) (Figure 3C).
Altogether, experimental results from the yeast model suggest that changing amino acids in Dis3 positions analogous to those mutated in MM patients affects S. cerevisiae growth through the impairment of the exosome ability to degrade its physiological substrates.
DIS3 is essential for survival of DT40 cell line
In contrast to yeast, the cellular role of vertebrate DIS3 and its potential redundancy with other exosome-associated nucleases, RRP6 and DIS3L, has not been intensively studied. Our previous siRNA-based experiments in human cells did not reveal a strong effect of hDIS3 depletion on cell growth rate, and the molecular phenotypes regarding potential substrates such as 5.8S rRNA were relatively mild (26). Therefore, to assess the importance of the DIS3 protein in the physiology of vertebrate cells, we constructed a conditional DIS3 knockout using DT40 chicken cells. The DT40 cell line has a high rate of homologous recombination, which allows for relatively straightforward generation of knockouts (50). Conditionality is achieved by insertion of LoxP sites into the integration cassettes and usage of a DT40 cell line derivative expressing tamoxifen-inducible Cre recombinase (51). We constructed vectors for chicken DIS3 deletion that contained the human DIS3 cDNA expression cassette flanked by LoxP sites. Thus, after tamoxifen-regulated induction of Cre-recombinase, cells would be devoid of DIS3 activity.
Two rounds of successful integrations into the DT40 genome resulted in construction of a cell line in which a large part of the endogenous DIS3 gene was deleted (Figure 4A) and hDIS3 was expressed, as verified by Southern hybridization and western blotting, respectively (Figure 4B and Supplementary Figure S1C). Modified heterozygotic and homozygotic DIS3-knockout cells were treated with 4-hydroxy-tamoxifen to excise the cassettes expressing hDIS3, and then subjected to subcloning. Despite numerous attempts, it was not possible to obtain clones with excised cassettes from the DIS3 homozygotic knockout line, while these clones were successfully obtained from the heterozygotic cell line. These results suggest that the DIS3 gene is essential for survival of the DT40 cell line.
Figure 4.

Verification of DIS3 deletion and integration of hDIS3 expression cassette into the genome of DT40 Cre1 chicken cell line. (A) Schematic representation of the fragment of DIS3 locus in DT40 Cre1 cells before (top) and after (bottom) integration of the hDIS3 expression cassette. Exons are numbered. Positions of the left and right arm, used in recombination, and the 5-kb fragment deleted by cassette integration (removing the majority of the chicken DIS3 RNB domain from the resulting protein), as well as elements of the integration cassette are indicated. NdeI and BsaI sites used in Southern blot analysis are marked with arrows. Open arrowheads indicate LoxP sites, which were used for tamoxifen-induced Cre-mediated excision of the hDIS3 expression cassette. Horizontal light-gray bar shows location of the probe used in Southern hybridization. Lengths of the DNA fragments generated by restriction enzymes are indicated by italicized underlined numbers. Note that the size of the NdeI-derived fragment differs in the case of the locus following integration, depending on the selection marker present (Puro or Bsr). (B) Results of Southern hybridization. Total genomic DNA was isolated from the parental DT40 Cre1 cell line (lanes 2, 3, 6), heterozygotic DIS3 knockout with Puro selection marker (lanes 4, 7) or homozygotic DIS3 knockout with both Puro and Bsr selection markers (lanes 5, 8). DNA was either nondigested (lane 2) or digested with NdeI (lanes 3–5) or BsaI (lanes 6–8), transferred onto membrane and hybridized with a PCR-based probe as marked in (A). Sizes of the molecular marker (GeneRulerTM 1 kb Plus DNA Ladder from Fermentas; lane 1) are indicated on the left. Lengths of the restriction fragments recognized by the probe (indicated on the right) are as expected.
MM-associated hDIS3 mutations result in accumulation of different RNA species in the human cellular model
The promising results of the biochemical analyses and experiments in yeast and DT40 cells prompted us to test the impact of hDIS3 mutations in humans using a proper cellular model. The most appropriate approach would be to identify MM cell lines bearing different hDIS3 variants analyzed in previous experiments. However, sequence analysis of several commercially available MM cell lines did not reveal the presence of mutations in hDIS3 (Supplementary Figure S2A). Most surprisingly, we were unable to confirm the sequence change detected by Chapman et al.(4) in H929 established cell lines from two different sources (Supplementary Figure S2A and B). Although the I845V mutation was identified in the SKMM1 cell line (Supplementary Figure S2A and C), we opted against using this line since the mutation does not influence the exoribonucleolytic activity of the enzyme (Figures 1C and 2). Therefore, we constructed our own cellular model that would allow us to analyze the influence of hDIS3 mutations on cell physiology and RNA metabolism. We combined the Flp-InTM T-RExTM system from Invitrogen with a compatible vector containing a bidirectional tetracycline-inducible promoter (52) (Supplementary Figure S3; see Supplementary Materials and Methods and Supplementary Figures S4 and S5 for details on cloning strategy and construction of cell lines). This experimental setup allowed us to generate HEK293 Flp-In T-REx cell lines stably co-expressing (i) sh-microRNA that silenced the endogenous hDIS3 copy and (ii) an exogenous hDIS3 variant tagged with a FLAG epitope and containing a recoded fragment to render it insusceptible to miRNA silencing (Supplementary Figure S3). We established four cell lines exogenously expressing hDIS3WT, the catalytic mutant hDIS3D487N or one of the two MM-associated hDIS3 variants that had produced the strongest phenotypes in previous experiments: hDIS3G766R and hDIS3R780K. Northern and western blot analyses confirmed that both inserts were efficiently expressed in all four cell lines (Supplementary Figure S6A and B). Importantly, exogenous proteins were completely absent from cells not subjected to induction, similarly to nontransfected HEK293 Flp-In T-Rex cells, indicating that the system that we used was relatively tight (Supplementary Figure S6B). Before beginning experiments, we first verified that the expression of other known human 3′–5′ exonucleases involved in RNA metabolism, both cooperating with the exosome core, hDIS3L and hRRP6, and functioning independently of the complex (hDIS3L2), remained unchanged in our experimental system (Supplementary Figure S6C). Furthermore, immunolocalization experiments confirmed that neither overexpression of hDIS3 nor the introduced mutations influenced the intracellular localization of the protein (Supplementary Figure S7A and B).
Using our model cell lines, we examined whether expression of mutant hDIS3 proteins led to aberrations in metabolism of RNA molecules representing various classes. We isolated total RNA from the cells, which were either untreated or subjected to doxycycline-mediated induction for 48 h. Notably, after induction, cells expressing hDIS3D487N and hDIS3R780K mutants grew at a slightly lower rate than those producing hDIS3WT or hDIS3G766R variants (data not shown).
First, we performed low-resolution northern blot analysis for selected transcripts synthesized by different RNA polymerases. Above all, we noticed significant accumulation of unprocessed 5.8S ribosomal RNA precursors (RNA polymerase I transcripts) following doxycycline-mediated induction in cells bearing mutated hDIS3 variants (but not the WT counterpart) (Figure 5A). In the case of RNA polymerase II transcripts, while GAPDH polyA+ mRNA levels, used for normalization, remained constant in all cell lines irrespective of induction, we observed a clear increase of histone H2A polyA− mRNA amounts in cells expressing hDIS3D487N, hDIS3G766R and hDIS3R780K mutant proteins (Figure 5A; see Supplementary Figure S8A for quantification). In regard to RNA polymerase III transcripts, although expression of hDIS3 mutants did not seem to have significant impact on RNase MRP RNA (Figure 5A and Supplementary Figure S8A), the levels of two other representative RNA species synthesized by this enzyme, namely RNase P RNA and 7SL RNA, were increased, particularly in cells producing hDIS3D487N and hDIS3R780K variants (Figure 5A and Supplementary Figure S8A). The phenotype observed for 7SL RNA was also confirmed by high-resolution northern blot analysis (Figure 5D).
Figure 5.


Model cell lines producing hDIS3 variants accumulate transcripts representing various RNA classes. (A) Low-resolution northern blot analysis of steady-state levels of RNA molecules synthesized by different RNA polymerases. Total RNA was isolated from cell lines expressing either WT or mutated hDIS3, either untreated (‘−dox’) or treated with doxycycline (‘+dox’). Following electrophoretic separation in a denaturing agarose gel, RNA was transferred onto a membrane, which was then sequentially hybridized with probes recognizing RNA pol I (5.8S rRNA precursor), RNA pol II (histone H2A mRNA, GAPDH mRNA) or RNA pol III transcripts (RNase P RNA, RNase MRP RNA, 7SL RNA), and signals were visualized by phosphorimaging. A significant increase in levels of 5.8S rRNA 3′-extented precursors was visible on induction, while a more modest accumulation was apparent for the histone H2A transcript, as well as RNase P and 7SL RNAs. For quantification of the results, see Supplementary Figure S8A. (B) High-resolution northern blot analysis of 5.8S rRNA and its precursor. The same RNA samples as in (A) were resolved in denaturing polyacrylamide gels for 1 h or for 2 h (‘longer run’; upper section). Following transfer, membranes were hybridized with probes for 5.8S rRNA or the region downstream its 3′ border (precursor probe); 5S rRNA was used as a loading control. Accumulation of slightly different precursors was observable depending on the mutant hDIS3 variant, without concomitant decrease of mature 5.8S rRNA levels. (C) High-resolution northern blot analysis of tRNAs. RNA samples as in (A) were separated in denaturing polyacrylamide gels and analyzed as above using probes for various tRNAs. Induction-dependent accumulation was observed for the majority of analyzed tRNAs and precursors of tRNATyr and tRNACys, particularly in cells producing hDIS3D487N and hDIS3R780K proteins. Hybridization with the probe for 5.8S rRNA served as positive control. For quantification of the results, see Supplementary Figure S8B. (D) High-resolution northern blot analysis of selected sn/snoRNAs. RNA samples as in (A) were analyzed as above, using probes specific to three different snRNA and U3 snoRNA. Induction-dependent accumulation was noticeable in the case of U5 snRNA; hybridizations with probes for 7SL RNA and 5.8S rRNA served as positive controls. For quantification of the results, see Supplementary Figure S8C. (E) Quantitative PCR analysis of different PROMPT regions and corresponding protein-coding transcripts in cell lines bearing various hDIS3 mutations. The graph shows results of quantification of three independent experiments; values on the left represent fold increase of transcripts in cells treated with doxycycline versus untreated cells. GAPDH mRNA, which was found to be relatively insensitive to hDIS3 mutations in northern blots [see panel (A)], was used for normalization. Abundance of all analyzed PROMPTs was significantly higher in cells producing D487N and R780K hDIS3 variants compared with the WT control. This effect was not seen for mRNAs synthesized under the control of respective promoters located downstream of three of the analyzed PROMPT regions or for the unrelated mitochondrial transcript, ATP6/8, which was used as a negative control.
As the degree of accumulation was the strongest for 5.8S rRNA precursors, we analyzed this in more detail by high-resolution northern blots, using 5S rRNA as loading control (Figure 5B). In general, we confirmed that the levels of these species were elevated exclusively on induction in cells with hDIS3 mutations (Figure 5B). Additionally, we found that (i) the patterns of observed precursors differed among the individual mutants and (ii) the patterns of accumulating precursors were more similar between hDIS3 D487N and R780K mutants than those in cells expressing hDIS3G766R (Figure 5B). Furthermore, despite the significant processing defects, levels of mature 5.8S rRNA were not markedly reduced (Figure 5B). Consequently, neither ribosome biogenesis nor polysome formation seemed to be significantly disturbed (Supplementary Figure S9A). Interestingly, the observed 5.8S rRNA precursors were incorporated into polysomes (Supplementary Figure S9B). It is also worth noting that their accumulation was not further increased by siRNA-mediated silencing of hRRP6 expression (Supplementary Figure S10).
Two recent publications underscored the importance of exosome and catalytic Dis3 activities in particular, in the global degradation of noncoding RNAs in yeast, and accumulation of tRNAs and their precursors was reported as one of the most prominent phenotypes resulting from Dis3 inactivation (37,53). These results, together with the increased levels of other RNA polymerase III transcripts in cells producing mutant hDIS3 (Figure 5A and Supplementary Figure S8A), prompted us to analyze the influence of hDIS3 mutations on selected tRNA molecules more carefully. We noted that almost all examined mature tRNA (except tRNATyr and tRNAPhe), as well as precursors of tRNATyr and tRNACys, accumulate in cells bearing exogenous hDIS3 with D487N or R780K mutations following doxycycline treatment (Figure 5C; for quantification see Supplementary Figure S8B). Although the effect was not as pronounced as for 5.8S rRNA precursors (Figure 5A–D), it was reproducible. It is again worth emphasizing that amino acid substitutions at positions 487 and 780 exerted stronger molecular phenotypes than the G766R change (Figure 5C and Supplementary Figure S8B).
We also investigated the impact of hDIS3 mutations on the accumulation of U3 snoRNA and three different snRNAs; however, we only noticed a phenotype for U5 snRNA that resembled the previously observed phenotype with tRNAs (Figure 5D; for quantification see Supplementary Figure S8C). We also analyzed PROMPTs, which are unstable transcripts previously shown to accumulate in cells subjected to siRNA-mediated depletion of human exosome subunits (26,39,40). We found that the levels of all analyzed PROMPTs were increased in all cell lines producing mutated hDIS3 variants (Figure 5E). In agreement with other analyses, accumulation of PROMPTs was far more pronounced in cells bearing hDIS3 D487N or R780K mutations than in the case of G766R substitution (Figure 5E). Additionally, we analyzed corresponding protein-coding transcripts synthesized from promoters localized downstream of three out of four PROMPTs and found that they were not affected by hDIS3 mutations, similar to the unrelated mitochondrially encoded transcript, ATP6/8 (Figure 5E).
In summary, hDIS3 mutations in our model cell lines resulted in accumulation of the majority of analyzed exosome substrates, including 5.8S processing intermediates, tRNAs, RNA polymerase III transcripts and PROMPTs. Although the degree of accumulation was variable among different exosome targets, in most cases we observed more significant effects in cells producing the hDIS3D487N and hDIS3R780K than the hDIS3G766R variant.
MM-associated hDIS3 mutations lead to growth inhibition of human cells and are synthetically lethal with PIN domain catalytic mutations
We noticed that cells expressing hDIS3 variants with D487N or R780K substitutions proliferated at somewhat slower rate following the doxycycline-mediated induction compared with the two other cell lines. Therefore, we performed additional growth and metabolic analyses by observing the untreated cells and their counterparts over a long induction period, 48 h of culture, followed by passage of cells into fresh medium containing doxycycline and another 48 h of culture. While all cell lines grew normally and comparably in medium lacking doxycycline, on longer sh-miRNA–mediated repression of endogenous hDIS3 and simultaneous expression of sh-miRNA-insensitive exogenous hDIS3 variants, the four cell lines behaved differently (Figure 6). hDIS3WT produced from the integrated construct complemented the silenced endogenous hDIS3, and the cell growth and metabolism were undisturbed (Figure 6A and B). In contrast, cells expressing hDIS3D487N and hDIS3R780K variants exhibited inhibition of growth and reduction of metabolic activity (Figure 6A and B), while G766R mutation in hDIS3 exerted only moderate effects (Figure 6A and B). These results were in line with biochemical and functional data (see Figures 1C, 2 and 5).
Figure 6.

MM-associated hDIS3 mutations adversely affect cell growth and metabolic activity in a human cellular model. (A) Cell-growth analysis. Equal amounts of cells from each line were seeded in culture dishes, subjected to doxycycline-mediated induction (48 h + 48 h) and analyzed by microscopy. The cells harboring mutated hDIS3 grew worse than the cell line with WT protein. The strength of the growth phenotype was as follows: hDIS3D487N > hDIS3R780K > hDIS3G766R. (B) Metabolic activity assay. Approximately equal numbers of cells from each stable cell line were grown in triplicate in 96-well plates, either untreated (‘−dox’) or treated with doxycycline (‘+dox’). AlamarBlue® reagent was added to the cells after the indicated time, and changes of metabolic status were evaluated by fluorescence measurements. In agreement with results from (A), hDIS3D487N and hDIS3G766R mutants gave the strongest and weakest phenotype, respectively.
Next, we were interested in checking whether, as in yeast, there is a synergistic effect of MM-associated RNB domain mutations and dysfunction of hDIS3 PIN domain endonuclease activity. We constructed analogous stable cell lines bearing an additional mutation in the catalytic site of PIN domain, D146N, which was previously shown to abolish hDIS3 endonucleolytic activity in the in vitro assays (26). Cell growth assays performed as above revealed that the growth of cell lines expressing hDIS3 variants with the D146N mutation was unaffected in the absence of doxycycline (Figure 7A). On the other hand, on induction, only cells expressing the hDIS3D146N single mutant grew relatively normally and comparably with its counterpart producing hDIS3WT, indicating that endonuclease activity alone has little impact on cell physiology (Figure 7A). In contrast, combination of the PIN domain catalytic mutation with any mutation in the RNB domain (D487N, G766R or R780K) strongly suppressed survival of cells (Figure 7A). A synergistic effect of inactivation of both nucleolytic activities of hDIS3 was most pronounced in cells expressing the hDIS3D146N G766R double mutant, particularly when noting that the cell line producing the hDIS3G766R mutant alone displayed mild growth inhibition compared with the WT control (Figure 7A). Importantly, the metabolic activity of the cells bearing mutations in the RNB domain was also additionally reduced on accompanying inactivation of PIN domain endonuclease (Figure 7B).
Figure 7.


Catalytic mutation in the hDIS3 PIN domain, responsible for endonucleolytic activity, leads to synergistic effects with MM-associated mutations in the RNB domain. (A) Cell-growth analysis. Equal amounts of cells from each line were seeded in culture dishes, grown in the absence (‘−dox’) or presence (‘+dox’) of doxycycline and analyzed by microscopy. The cell lines producing hDIS3 with mutations in both catalytic domains grew significantly worse than cells producing hDIS3 with mutations only in the RNB domain. Nontransfected HEK293 Flp-In T-Rex cells were analyzed in parallel as a control. (B) Metabolic activity assay. Approximately equal numbers of cells from each stable cell line, or nontransfected HEK293 Flp-In T-Rex cells, were grown in triplicate in 96-well plates, either untreated (‘−dox’) or treated with doxycycline (‘+dox’). AlamarBlue® reagent was added after 72 h, and metabolic status was assessed by fluorescence measurements. In agreement with results from (A), cells producing hDIS3 double mutants displayed lower metabolic activity than cells expressing protein variants with an intact PIN domain. (C) Catalytic mutation of PIN domain does not enhance accumulation of 3′-extended 5.8S rRNA precursor molecules. High-resolution northern blot analysis of 5.8S rRNA and its precursor. Total RNA was isolated from cell lines producing WT hDIS3 or hDIS3 PIN or/and RNB domain mutants, which were all subjected to doxycycline treatment. RNA samples were resolved in denaturing polyacrylamide gel for 2 h; following RNA transfer, nylon membranes were hybridized with probes for mature 5.8S rRNA or to the region located downstream its 3′ border (precursor probe); 5S rRNA was used as a loading control. (D and E) Quantitative PCR analysis of different PROMPT regions (D) and corresponding protein-coding transcripts (E) in cell lines producing hDIS3 variants. The graphs show results of quantification of three independent experiments; values on the left represent fold increase of transcripts in cells expressing mutated versus WT hDIS3. GAPDH mRNA was used for normalization. Abundance of all analyzed PROMPTs was highly increased in cells producing hDIS3 double mutants compared with cells expressing proteins harboring mutations only in the RNB domain (D); the same trend was also observed for mRNAs synthesized under the control of respective promoters located downstream of three of the analyzed PROMPT regions, but not for the unrelated mitochondrial transcript, ATP6/8, which was used as a negative control (E).
We then analyzed molecular phenotypes resulting from combination of MM-associated hDIS3 mutations in the RNB domain with disruption of PIN domain endonucleolytic activity by comparing them with the respective single mutants. As shown in Figure 7C, inactivation of the PIN domain catalytic activity alone did not lead to accumulation of 5.8S rRNA precursors. Interestingly, we were not able to observe any synergistic effect of D146N substitution in the background of RNB domain mutations, as the levels of precursor molecules were not further elevated in cells expressing hDIS3D146N D487N, hDIS3D146N G766R and hDIS3D146N R780K compared with the cells producing respective hDIS3 versions with an intact PIN domain (Figure 7C). This indicates that such 3′-extended 5.8S rRNA species are normally degraded mainly by hDIS3 exoribonucleolytic activity. On the contrary, our results clearly demonstrated that while the levels of different PROMPTs were comparable between cell lines producing either hDIS3 with D146N substitution or its WT counterpart, the cells expressing hDIS3D146N D487N, hDIS3D146N G766R and hDIS3D146N R780K double mutants accumulated much higher amounts of PROMPTs than the respective single mutants (Figure 7D), suggesting the cooperative action of both nucleolytic activities of hDIS3. Intriguingly, we noticed a highly similar trend for protein-coding transcripts corresponding to three out of four tested PROMPTs (but not for mitochondrially encoded ATP6/8 mRNA, which we would not expect to be affected by exosome dysfunction), although the fold differences between double and single mutants were not so prominent as in the case of PROMPTs (Figure 7E).
In conclusion, although the molecular events linking the inactivation of hDIS3 nucleolytic properties and its inability to efficiently degrade transcripts with reduced cell viability remain to be elucidated, the negative synthetic interaction between MM-associated RNB domain mutations and PIN domain dysfunction may suggest the latter as a promising drug target for cancers bearing mutations that affect hDIS3 RNB domain exonucleolytic activity.
DISCUSSION
In this study, we showed that MM-associated hDIS3 mutations have negative effects on hDIS3 protein function, cell physiology and RNA metabolism, leading to vulnerabilities that can be used for anti-cancer drug development. Moreover, our analyses prove that DIS3 has an essential function in RNA metabolism in vertebrates.
Inhibition of hDIS3 exonucleolytic activity by MM-associated mutations
Results of biochemical assays indicate that in the majority of cases, amino acids substituted in hDIS3 in MM patients play an important role in regulating hDIS3 exoribonucleolytic activity, which is generally in agreement with data from literature and structural information. Five out of six analyzed mutations are positioned in the RNB domain, strictly associated with exoribonucleolytic activity (Figure 1A; Supplementary Figure S11). The only exception is isoleucine at position 845 (I845), which is located in a less conserved region downstream of the RNB domain; its occurrence is limited exclusively to the human protein (Figure 1A; Supplementary Figure S11). Accordingly, activity of hDIS3I845V was unaffected, irrespective of whether tested on single- or partially double-stranded RNA substrates.
Although our results do not allow us to directly propose the possible mechanism of inhibition of hDIS3 exoribonucleolytic activity resulting from MM-associated mutations, some clues can be obtained following comparison of the solved 3D structures of E. coli RNase II and yeast Dis3 RNB domains {the latter in the context of exosome core, according to the recently published structure of 11-subunit exosome [(25); Supplementary Figure S12A]}. For instance, S541 in S. cerevisiae Dis3 (an equivalent of S477 in hDIS3) and threonine in the corresponding position of E. coli RNase II (Supplementary Figure S11) are located in the proximity of RNB domain catalytic center (Supplementary Figure S12B). We may hypothesize that substitution with large amino acid, like arginine (as in the case of the S477R hDIS3 mutation), may affect corresponding β-sheet formation and most likely result in a steric conflict and/or abolish hydrogen bonds required for correct structure, therefore interfering with magnesium coordination and decreasing enzymatic activity. On the other hand, perfectly conserved G833 in yeast Dis3 (a counterpart of G766 in hDIS3) is situated in the ‘neck region’ conserved between RNase II and Dis3, which is responsible for binding of the 3′-end of RNA (Supplementary Figure S12C). A potentially important function of glycine in this particular position is further underscored by the fact that the G833D mutation is known to suppress degradation of hypomodified 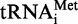 in S. cerevisiae (43,54). Introduction of a negatively charged amino acid might disrupt the interaction between protein and RNA. Substitution of glycine with a bigger amino acid, such as arginine (as in the case of the human G766R mutation), although positively charged, may result in a steric hindrance. Additionally, this particular glycine is located at the top of a loop, and flexibility of this small amino acid may be crucial for the structural integrity. In turn, R780/R847 in human/yeast Dis3 is in the same position as R500 in E.coli RNase II; this amino acid is localized in the active site of the bacterial enzyme [(55); Supplementary Figure S12D] and its substitution with alanine leads to an over 40 000-fold reduction of the exoribonucleolytic activity (56). We may thus infer that, similar to E. coli RNase II, the role of arginine in this specific location is to increase susceptibility of the phosphodiester bonds present in RNA substrates to cleavage. All the findings mentioned above nicely corroborate our results demonstrating that the exoribonucleolytic activity of hDIS3 is affected most significantly in protein variants bearing S477R, G766R or R780K mutation.
in S. cerevisiae (43,54). Introduction of a negatively charged amino acid might disrupt the interaction between protein and RNA. Substitution of glycine with a bigger amino acid, such as arginine (as in the case of the human G766R mutation), although positively charged, may result in a steric hindrance. Additionally, this particular glycine is located at the top of a loop, and flexibility of this small amino acid may be crucial for the structural integrity. In turn, R780/R847 in human/yeast Dis3 is in the same position as R500 in E.coli RNase II; this amino acid is localized in the active site of the bacterial enzyme [(55); Supplementary Figure S12D] and its substitution with alanine leads to an over 40 000-fold reduction of the exoribonucleolytic activity (56). We may thus infer that, similar to E. coli RNase II, the role of arginine in this specific location is to increase susceptibility of the phosphodiester bonds present in RNA substrates to cleavage. All the findings mentioned above nicely corroborate our results demonstrating that the exoribonucleolytic activity of hDIS3 is affected most significantly in protein variants bearing S477R, G766R or R780K mutation.
Contrary to the mutations mentioned above, V504G substitution did not have any impact on hDIS3 exoribonucleolytic activity. It might be a bit surprising, as valine in this position is perfectly conserved (Supplementary Figure S11). However, superimposition of yeast Dis3 and E. coli RNB domain structures reveals that its counterpart in S. cerevisiae Dis3 (V568) is located at the periphery of the protein (Supplementary Figure S12E). Similarly, A588 in yeast Dis3 (equivalent of A524 in hDIS3) is located far from the catalytic center, in a large 4-stranded β-sheet, inside a globular hydrophobic structure (Supplementary Figure S12F). Furthermore, it is the least conserved amino acid, absent from E. coli RNase II/R, which both contain a basic amino acid residue in this position (K or R) (Supplementary Figure S11). Although we were not able to assess the activity of the hDIS3A524P protein variant due to its insolubility, analyses performed in a yeast model indirectly indicate that the dysfunction of such mutant is not so severe as in the case of hDIS3G766R or hDIS3R780K versions.
Together these results show that some MM-associated hDIS3 mutations exert strong effects on the ability of the enzyme to exonucleolytically digest RNA substrates, which is in agreement with the currently available structural information. However, it is important to point out that other hDIS3 mutations, like I845V and V504G, do not affect the enzyme activity, what may suggest that they represent random mutational noise, since global sequencing of tumor samples leads to the discovery of thousands of single nucleotide polymorphisms and not all of them are relevant to cancer development. Nevertheless, this does not diminish the significance of other detected amino acid substitutions, as there are currently 60 described DIS3 mutations in the COSMIC database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic) and a large fraction of them is located in the conserved region of the RNB domain.
Importance of hDIS3 and intactness of its RNB domain for cell physiology
Results of multiple analyses performed in this study using different models highlight the importance of hDIS3 and intactness of its RNB domain for cell physiology. First, knockout of both alleles of DIS3 gene in the chicken DT40 cell line was found to be lethal, which demonstrated for the first time that in vertebrates the gene is indispensable for survival, thus confirming findings in S. cerevisiae. We used this model to analyze the influence of MM-associated DIS3 mutations in the RNB domain on cell viability and discovered that, in general, amino acid changes leading to more significantly decreased enzymatic activity of hDIS3 protein in vitro were reflected by more severe molecular phenotypes and growth defects of corresponding yeast mutant strains. This observation was in turn corroborated by the results of assays performed using our experimental system based on HEK293-derived human cell lines.
Among the strongest molecular phenotypes caused by MM mutations are increased levels of extended 5.8S precursor molecules, improperly processed at the 3′-end, and an intermediate in rRNA maturation pathway, 5′-ETS. In both yeast and human cells, accumulation was particularly apparent in the case of mutations most adversely affecting the activity of the protein. Intriguingly, in neither of the two models was accumulation of 5.8S rRNA precursor accompanied by concomitant significant decrease in the levels of mature 5.8S rRNA. Furthermore, we found that such precursors were successfully incorporated into polysomes in human cells, indicating that their overabundance may not influence protein synthesis. Therefore, the significance of accumulation of these species for cell physiology remains to be elucidated. Moreover, not only full-length 5.8S precursors and 5′-ETS but also their degradation intermediates accumulated in the yeast mutants, confirming previous observations that hDIS3 exoribonucleolytic activity plays a crucial role in their removal from the cell (18). It also appears that RRP6 exonuclease cannot fully take over hDIS3 functions in the degradation of such RNA species in the nucleus, as neither the deletion of RRP6 gene in yeast nor RNA interference (RNAi)-based suppression of hRRP6 expression is human cells had an additive effect on their accumulation. However, our results do not provide explanation as to whether aberrations in ribosomal RNA maturation pathway are directly associated with cell growth defects.
Further analyses of molecular phenotypes resulting from MM-associated hDIS3 mutations in human cells revealed that inhibition of hDIS3 exoribonucleolytic activity results in decreased degradation rate of RNA molecules belonging to other RNA classes, such as tRNAs and their precursors, snRNAs and other small RNAs, including RNase P RNA and 7SL RNA. One of the most interesting and reproducible phenotypes was the strong accumulation of PROMPTs, unstable transcripts synthesized by RNA Pol II upstream many promoters of protein-coding genes. Moreover, accumulation of PROMPTs was further enhanced when MM-associated RNB domain mutations were combined with hDIS3 D146N (endo−) mutation within PIN domain, indicating that degradation of these RNA species requires both exonucleolytic and endonucleolytic activities of hDIS3. It should be also pointed out that DIS3 mutations in yeast (in which Dis3p is present both in the nucleus and the cytoplasm) lead to the accumulation of nuclear noncoding RNAs somewhat related to human PROMPTs, namely CUTs. This indicates that the level of ‘transcriptional noise’ is probably much higher in the nucleus of the cells producing mutated hDIS3 variants. Whether and how this may affect physiology, metabolic activity and survival potential of the cells remains to be addressed. Furthermore, it is essential to apply high-throughput sequencing analyses to obtain deeper insights into the transcriptome changes resulting from MM-associated hDIS3 mutations.
Moreover, the role of hDIS3 mutations in cancer development remains to be determined. One possibility is the involvement of hDIS3 as a part of the exosome complex, known to be involved in AID-mediated immunoglobulin hypermutations and class switches (48), which lead to DNA translocations associated with MM development. The mutations may also have specific direct effects on gene expression; alternatively, as the exosome is the main enzyme degrading unstable transcripts from noncoding regions and some such transcripts affect chromatin structure, their overabundance resulting from hDIS3 dysfunction may indirectly affect expression of genes important for carcinogenesis. All of that remains to be experimentally verified using true MM models, but since thus far there is no single established MM cell line with hDIS3 mutation affecting activity of the protein, this is a challenging task.
hDIS3 PIN domain as a potential drug target for cancers bearing mutations in hDIS3 RNB domain
Synthetic lethality has recently emerged as one of the most promising strategies for development of targeted cancer therapies (57). Such a strategy enables the targeting of cancer cells with a potentially low effect on untransformed cells. For example, breast cancer cells with mutations in the BRCA1 and BRCA2 are sensitive to inhibition of poly [ADP-ribose] polymerase (PARP1), whereas inhibition of PARP1 has little influence on survival and proliferation of normal cells with WT BRCA (58,59). Lead compounds based on PARP1 inhibitors are in advanced clinical trials (60).
In the yeast system, both deletion of accessory nuclear exosome catalytic subunit Rrp6 and inactivation of Dis3 PIN endonuclease domain are synthetically lethal with mutations abolishing Dis3 exonucleolytic activity. Since we show here that, as in yeast, vertebrate DIS3 is essential for cell survival, it was possible that similar synthetic interactions could exist in the case of hDIS3. Results presented herein indicate that MM-associated mutations in hDIS3 RNB domain, inhibiting its exonuclease activity, are indeed synthetically lethal with inactivation of hDIS3 endonucleolytic activity. This is valid for both the yeast and cellular model described in this publication. In human cells, the combination of MM mutations with mutations in the PIN domain had a cumulative effect on cell growth inhibition and accumulation of exosome targets.
In contrast to inactivation of the DIS3 PIN domain, deletion or depletion of RRP6 was not synthetically lethal with MM-associated hDIS3 mutations.
In summary, our data suggest the hDIS3 PIN domain but not hRRP6 as a drug target for cancers bearing hDIS3 mutations. Importantly, mutations inactivating the hDIS3 PIN domain alone have little effect on cell physiology, since research performed by us and others did not reveal any detectable phenotype of hDIS3 PIN domain dysfunction without the presence of other mutations. This suggests that the active site of the hDIS3 PIN domain endonuclease may be a particularly promising drug target for cancers bearing mutations affecting hDIS3 RNB domain exonucleolytic activity. This is especially important for MM, as it remains incurable, although we cannot exclude the possibility that due to differences in physiology, synthetic interactions between mutations in hDIS3 PIN and RNB domains might not occur in MM cells. The DIS3 PIN domain structure is solved and the in vitro assays for its endoribonucleolytic activity have been developed, what is an additional advantage that should facilitate identification of its inhibitors based on classical medical chemistry or virtual screening for putative lead compounds.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [61–65].
FUNDING
National Science Centre [NCN Maestro: UMO-2011/02/A/NZ1/00001 to A.D.]; National Centre for Research and Development [NCBR PBS: 176911 to A.D., NCBR LIDER: LIDER/35/46/L-3/11/NCBR/2012 to R.T.]; Polish-Swiss Research Programme [PSPB-183/2010 to A.D.]; Scholarship for outstanding young scientists from the Polish Ministry of Science and Higher Education (to R.T. and R.J.S.);. Experiments were carried out with the use of CePT infrastructure financed by the European Union—the European Regional Development Fund [Innovative economy 2007–13, Agreement POIG.02.02.00-14-024/08-00]. Funding for open access charge: National Science Centre Poland [NCN Maestro: UMO-2011/02/A/NZ1/00001 to A.D.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors wish to thank Kamila Klosowska for her assistance in experiments with DT40 cells, Aleksander Chlebowski for his help in microscopy analysis and all the members of the laboratory for their support. We thank Prof. Zbigniew Dominski and Michal Lubas for providing reagents. We acknowledge Prof. Jakub Golab and Dr Dominika Nowis for helpful discussions and critical reading of the manuscript. Dominika Nowis is additionally acknowledged for her help with MM cell lines. We also thank Dr Jonathan J. Keats for sharing unpublished data with us. Author contributions: A.D. conceived and directed the studies. R.T. designed most of the experiments, prepared the majority of the plasmid constructs and performed all biochemical assays and experiments in the human cellular model. K.D. performed all experiments in yeast and participated in the growth assays in human cell lines. I.K. carried out in silico analyses of hDIS3 mutations. I.K. and J.G. performed experiments on DT40 cells. K.S. expressed and purified recombinant proteins; R.J.S. participated in the design of experiments and supervised studies in DT40 cells. J.G. carried out analysis of hDIS3 mutations in multiple myeloma cell lines. E.P.O. and K.K. established stable human cell lines. R.T. and A.D. wrote the manuscript, with contributions from I.K. and R.J.S.
REFERENCES
- 1.Laubach J, Richardson P, Anderson K. Multiple myeloma. Annu. Rev. Med. 2011;62:249–264. doi: 10.1146/annurev-med-070209-175325. [DOI] [PubMed] [Google Scholar]
- 2.Nahi H, Sutlu T, Jansson M, Alici E, Gahrton G. Clinical impact of chromosomal aberrations in multiple myeloma. J. Intern. Med. 2011;269:137–147. doi: 10.1111/j.1365-2796.2010.02324.x. [DOI] [PubMed] [Google Scholar]
- 3.Palumbo A, Anderson K. Multiple myeloma. N. Engl. J. Med. 2011;364:1046–1060. doi: 10.1056/NEJMra1011442. [DOI] [PubMed] [Google Scholar]
- 4.Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011;471:467–472. doi: 10.1038/nature09837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker BA, Wardell CP, Melchor L, Hulkki S, Potter NE, Johnson DC, Fenwick K, Kozarewa I, Gonzalez D, Lord CJ, et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood. 2012;120:1077–1086. doi: 10.1182/blood-2012-03-412981. [DOI] [PubMed] [Google Scholar]
- 6.Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, Boca SM, Carter H, Samayoa J, Bettegowda C, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rose AE, Poliseno L, Wang J, Clark M, Pearlman A, Wang G, Vega Y, Saenz de Miera EC, Medicherla R, et al. Integrative genomics identifies molecular alterations that challenge the linear model of melanoma progression. Cancer Res. 2011;71:2561–2571. doi: 10.1158/0008-5472.CAN-10-2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim J, Kuroki T, Ozaki K, Kohsaki H, Yamori T, Tsuruo T, Nakamori S, Imaoka S, Endo M, Nakamura Y. Isolation of murine and human homologues of the fission-yeast dis3+ gene encoding a mitotic-control protein and its overexpression in cancer cells with progressive phenotype. Cancer Res. 1997;57:921–925. [PubMed] [Google Scholar]
- 10.Liang L, Qu L, Ding Y. Protein and mRNA characterization in human colorectal carcinoma cell lines with different metastatic potentials. Cancer Invest. 2007;25:427–434. doi: 10.1080/07357900701512258. [DOI] [PubMed] [Google Scholar]
- 11.Camps J, Pitt JJ, Emons G, Hummon AB, Case CM, Grade M, Jones TL, Nguyen QT, Ghadimi BM, Beissbarth T, et al. Genetic amplification of the NOTCH modulator LNX2 upregulates the WNT/β-catenin pathway in colorectal cancer. Cancer Res. 2013;73:2003–2013. doi: 10.1158/0008-5472.CAN-12-3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pils D, Tong D, Hager G, Obermayr E, Aust S, Heinze G, Kohl M, Schuster E, Wolf A, Sehouli J, et al. A combined blood based gene expression and plasma protein abundance signature for diagnosis of epithelial ovarian cancer - a study of the OVCAD consortium. BMC Cancer. 2013;13:178. doi: 10.1186/1471-2407-13-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reis FP, Pobre V, Silva IJ, Malecki M, Arraiano CM. The RNase II/RNB family of exoribonucleases: putting the ‘Dis’ in disease. Wiley Interdiscip. Rev. RNA. 2013;4:607–615. doi: 10.1002/wrna.1180. [DOI] [PubMed] [Google Scholar]
- 14.Liu Q, Greimann JC, Lima CD. Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell. 2006;127:1223–1237. doi: 10.1016/j.cell.2006.10.037. [DOI] [PubMed] [Google Scholar]
- 15.Dziembowski A, Lorentzen E, Conti E, Séraphin B. A single subunit, Dis3, is essentially responsible for yeast exosome core activity. Nat. Struct. Mol. Biol. 2007;14:15–22. doi: 10.1038/nsmb1184. [DOI] [PubMed] [Google Scholar]
- 16.Tomecki R, Drazkowska K, Dziembowski A. Mechanisms of RNA degradation by the eukaryotic exosome. Chembiochem. 2010;11:938–945. doi: 10.1002/cbic.201000025. [DOI] [PubMed] [Google Scholar]
- 17.Allmang C, Kufel J, Chanfreau G, Mitchell P, Petfalski E, Tollervey D. Functions of the exosome in rRNA, snoRNA and snRNA synthesis. EMBO J. 1999;18:5399–5410. doi: 10.1093/emboj/18.19.5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lebreton A, Tomecki R, Dziembowski A, Séraphin B. Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature. 2008;456:993–996. doi: 10.1038/nature07480. [DOI] [PubMed] [Google Scholar]
- 19.Schaeffer D, Tsanova B, Barbas A, Reis FP, Dastidar EG, Sanchez-Rotunno M, Arraiano CM, van Hoof A. The exosome contains domains with specific endoribonuclease, exoribonuclease and cytoplasmic mRNA decay activities. Nat. Struct. Mol. Biol. 2009;16:56–62. doi: 10.1038/nsmb.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneider C, Leung E, Brown J, Tollervey D. The N-terminal PIN domain of the exosome subunit Rrp44 harbors endonuclease activity and tethers Rrp44 to the yeast core exosome. Nucleic Acids Res. 2009;37:1127–1140. doi: 10.1093/nar/gkn1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonneau F, Basquin J, Ebert J, Lorentzen E, Conti E. The yeast exosome functions as a macromolecular cage to channel RNA substrates for degradation. Cell. 2009;139:547–559. doi: 10.1016/j.cell.2009.08.042. [DOI] [PubMed] [Google Scholar]
- 22.Malet H, Topf M, Clare DK, Ebert J, Bonneau F, Basquin J, Drazkowska K, Tomecki R, Dziembowski A, Conti E, et al. RNA channelling by the eukaryotic exosome. EMBO Rep. 2010;11:936–942. doi: 10.1038/embor.2010.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wasmuth EV, Lima CD. Exo- and endoribonucleolytic activities of yeast cytoplasmic and nuclear RNA exosomes are dependent on the noncatalytic core and central channel. Mol. Cell. 2012;48:133–144. doi: 10.1016/j.molcel.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drazkowska K, Tomecki R, Stodus K, Kowalska K, Czarnocki-Cieciura M, Dziembowski A. The RNA exosome complex central channel controls both exonuclease and endonuclease Dis3 activities in vivo and in vitro. Nucleic Acids Res. 2013;41:3845–3858. doi: 10.1093/nar/gkt060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makino DL, Baumgärtner M, Conti E. Crystal structure of an RNA-bound 11-subunit eukaryotic exosome complex. Nature. 2013;495:70–75. doi: 10.1038/nature11870. [DOI] [PubMed] [Google Scholar]
- 26.Tomecki R, Kristiansen MS, Lykke-Andersen S, Chlebowski A, Larsen KM, Szczesny RJ, Drazkowska K, Pastula A, Andersen JS, Stepien PP, et al. The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J. 2010;29:2342–2357. doi: 10.1038/emboj.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Staals RH, Bronkhorst AW, Schilders G, Slomovic S, Schuster G, Heck AJ, Raijmakers R, Pruijn GJ. Dis3-like 1: a novel exoribonuclease associated with the human exosome. EMBO J. 2010;29:2358–2367. doi: 10.1038/emboj.2010.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lykke-Andersen S, Tomecki R, Jensen TH, Dziembowski A. The eukaryotic RNA exosome: same scaffold but variable catalytic subunits. RNA Biol. 2011;8:61–66. doi: 10.4161/rna.8.1.14237. [DOI] [PubMed] [Google Scholar]
- 29.Malecki M, Viegas SC, Carneiro T, Golik P, Dressaire C, Ferreira MG, Arraiano CM. The exoribonuclease Dis3L2 defines a novel eukaryotic RNA degradation pathway. EMBO J. 2013;32:1842–1854. doi: 10.1038/emboj.2013.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang HM, Triboulet R, Thornton JE, Gregory RI. A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28-let-7 pathway. Nature. 2013;497:244–248. doi: 10.1038/nature12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lubas M, Damgaard CK, Tomecki R, Cysewski D, Jensen TH, Dziembowski A. Exonuclease hDIS3L2 specifies an exosome-independent 3′-5′ degradation pathway of human cytoplasmic mRNA. EMBO J. 2013;32:1855–1868. doi: 10.1038/emboj.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Astuti D, Morris MR, Cooper WN, Staals RH, Wake NC, Fews GA, Gill H, Gentle D, Shuib S, Ricketts CJ, et al. Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat. Genet. 2012;44:277–284. doi: 10.1038/ng.1071. [DOI] [PubMed] [Google Scholar]
- 33.Lebreton A, Séraphin B. Exosome-mediated quality control: substrate recruitment and molecular activity. Biochim. Biophys. Acta. 2008;1779:558–565. doi: 10.1016/j.bbagrm.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 34.Schmid M, Jensen TH. The exosome: a multipurpose RNA-decay machine. Trends Biochem. Sci. 2008;33:501–510. doi: 10.1016/j.tibs.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee D, Gao M, O'Connor JP, Raijmakers R, Pruijn G, Lutz CS, Wilusz J. The mammalian exosome mediates the efficient degradation of mRNAs that contain AU-rich elements. EMBO J. 2002;21:165–174. doi: 10.1093/emboj/21.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gherzi R, Lee KY, Briata P, Wegmüller D, Moroni C, Karin M, Chen CY. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol. Cell. 2004;14:571–583. doi: 10.1016/j.molcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 37.Gudipati RK, Xu Z, Lebreton A, Séraphin B, Steinmetz LM, Jacquier A, Libri D. Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol. Cell. 2012;48:409–421. doi: 10.1016/j.molcel.2012.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Régnault B, Devaux F, Namane A, Séraphin B, et al. Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell. 2005;121:725–737. doi: 10.1016/j.cell.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 39.Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. RNA exosome depletion reveals transcription upstream of active human promoters. Science. 2008;322:1851–1854. doi: 10.1126/science.1164096. [DOI] [PubMed] [Google Scholar]
- 40.Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH. Interaction profiling identifies the human nuclear exosome targeting complex. Mol. Cell. 2011;43:624–637. doi: 10.1016/j.molcel.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 41.Orban TI, Izaurralde E. Decay of mRNAs targeted by RISC requires XRN1, the Ski complex, and the exosome. RNA. 2005;11:459–469. doi: 10.1261/rna.7231505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allmang C, Mitchell P, Petfalski E, Tollervey D. Degradation of ribosomal RNA precursors by the exosome. Nucleic Acids Res. 2000;28:1684–1691. doi: 10.1093/nar/28.8.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J. Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev. 2004;18:1227–1240. doi: 10.1101/gad.1183804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Isken O, Maquat LE. Quality control of eukaryotic mRNA: safeguarding cells from abnormal mRNA function. Genes Dev. 2007;21:1833–1856. doi: 10.1101/gad.1566807. [DOI] [PubMed] [Google Scholar]
- 45.Wang X, Jia H, Jankowsky E, Anderson JT. Degradation of hypomodified tRNAiMet in vivo involves RNA-dependent ATPase activity of the DExH helicase Mtr4p. RNA. 2008;14:107–116. doi: 10.1261/rna.808608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Assenholt J, Mouaikel J, Andersen KR, Brodersen DE, Libri D, Jensen TH. Exonucleolysis is required for nuclear mRNA quality control in yeast THO mutants. RNA. 2008;14:2305–2313. doi: 10.1261/rna.1108008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parker R. RNA degradation in Saccharomyces cerevisiae. Genetics. 2012;191:671–702. doi: 10.1534/genetics.111.137265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144:353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lorentzen E, Basquin J, Tomecki R, Dziembowski A, Conti E. Structure of the active subunit of the yeast exosome core, Rrp44: diverse modes of substrate recruitment in the RNase II nuclease family. Mol. Cell. 2008;29:717–728. doi: 10.1016/j.molcel.2008.02.018. [DOI] [PubMed] [Google Scholar]
- 50.Buerstedde JM, Takeda S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell. 1991;67:179–188. doi: 10.1016/0092-8674(91)90581-i. [DOI] [PubMed] [Google Scholar]
- 51.Arakawa H. Excision of floxed-DNA sequences by transient induction of Mer-Cre-Mer. Subcell. Biochem. 2006;40:347–349. doi: 10.1007/978-1-4020-4896-8_22. [DOI] [PubMed] [Google Scholar]
- 52.Sammarco MC, Grabczyk E. A series of bidirectional tetracycline-inducible promoters provides coordinated protein expression. Anal. Biochem. 2005;346:210–216. doi: 10.1016/j.ab.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 53.Schneider C, Kudla G, Wlotzka W, Tuck A, Tollervey D. Transcriptome-wide analysis of exosome targets. Mol. Cell. 2012;48:422–433. doi: 10.1016/j.molcel.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schneider C, Anderson JT, Tollervey D. The exosome subunit Rrp44 plays a direct role in RNA substrate recognition. Mol. Cell. 2007;27:324–331. doi: 10.1016/j.molcel.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Frazão C, McVey CE, Amblar M, Barbas A, Vonrhein C, Arraiano CM, Carrondo MA. Unravelling the dynamics of RNA degradation by ribonuclease II and its RNA-bound complex. Nature. 2006;443:110–114. doi: 10.1038/nature05080. [DOI] [PubMed] [Google Scholar]
- 56.Barbas A, Matos RG, Amblar M, López-Viñas E, Gomez-Puertas P, Arraiano CM. Determination of key residues for catalysis and RNA cleavage specificity: one mutation turns RNase II into a “SUPER-ENZYME”. J. Biol. Chem. 2009;284:20486–20498. doi: 10.1074/jbc.M109.020693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Porcelli L, Quatrale AE, Mantuano P, Silvestris N, Brunetti AE, Calvert H, Paradiso A, Azzariti A. Synthetic lethality to overcome cancer drug resistance. Curr. Med. Chem. 2012;19:3858–3873. doi: 10.2174/092986712802002563. [DOI] [PubMed] [Google Scholar]
- 58.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 59.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 60.Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, Assiotis I, Rodrigues DN, Filho JS, Moreno V, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013;229:422–429. doi: 10.1002/path.4140. [DOI] [PubMed] [Google Scholar]
- 61.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Séraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 62.Bryksin AV, Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. Biotechniques. 2010;48:463–465. doi: 10.2144/000113418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Arakawa H, Lodygin D, Buerstedde JM. Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 2001;1:7. doi: 10.1186/1472-6750-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song J, Liang C, Chen X. Transduction of avian cells with recombinant baculovirus. J. Virol. Methods. 2006;135:157–162. doi: 10.1016/j.jviromet.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 65.Li MZ, Elledge SJ. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods. 2007;4:251–256. doi: 10.1038/nmeth1010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.