


Abstract
Human APOBEC3A (A3A) is a single-domain cytidine deaminase that converts deoxycytidine residues to deoxyuridine in single-stranded DNA (ssDNA). It inhibits a wide range of viruses and endogenous retroelements such as LINE-1, but it can also edit genomic DNA, which may play a role in carcinogenesis. Here, we extend our recent findings on the NMR structure of A3A and report structural, biochemical and cell-based mutagenesis studies to further characterize A3A’s deaminase and nucleic acid binding activities. We find that A3A binds ssRNA, but the RNA and DNA binding interfaces differ and no deamination of ssRNA is detected. Surprisingly, with only one exception (G105A), alanine substitution mutants with changes in residues affected by specific ssDNA binding retain deaminase activity. Furthermore, A3A binds and deaminates ssDNA in a length-dependent manner. Using catalytically active and inactive A3A mutants, we show that the determinants of A3A deaminase activity and anti-LINE-1 activity are not the same. Finally, we demonstrate A3A’s potential to mutate genomic DNA during transient strand separation and show that this process could be counteracted by ssDNA binding proteins. Taken together, our studies provide new insights into the molecular properties of A3A and its role in multiple cellular and antiviral functions.
INTRODUCTION
The human APOBEC3 (A3) proteins are host restriction factors that exhibit activity against retroviruses and endogenous retroelements and play an important role in the innate immune response to human pathogens (1–4). These proteins function as deoxycytidine deaminases that convert dC to dU in single-stranded DNA (ssDNA) and behave as DNA mutators (5,6). The A3 family consists of seven members, having either one (A3A, A3C and A3H) or two (A3B, A3D, A3F and A3G) zinc (Zn)-binding domains with the conserved motif HX1EX23–24CX2-4C (X is any amino acid) (1,2,7). The cysteine and histidine residues coordinate a zinc metal ion (Zn2+), and the glutamic acid is implicated in proton transfer during catalysis (1,8). The deamination target site specificity of A3 proteins is determined by the nucleotides (nt) flanking dC residues in the substrate (6,9).
A3A is a single-domain deaminase that acts on the cytosine bases in TC-containing ssDNA (10–16); moreover, it can also deaminate methyl cytosine residues (17–19). It is expressed in peripheral blood mononuclear cells, specifically in the cells of the CD14+ lineage that includes monocytes and macrophages (10,12,20–22). In addition, A3A is also expressed in psoriatic keratinocytes and in normal keratinocytes, but only on treatment with an activator of protein kinase C (23,24). Although A3A displays multiple biological activities, deamination is not necessarily required in each case. Early studies showed that A3A has robust activity against long interspersed element-1 (LINE-1) and Alu retrotransposition in cell-based assays (10,13,25–30) (reviewed in refs. 31,32). Surprisingly, no DNA mutations that could be ascribed to A3A’s deaminase activity were detected in LINE-1 and other retrotransposon DNA sequences, suggesting that alternative mechanisms, independent of A3A’s catalytic activity, might be involved (10,26,27). A deaminase-independent mechanism was also indicated for A3A-mediated inhibition of parvovirus replication (10,33). However, A3A’s editing function was found to be required for its ability to mutate human papilloma virus DNA (34), to degrade foreign DNA in human cells (12,15) and to block replication of Rous sarcoma virus (35) and human T-lymphotropic virus type 1 (HTLV-1) (36).
Initially it was reported that A3A does not inhibit HIV-1 infectivity in single-cycle and replication assays in T cells and other established cell lines (10,11,28,37–39). However, several groups have provided evidence that A3A inhibits HIV-1 replication in cells of myeloid origin (e.g. monocyte-derived macrophages) (20,40,41), where its expression is stimulated by addition of interferon-alpha (10,20–22,42). Moreover, A3A-specific editing of HIV-1 minus-strand DNA during reverse transcription was found to occur in infected myeloid cells (40), suggesting a role for A3A deaminase activity in HIV-1 restriction.
A3A appears to play an important role as a cellular defense protein, but its DNA mutator activity could also be detrimental to the cell in which it is expressed. Moreover, as A3A localizes to the cytoplasm and to the nucleus in certain cell lines, it has access to genomic DNA (10,26,27,38,43). Studies using cell-based assays showed that A3A has the potential to mutate genomic and mitochondrial DNA (15,44,45). In addition, cellular expression of A3A was shown to induce strand breaks, trigger the DNA damage response and cause cell cycle arrest (44,46). Thus, it seems likely that cellular regulatory mechanisms may be in place to modulate A3A function and prevent these harmful effects. The human TRIB3 protein was shown to lower the levels of genomic DNA editing by A3A (47). More recently, however, it was reported that in myeloid cells, A3A is found exclusively in the cytoplasm, suggesting that cytoplasmic retention in these cells prevents damage to nuclear DNA (48).
To elucidate the molecular mechanisms that govern the biological functions of A3A, we recently solved the A3A solution structure at high resolution using NMR spectroscopy (16). The overall structure of A3A was found to be similar to structures described for A3C (49) and for the C-terminal domain of A3G (A3G-CD2) (50–54) (reviewed in ref. 55), despite some differences, mainly in the loop regions. We also defined the surface for A3A interaction with short ssDNA substrates (≥15 nt) and performed a detailed analysis of substrate binding and specificity. Our results showed that the binding and deamination activity of A3A with ssDNA oligonucleotides containing TTCA and ACCCA motifs are similar, consistent with the A3A editing site preference in cell-based assays, i.e. predominantly in TC- and CC-containing regions (12,15,34,36).
Here, we report the biochemical and biological properties of A3A, taking advantage of the structural information in our earlier study (16). Initially, we probed the interaction of A3A with a 9-nt ssRNA by NMR. In contrast to previous results obtained with a 9-nt ssDNA of the same sequence (16), we found that ssRNA is not a substrate for A3A deaminase activity and is bound more weakly to the enzyme. Using structure-guided mutagenesis, point mutations were introduced into residues identified as important for specific binding to ssDNA substrates and the effect on deaminase activity was measured. In addition, longer ssDNAs (≥20 nt) were used to determine the oligonucleotide length dependence for ssDNA deamination and nucleic acid binding. Finally, we explored the role of deaminase and nucleic acid binding properties of A3A in relation to its biological activities, i.e. inhibition of LINE-1 retrotransposition and HIV-1 reverse transcription as well as genomic DNA editing, which can lead to tumor formation.
MATERIALS AND METHODS
Materials
Unlabeled DNA oligonucleotides used for electrophoretic mobility shift assays (EMSA), deaminase assays and cloning were obtained from Lofstrand Labs Ltd (Gaithersburg, MD, USA). All RNA oligonucleotides and DNA oligonucleotides labeled with AlexaFluor 488® were purchased from Integrated DNA Technologies (Coralville, IA, USA). Oligonucleotides used for deaminase assays and EMSA were either gel-purifed or were subjected to purification using high-pressure liquid chromatography (HPLC) and are listed in Table 1. Oligonucleotide concentrations were determined by absorbance at 260 nm, using the extinction coefficient provided by the manufacturer. [γ-33P]ATP and [γ-32P]ATP (each 3000 Ci/mmol) were purchased from PerkinElmer (Waltham, MA, USA). HIV-1 reverse transcriptase (RT) was obtained from Worthington Biochemical Corp (Lakewood, NJ, USA). T4 polynucleotide kinase and Escherichia coli (E. coli) uracil DNA glycosylase (UDG) were purchased from New England Biolabs (Beverly, MA, USA). Nuclease-free water, SUPERaseIn, Proteinase K and gel loading buffer were obtained from Ambion® (Life Technnologies Corp., Grand Island, NY, USA). Rabbit anti-A3G antibody (ApoC17) (56), a highly cross-reactive antiserum used for detection of A3A proteins, was a generous gift from Dr. Klaus Strebel (NIAID, NIH). Anti-tubulin antibody was obtained from Abcam (Cambridge, MA, USA). HIV-1 nucleocapsid protein (NC) was kindly provided by Dr. Robert J. Gorelick (SAIC-Frederick, Inc.). T4 Gene 32 and E. coli SSB proteins were purchased from Affymetrix, Inc. (Santa Clara, CA, USA).
Table 1.
List of oligonucleotides used for EMSA and deaminase assays
| Name | Purpose | Length (nt) | Sequencea |
|---|---|---|---|
| JL-895 | EMSA, Deamination | 40 | 5′-ATT ATT ATT ATT ATT ATT ATT TCA TTT ATT TAT TTA TTT A-3′ |
| JL-913 | Deamination | 40 | 5′-(Alexa488) ATT ATT ATT ATT ATT ATT ATT TCA TTT ATT TAT TTA TTT A-3′ |
| JL-935 | EMSA, Deamination | 40 | 5′-ATT ATT ATT ATT ATT ATT ATT TUA TTT ATT TAT TTA TTT A-3′ |
| JL-974 | EMSA | 30 | 5′-TTA TTA TTA TTA TTT CAT TTA TTT ATT TAT-3′ |
| JL-975 | EMSA | 20 | 5′-ATT ATT ATT TCA TTT ATT TA-3′ |
| JL-984 | EMSA, Deamination | 40 | 5′-TAA ATA AAT AAA TAA ATG AAA TAA TAA TAA TAA TAA TAA T-3′ |
| JL-986 | EMSA, Deamination | 60 | 5′-ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT TCA TTT ATT TAT TTA TTT ATT TAT TTA-3′ |
| JL-987 | EMSA | 70 | 5′-ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT TCA TTT ATT TAT TTA TTT ATT TAT TTA TTT A-3′ |
| JL-988 | EMSA, Deamination | 80 | 5′-ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT TCA TTT ATT TAT TTA TTT ATT TAT TTA TTT ATT TA-3′ |
| JL1152 | Deamination | 20 | 5′-(Alexa488) ATT ATT ATT TCA TTT ATT TA-3′ |
| JL1153 | Deamination | 60 | 5′-(Alexa488) ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT ATT TCA TTT ATT TAT TTA TTT ATT TAT TTA-3′ |
| JL-1045 | EMSA | 40 | 5′-auu auu auu auu auu auu auu uca uuu auu uau uua uuu a-3′ |
| JL-970 | EMSA | 40 | 5′-uaa aua aau aaa uaa aug aaa uaa uaa uaa uaa uaa uaa u-3′ |
| JL1181 | Deamination | 9 | 5′-auu uca uuu-3′ |
| JL-1088 | Deamination | 40 | 5′-TAA ATA AAT AAA TAA ATC AAA TAA TAA TAA TAA TAA TAA T-3′ |
| JL-1089 | Deamination | 40 | 5′-TAA ATA AAT AAA TAA AAC TAA TAA TAA TAA TAA TAA TAA T-3′ |
| JL-1090 | Deamination | 40 | 5′-TAA ATA AAT AAA TAA TAC TTA TAA TAA TAA TAA TAA TAA T-3′ |
| JL-1091 | Deamination | 40 | 5′-TAA ATA AAT AAA TTT TAC TTT AAA TAA TAA TAA TAA TAA T-3′ |
| JL-1095 | Deamination | 40 | 5′-(Alexa488) ATT ATT ATT ATT ATT ATT ATA CCC AAA ATT TAT TTA TTT A-3′ |
aUpper and lower case letters represent DNA and RNA oligonucleotides, respectively.
A3A expression in E. coli and purification
Synthetic A3A genes with a C-terminal His6-tag (LEHHHHHH) were inserted into the NdeI–XhoI site of the pET21 plasmid (Novagen) for expression in E. coli Rosetta 2 (DE3) and purified as described previously (16). Recombinant proteins were estimated to be >95% pure, as determined by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and 2D 1H-15N heteronuclear single quantum coherence (HSQC) spectroscopy (16). The protein concentration was determined using the BCA protein assay kit (Pierce, Thermo Fisher Scientific Inc., Rockland, IL, USA) and verified by amino acid analysis of an acid hydrolysate (Keck Biotechnology Resource Laboratory, Yale University, New Haven, CT, USA). To check for nuclease contamination in each A3A preparation, different amounts of A3A protein were incubated with 32P-labeled DNA or RNA oligonucleotides. Analysis by PAGE indicated that the A3A preparations were virtually nuclease-free (data not shown). The E72Q, Y130F and D131E mutant proteins were constructed, expressed and purified as described above (16). These proteins were shown to exhibit a wild-type (WT) fold by 1H-15N HSQC NMR spectroscopy (Supplementary Figure S1).
Real-time NMR studies of ssRNA deamination
A3A (final concentration, 0.07 mM) was added to a HPLC -purified 9-nt ssRNA oligonucleotide (1.12 mM), AUUUCAUUU (JL1181, Table 1), in 25 mM sodium phosphate buffer (pH 6.9) containing 10 mM DTT. Deamination was monitored by acquiring a time series of 2D 1H-13C HSQC spectra at 900 MHz, 25°C (16).
Mapping the A3A ssRNA binding site and titration by NMR
To monitor ssRNA binding to A3A, aliquots of a 20 mM AUUUCAUUU stock solution were added to 0.074 mM 13C/15N-labeled A3A in 25 mM sodium phosphate buffer (pH 6.9) containing 10 mM DTT. A series of 2D 1H-15N HSQC spectra were acquired at 900 MHz, 25°C. 1H,15N-combined chemical shift changes were calculated using  , with ΔδHN and ΔδN, the 1HN and 15N chemical shift differences observed for A3A before and after adding nucleotides (∼90% saturation). The residues showing large (>0.05 p.p.m) 1H,15N-combined chemical shift changes were mapped on the A3A NMR structure (16). Binding isotherms were obtained by plotting 1HN proton chemical shift changes observed for F54, W98, S103 and W104 versus nucleotide concentration. Dissociation constants (Kd) were calculated by non-linear best fit of the data using KaleidaGraph (Synergy Software, Reading, PA, USA), with subsequent averaging of the Kd values of all four amino acid resonances. All of the structure figures were generated using MOLMOL (57).
, with ΔδHN and ΔδN, the 1HN and 15N chemical shift differences observed for A3A before and after adding nucleotides (∼90% saturation). The residues showing large (>0.05 p.p.m) 1H,15N-combined chemical shift changes were mapped on the A3A NMR structure (16). Binding isotherms were obtained by plotting 1HN proton chemical shift changes observed for F54, W98, S103 and W104 versus nucleotide concentration. Dissociation constants (Kd) were calculated by non-linear best fit of the data using KaleidaGraph (Synergy Software, Reading, PA, USA), with subsequent averaging of the Kd values of all four amino acid resonances. All of the structure figures were generated using MOLMOL (57).
Construction of A3A mutants for cell-based assays
The A3A coding sequence (Genbank Accession # NM_145699) was inserted into a pCDNA 3.1(−) vector (Invitrogen, Life Technologies Corp.) for expression in mammalian cells. All of the A3A mutants were constructed using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). The primers for mutant construction were designed using the online QuikChange Primer Design Program provided by the manufacturer. Large-scale plasmid preparation was performed using the HiSpeed Plasmid Maxi Prep kit (Qiagen Inc., Germantown, MD, USA). For each plasmid, the sequence of the insert was verified by DNA sequencing performed by ACGT, Inc. (Wheeling, IL, USA).
Cell culture, transfection procedure and preparation of mammalian cell extracts
The 293T cells were propagated in Dulbecco’s modified Eagle’s medium (Invitrogen) containing 10% fetal bovine serum (Hyclone, now Thermo Scientific), 2 mM glutamine, penicillin (50 IU/ml) and streptomycin (50 µg/ml). The cells (∼2.4 × 105/well in a 12-well plate) were transfected with 0.75 μg of either empty pCDNA 3.1(−) vector or an A3A-containing plasmid (WT or mutant), using GenJet™ In Vitro DNA Transfection Reagent (SignaGen® Laboratories, Gaithersburg, MD, USA) according to the manufacturer’s instructions. Following incubation for 48 h at 37°C in an incubator with 5% CO2, the cells were harvested and lysed using M-PER mammalian protein extraction reagent (Thermo Fisher Scientific Inc., Rockford, IL, USA), as described in the manufacturer’s manual, in the presence of Complete Protease Inhibitor Cocktail (Roche Diagnostics Corp., Indianapolis, IN, USA). After cell lysis, NaCl (final concentration, 100 mM) and glycerol (final concentration, 10% vol/vol) were added and the resulting mixture was centrifuged at 13 000 g for 10 min. The total concentration of protein in the supernatant was determined using the BCA protein assay kit. To check A3A expression levels in each sample, ∼20 μg of total protein was subjected to western blot analysis, using the Western Breeze® chemiluminescent Western blot immunodetection kit (Invitrogen) and either anti-A3G (for A3A expression) or anti-tubulin (loading control) as primary antibodies.
Deaminase assays
Deaminase assays were performed using a protocol adapted from the procedure described by Iwatani et al. (58). The substrate was a 40-nt Alexa-Fluor 488-labeled TTCA-containing ssDNA (JL913) (Table 1), unless specified otherwise. Activity of cell extracts was evaluated in reactions (final volume, 40 µl) containing the ssDNA substrate (final concentration, 180 nM), reaction buffer [10 mM Tris–HCl buffer (pH 8.0), 50 mM NaCl, 1 mM dithiothreitol (DTT), 1 mM EDTA and 4 U of UDG] and increasing amounts of extract (total protein expressed in μg), as specified; incubation was for 1 h at 37°C. Reactions were terminated by further incubation at 37°C with 10 µl of 1 N NaOH for 15 min, followed by neutralization with 10 µl of 1 N HCl. Analysis was performed by electrophoresis on a 10% denaturing polyacrylamide gel as detailed in Byeon et al. (16). Gels were scanned in fluorescence mode on a Typhoon 9400 Imager (GE Healthcare) and the data were quantified using ImageQuant software. The values for percent deaminase product formed in each reaction were calculated by dividing the product signal intensity by the total signal intensity in the lane and multiplying by 100. The average of two independent experiments is shown in the figures.
Deaminase activity of purified A3A using a UDG-dependent gel-based assay was measured as described previously (16). The data are the average values from three independent experiments. The catalytic C in TTCA and CCCA motifs is underlined throughout the text.
EMSA
EMSA assays were performed with ss and double-stranded (ds) oligonucleotides (Table 1), using the protocol provided in Byeon et al. (16). Radioactivity was quantified by using the Typhoon 9400 Imager and ImageQuant software. The values for percent A3A-bound nucleic acid were calculated by dividing the intensity of the shifted band (A3A-bound nucleic acid) by the total intensity in the lane (the sum of the intensities of the shifted band, the free nucleic acid band and the area between the shifted and free nucleic acid bands). The % A3A-bound nucleic acid values of at least three independent experiments were averaged (standard deviation 5–10%).
Retrotransposition assay
The assay uses a vector that is similar to the replication-dependent LINE-1 vector originally designed by D. Garfinkel, T. Heidmann and J. Moran (59–61). The plasmid used in this study was constructed as follows: (i) a firefly luciferase (luc) expression cassette driven by a cytomegalovirus immediate early promoter was embedded in the 3′UTR of the LINE-1 transcription unit such that the luc cassette and LINE-1 are transcribed in opposite orientations; and (ii) a rabbit γ-globin intron was inserted into the luc sequence in the sense orientation of the LINE-1 transcript, thereby disrupting the luc open reading frame on the plasmid construct and preventing expression of luciferase from the transfected plasmid. After transfection into 293T cells, the LINE-1 sequence is transcribed, the intron is removed by splicing and the transcript is reverse transcribed and integrated. As a consequence, the now uninterrupted luc gene directs the synthesis of firefly luciferase, whose activity serves as a measure of the retrotransposition events that occurred.
For the retrotransposition assay, a constant amount of the LINE-1 plasmid (0.75 μg) was transfected into 293T cells (see above) with 0.1 or 0.5 µg of pCDNA 3.1(−) plasmids encoding A3A WT or mutant proteins in 12-well plates. The total amount of pCDNA 3.1(−) plasmid was set at 0.75 µg for each transfection by adding either 0.65 or 0.25 µg of empty vector, respectively. For controls without A3A, 0.75 µg of empty vector was used. Three days after transfection, the cells were lysed and luciferase activity was determined using the Bright-Glo™ luciferase assay system (Promega Corp., Madison, WI, USA), according to the manufacturer’s instructions, 100% luciferase activity corresponds to no inhibition, whereas 0% activity signifies complete inhibition of retrotransposition. Results for retrotransposition assays shown in the figures represent the average of two or three independent experiments.
HIV-1 minus-strand transfer assay
Assay of the minus-strand transfer step in reverse transcription (62–64) was performed using the reaction conditions described previously (65,66), except that HIV-1 NC was not added. Briefly, acceptor RNA (148 nt) was heat annealed to 33P-labeled (−) strong-stop DNA [(−) SSDNA] (128 nt) in buffer containing 50 mM Tris–HCl (pH 8.0) and 75 mM KCl. The annealed duplex contains a 54-nt 5′ ssRNA overhang, which serves as the template for extension of the annealed (−) SSDNA [see diagram in Figure 1C in (66)]. Varying amounts of purified A3A (WT or E72Q mutant), as specified, were added and the mixture was incubated at 37°C for 10 min, after which HIV-1 RT, dNTPs and MgCl2 (final concentration, 1 mM) were added with further incubation at 37°C for 1 h. Termination of the reactions, electrophoresis in denaturing gels and PhosphorImager analysis were performed as described (67).
Figure 1.

(A) Sequence alignment of A3A (residues 1–199) with A3G-CD2 (residues 194–384). The Zn-binding residues and the catalytic glutamic acid residue are highlighted in blue and the loop 7 region (residues 127–135) of A3A is marked. The A3A residues mutated in this study are labeled in green asterisks. The sequence alignment was performed using Lasergene software (DNASTAR, Inc., Madison, WI, USA). (B) and (C) A3A ribbon structures with residues that were mutated and tested (Figures 3B, 5 and 6C) highlighted in green. The coordinated Zn2+ ion is shown as a brown ball.
RESULTS
NMR analysis to assess ssRNA deamination and binding to A3A
Human A3 proteins are thought to use ssDNA as the exclusive substrate for deaminase activity. The inability to act on ssRNA has been shown experimentally for purified A3G (58) and in early work, Madsen et al. (24) reported that A3A (known as phorbolin-1 at that time) could not deaminate a mononucleoside or apolipoprotein B cRNA. In our recent study of the A3A NMR solution structure (16) (Figure 1), we followed deamination of ssDNA using a real-time NMR assay. Here, we adopted a similar approach to determine whether A3A can deaminate a 9-nt ssRNA with the sequence AUUUCAUUU (Table 1, JL1181) (Figure 2A). The intensity of the 1H-13C HSQC resonance from the only cytosine in the ssRNA sample (1.12 mM), which resonates at 5.945 p.p.m. (1H) and 98.84 p.p.m. (13C), did not decrease after addition of A3A (0.07 mM) and incubation for 1.5 h (left) and 9.6 h (right). This result is in stark contrast to our previous observation with the corresponding 9-nt ssDNA, ATTTCATTT, which was completely deaminated within 1 min under similar conditions (16). Although unlikely, secondary structure formation could render the cytosine inaccessible to the enzyme; we evaluated this possibility by 1H 1D NMR. No imino resonances compatible with base pairing in the RNA oligonucleotide were detected, confirming the ss nature of the RNA.
Figure 2.

Deamination and binding assays with A3A and a 9-nt ssRNA. (A) Deamination of the single cytosine in the ssRNA oligonucleotide, AUUUCAUUU (JL1181, Table 1), was monitored by following the 13C-5-1H resonances of cytosine and uracil in 2D 1H-13C HSQC spectra. The total reaction time is indicated at the top right. Binding isotherms for representative HN resonances (B) and mapping of the binding site onto the A3A structure (C) for ssRNA (AUUUCAUUU) interacting with A3A, monitored by 1H-15N HSQC spectroscopy. (D) Binding site of ssDNA ATTTCATTT mapped onto the A3A structure (16) for comparison. In (C) and (D), A3A residues whose resonances exhibit large 1H,15N-combined chemical shift changes on nucleotide addition are colored red (> 0.05 p.p.m.) and orange (0.028–0.050 p.p.m.).
1H-15N HSQC spectra were used to study binding of the 9-nt ssRNA to A3A. Figure 2B shows titration isotherms obtained by monitoring changes of amide resonances for several representative A3A residues that are perturbed by the ssRNA binding. Similar titration isotherms were observed for these resonances, yielding a Kd value of 0.42 ± 0.07 mM. This indicates that RNA binding is specific, albeit approximately one order of magnitude weaker than binding of the corresponding 9-nt DNA (Kd = 0.06 ± 0.01 mM) (16). In gel-shift assays with a 40-nt ssRNA, the binding efficiency appears to be ∼3-fold higher for ssRNA than for the corresponding 40-nt ssDNA (16). This result is most likely caused by both specific and non-specific binding to a long RNA as well as differences in the EMSA and NMR conditions (e.g. differences in oligonucleotide length).
The RNA binding site (Figure 2C) was mapped onto the A3A NMR structure (PDB code 2m65) and compared with the previously determined DNA binding site (Figure 2D) (16). Although the binding interfaces are similar, interesting differences were observed. Clearly, the number of residues that are located on the binding surface when A3A is bound to RNA is smaller than when it is bound to DNA. Most noticeably, the K30, N57 and Q58 amide resonances exhibited little perturbation on RNA binding, whereas they experienced large changes on DNA binding. This is significant as these amino acids reside at the bottom of the deep binding pocket that accommodates the reactive cytosine base in ssDNA (16). Thus, the data suggest that a cytosine ring in an RNA substrate cannot be positioned accurately in the catalytic site for deamination.
Differential role of A3A amino acids in nucleic acid binding and deaminase activity
The NMR study allowed us to identify A3A residues that are affected by specific binding of short ssDNA substrates containing a single TTCA deaminase recognition site (16). These residues include amino acids that may play a direct role in deamination and those that may interact with the neighboring nucleotides on both sides of the reactive dC (TTCA). Non-catalytic residues that exhibit the largest chemical shift perturbations include W104, G105, L135 and E138. The two-residue WG sequence is unique to A3A and the C-terminal domain of A3B. In addition, the G65 and F66 residues, located at the center of a flexible loop (loop 3, residues 57–70) that undergoes conformational exchange in free A3A, also show large chemical shift perturbations on binding of ssDNA substrates (16).
To determine whether any of these residues (Figure 1A and B) are important for A3A catalytic function, we performed alanine scanning mutagenesis and tested the mutant cell lysates in deaminase assays (Figure 3). All mutant proteins, except G105A, exhibited deaminase activity, albeit to varying degrees (Figure 3A). The protein expression levels of all variants were similar (Figure 3B). G105A lacked activity, even when high amounts of protein were present in the reaction. These results suggest that although all of the residues are affected by specific binding to ssDNA substrates, not surprisingly, the individual contributions to A3A’s deaminase activity are not equivalent. Data on other A3A residues that interact with nucleic acids are presented below.
Figure 3.

Deaminase activity of A3A mutants. Residues were selected for mutagenesis based on our earlier NMR results (16). (A) Deaminase activity. (B) Expression of A3A WT and mutants in 293T cells detected by western blot analysis, as described in ‘Materials and Methods’.
A3A binds and deaminates ssDNA oligonucleotides in a length-dependent manner
As we previously mapped the nucleic acid binding sites for short (≤15 nt) ssDNA substrates (16), it was of interest to further investigate A3A’s binding properties by using longer ss and ds nucleic acids (≥20 nt). Initially, we determined whether A3A binds to ssDNA in a length-dependent manner (Figure 4A). Gel analysis (EMSA) showed that the amount of bound ssDNA increased with increasing oligonucleotide length from 20 to 80 nt. For oligonucleotides ≥60 nt, at the highest protein concentration used (100 µM), 73–82% of the input DNA was complexed to protein. Like A3G (58,68), A3A interacts poorly with ds nucleic acids with a rank order of binding of dsDNA>DNA-RNA∼dsRNA (Supplementary Figure S2).
Figure 4.

A3A EMSA and deaminase assays with ssDNAs of different lengths. (A) Binding of A3A to TTCA-containing DNA oligonucleotides of varying lengths (20 nM) (Table 1): 80 nt (JL988); 70 nt (JL987); 60 nt (JL986); 40 nt (JL895); 30 nt (JL974); and 20 nt (JL975). Lanes: 1, 6, 11, 16, 21, 26, no A3A; 2, 7, 12, 17, 22, 27, 25 µM; 3, 8, 13, 18, 23, 28, 50 µM; 4, 9, 14, 19, 24, 29, 75 µM; 5, 10, 15, 20, 25, 30, 100 µM. The percentage of oligonucleotide bound to A3A is indicated at the bottom of the gel image as percent shifted. The positions of free oligonucleotide and shifted bands (A3A-bound nucleic acid) are also indicated. The calculations were performed as described in ‘Materials and Methods’. (B) Deaminase assays using 100 nM and 200 nM purified WT A3A and ssDNAs with different lengths (180 nM) (Table 1): JL1152 (20 nt, white bar), JL913 (40 nt, black bar) and JL1153 (60 nt, gray bar). (C) Binding of an 80-nt TTCA-containing ssDNA (JL988) by 60 μM (lanes 2, 4, 6, 8 and 10) or 80 μM (lanes 3, 5, 7, 9 and 11) A3A. Lanes: 1 and 12, no A3A; 2, 3, 6 and 7, WT A3A; 4 and 5, E72Q; 8 and 9, Y130F; 10 and 11, D131E. The positions of free and protein-bound DNA and the percent shifted are indicated.
To determine whether substrate length influences enzymatic activity, A3A was incubated with 20-, 40- and 60-nt ssDNA substrates (Figure 4B). There was no significant difference in the amount of deaminase product when 200 nM A3A was present in the reaction. However, with 100 nM A3A, activity was highest with the 20-nt ssDNA and was reduced by ∼20 and ∼25% when the 40- and 60-nt substrates were used, respectively, presumably because binding to the larger substrates was not saturated with the lower amount of A3A. Interestingly, in NMR experiments with ssDNAs that were 6, 9 or 15 nt in length, the apparent second-order rate constants (kcat/KM) for the deaminase activity of the 9- and 15-nt substrates were identical. Moreover, these rate constants were ∼3-fold higher than the value obtained with the 6-mer (16), mainly due to the tighter binding of the larger ssDNAs (smaller KM). This suggests that the minimum substrate length for optimal deaminase activity is between 6 and 9 nt.
Relationship between A3A deaminase activity and inhibition of LINE-1 retrotransposition
An important aspect of this study is to relate our findings on A3A’s binding and deaminase properties to its biological functions. We first consider this issue in regard to LINE-1 retrotransposition. Although A3A has been reported to be a potent inhibitor of LINE-1 retrotransposition in cell-based assays (reviewed in refs. 31,32), the underlying mechanism is still unclear and questions remain as to whether A3A deaminase activity is necessary for this activity. To investigate this issue, we changed A3A residues that might be important for deamination and/or inhibition of LINE-1 activity. A3A residues that were mutated are highlighted with green asterisks in Figure 1A and are marked on the A3A NMR structure in Figure 1C. Deaminase assays were performed with extracts from 293T cells that were transfected with vectors carrying the A3A genes (WT or mutants). The amounts of expressed protein for WT and all of the A3A mutants were similar (Figures 5C and 6C and F). No A3A was present in cells with the empty vector (Figure 5C) and the extract was, therefore, devoid of deaminase activity (Figure 6D).
Figure 5.
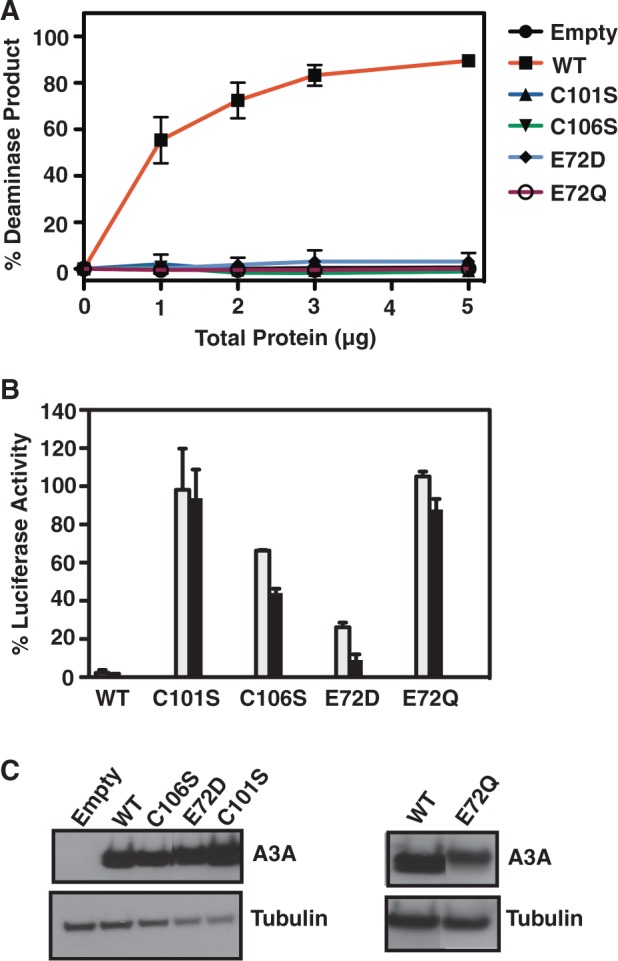
Deaminase and retrotransposition activities of A3A WT and active site mutants. (A) Deaminase assays using 293T cell extracts expressing WT and active site mutants of A3A. (B) Retrotransposition assays to monitor the effect of active site mutations of A3A on LINE-1 activity. A description of the assays is given in ‘Materials and Methods’. The gray and black bars represent transfection of 0.1 and 0.5 μg of A3A WT or mutant plasmids, respectively. (C) Western blot analysis of A3A WT and active site mutant expression in 293T cells.
Figure 6.

Deaminase and retrotransposition activities of A3A WT and non-active site mutants. (A and D) Deaminase and (B and E) retrotransposition assays were performed as described in ‘Materials and Methods’. (C and F) Western blot analysis of A3A WT and non-active site mutant expression in 293T cells. In the Loop 7 mutant, A3A residues were changed to the corresponding residues in A3G: Y132D/D133Q/P134G/L135R (Figure 1).
As controls, the activities of A3A variants with changes in the highly conserved active site cysteine and glutamic acid residues were tested as well (Figure 5). Substitution of cysteines 101 and 106 by serine (C101S and C106S), which abolishes Zn binding, led to complete loss of deaminase activity, as expected (Figure 5A). Similarly, substitution of glutamic acid 72 by aspartic acid (E72D) or glutamine (E72Q) also resulted in abrogation of deaminase activity (Figure 5A). These results are in accord with previous mutational studies of active site residues (10,13).
Whether A3A deaminase activity is necessary for the inhibition of LINE-1 retrotransposition was also tested using these mutants (Figure 5B). As expected, WT A3A displayed potent anti-retrotransposition activity (∼2% luciferase activity), in agreement with earlier studies (10,13,25–30). The E72Q and C101S mutants had essentially no inhibitory activity; in contrast, the C106S mutant exhibited moderate anti-LINE-1 activity, whereas the E72D mutant strongly inhibited retrotransposition. The behavior of the E72D mutant contrasts with the lack of inhibitory activity displayed by other Glu mutants, E72Q (Figure 5B) (10) and E72A (13,27).
Deaminase and retrotransposition activities of non-active site mutants, including those in loop regions (Figure 1A and C), were also determined (Figure 6). Although A3 loop residues do not directly participate in catalysis, loop 7 residues in A3G-CD2 were shown to play a role in the preferential deamination of CCC-containing ssDNA (6,51,69,70). Consistent with these observations, mutation of A3A loop 7 residues (Y130, D131, Y132), either singly or in pairs, completely abolished deaminase activity (Figure 6A). Moreover, exchanging the entire A3A loop 7 sequence for that of A3G-CD2 (Figure 1A) (‘Loop 7’ mutant) caused complete loss of deaminase activity (Figure 6A). Interestingly, although P134 is part of a surface that interacts with the 5′ T in the TTCA recognition motif, the P134A loop 7 mutation had only a small effect on deamination (Figure 6D). In addition to P134A, other non-catalytic site mutants (R29A, I26G, R191D and F102) also retained activity with ssDNA substrates, albeit at different levels (Figure 6D). This behavior reflects their different locations in the A3A protein structure, which could influence the mode of interaction with nucleic acids.
With the exception of D131E, which displayed a high level of anti-LINE-1 activity, all of the other loop 7 mutants (Figure 6A) were able to only weakly inhibit retrotransposition (Figure 6B). The dual loss of activity for most loop 7 mutants may be related to the possible loss of the structural integrity of the mutant proteins. Several deaminase-active mutants displayed anti-retrotransposition activities that were as high as that of WT (F102A and P134A; Figure 6E) or had substantial inhibitory activity (R69A, I26G and R191D; Figure 6E). However, taken together, the data in Figures 5 and 6 indicate that in most cases, total loss of deaminase activity is not necessarily associated with an inability to inhibit LINE-1 retrotransposition.
Previous studies of A3A inhibition of retrotransposition (see above) suggested that a deaminase-independent mechanism might be involved. To further investigate this hypothesis, we performed retrotransposition assays using different amounts of WT and mutant A3A plasmids. Interestingly, the inhibitory activity increased when the amount of transfected DNA was increased (0.1 and 0.5 μg). The dose dependence was particularly striking for the E72D (Figure 5B), D131E and Y130F (Figure 6B) mutants that displayed no deaminase activity, even at high total protein concentrations (Figure 5A and 6A, respectively). This gain-in-function (enhanced anti-retrotransposition activity) for most of the deaminase-negative mutants suggests the possible existence of additional mechanism(s) that do not require A3A catalytic activity.
For example, deaminase-independent inhibition of LINE-1 retrotransposition by A3A might involve binding to ssDNA intermediate(s). If that were the case, we would expect that the level of anti-LINE-1 activity would be related to the efficiency of ssDNA binding. To probe this possibility, we evaluated binding to an 80-nt ssDNA (Figure 4C), using three A3A mutant proteins, E72Q, Y130F and D131E, which were all deaminase-negative (Figures 5A and 6A), but exhibiting different levels of anti-LINE-1 activity (Figures 5B and 6B). Surprisingly, all of the mutants displayed binding levels similar to that of WT, even though they had non-detectable (E72Q), modest (Y130F) or strong (D131E) anti-LINE-1 activities. Thus, it would appear there is no direct correlation between ssDNA binding activity and the ability to inhibit LINE-1 retrotransposition.
A3A does not block nucleotide incorporation by HIV-1 RT
Another biological function of A3A is its ability to inhibit HIV-1 replication in myleoid cells (20,40,41). A3A was shown to inhibit synthesis of viral DNA during HIV-1 reverse transcription, and it was suggested that this activity depends on A3A’s ability to edit viral ssDNA (40). In the case of A3G, in vitro assays (71–74) as well as assays in virus-infected cells (75–79) provided evidence that A3G can inhibit reverse transcription by deaminase-dependent and deaminase-independent mechanisms. Based on A3G’s biochemical properties, i.e. its slow dissociation from bound nucleic acid (71,74) and its ability to oligomerize (54,80–83), it was proposed that deaminase-independent inhibition could result from A3G binding to the ss template, causing a ‘roadblock’ to RT-catalyzed DNA elongation (63,71,74).
To investigate whether A3A can also interfere with RT movement along the template, we used a modified version of our reconstituted minus-strand transfer assay (65,66), as described in ‘Materials and Methods’. As shown in Figure 7, addition of 5 μM or even 10 μM A3A was not sufficient to reduce the levels of the extension product to any significant extent (compare with RT only reaction), independent of whether WT A3A or the E72Q deaminase-negative mutant was used. This indicates that A3A is unable to directly block RT-catalyzed DNA extension.
Figure 7.
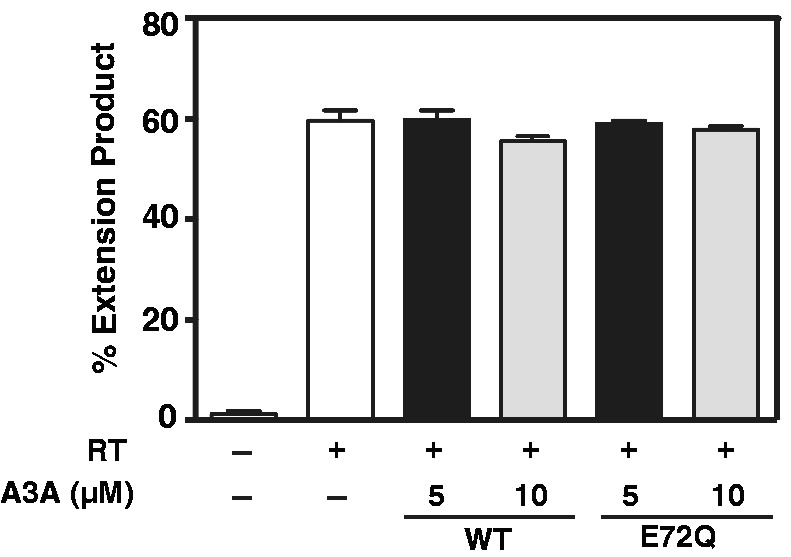
Effect of A3A on HIV-1 RT-catalyzed extension of a (−) SSDNA oligonucleotide. The bar graph shows the percent of DNA extension product without (−) or with (+) RT and/or A3A. The positive control reaction with RT only is shown as a white bar. Reactions with RT contain either 5 µM (black bar) or 10 µM (gray bar) A3A (WT and E72Q mutant).
A3A has the potential to mutate DNA during transcription and SSB proteins could mitigate this effect
As discussed above, A3A functions as a host restriction factor and plays a role in the host innate immune response. However, it appears to also have an opposing biological activity. Thus, its mutagenic potential may exert a detrimental effect on the cells where it is expressed, especially during cellular processes such as transcription, which require transient opening of the duplex (strand separation). To investigate this possibility, we designed short (40 nt) dsDNA substrates, containing unpaired regions at the center to mimic transcription bubbles (TBs) of different sizes [1 nt (TB-1), 3 nt (TB-3), 5 nt (TB-5) and 9 nt (TB-9)] (Figure 8A) and tested their susceptibility to deamination by WT A3A (Figure 8B).
Figure 8.

Deamination of dC in ss regions of a 40-bp DNA duplex and the effect of SSB proteins. (A) Schematic representation of a series of TBs in a ds nucleic acid. Unpaired bases are located in the center of a 40-bp DNA duplex that contains the TTCA sequence in the ss region of one strand. (B) Deaminase assay performed using duplexes (40 bp) containing TBs with different lengths of unpaired bases (1–9 nt). These duplexes were generated by heat annealing the ssDNA substrate (JL913) to oligonucleotides containing 1 nt (JL1088; TB-1), 3 (JL1089; TB-3), 5 (JL1090; TB-5) and 9 (JL1091; TB-9) that are not complementary to the corresponding residues in the other DNA strand. (C) Deaminase assay using the ssDNA substrate (JL913; 180 nM) after preincubation with SSB proteins (HIV-1 NC, T4 Gene 32 or E. coli SSB; each protein at 500 nM) for 15 min at 37°C before addition of A3A and incubation for 1 h. Bars: 1, 5 and 9, no proteins (negative control); 2, 6 and 10, A3A only (positive control); 3, 7, 11 and 4, 8, 12, A3A and ssDNA preincubated with 2.5 or 5 µM SSB protein, respectively.
As expected, a dsDNA lacking any unpaired bases (Duplex) was essentially not deaminated (∼3%), even at the highest concentration of A3A used (100 nM). A partially dsDNA that contained a single unpaired dC nucleotide (TB-1) exhibited somewhat higher levels of deamination (16 and 23% with 50 and 100 nM A3A, respectively) Sequential addition of unpaired nucleotides surrounding the central dC (TB-3 to TB-9) led to increasing amounts of deaminase products, up to ∼90% with TB-9 at 100 nM A3A. Moreover, deamination increased as the concentration of A3A was raised from 50 to 100 nM. The results indicate that access of A3A to the reactive dC in the transcription bubble becomes more facile with increasing ss character and the more flexible structure of the larger bubbles. These data clearly support the notion that A3A is capable of mutating genomic DNA during transcription, in agreement with previous studies by Love et al. (14) using a cell-based transcription system.
The data presented in Figure 8B raise an important question: is there a cellular mechanism that could prevent possible A3A-induced damage to genomic DNA? One possibility is that SSB proteins, which coat ssDNA regions of genomic DNA in the nucleus and are involved in maintenance of genome stability (84), block A3A access to target sequences on unpaired DNAs. Readily available SSB proteins including HIV-1 NC, T4 Gene 32 and E. coli SSB were used to model this scenario in vitro. Deaminase assays were performed after preincubating the ssDNA substrate with different concentrations of these SSB proteins. Negative controls (no protein) and positive controls (A3A only) provided basal and maximal activity for the final experimental conditions (Figure 8C). The maximal activity of A3A (positive control, Lanes 2, 6 and 10) remained essentially the same under different buffer conditions. When ssDNA was bound to HIV-1 NC, no change in deamination was observed, whereas T4 Gene 32 or E. coli SSB severely reduced A3A’s ability to deaminate the ssDNA substrate. These results most likely reflect rapid on/off nucleic acid binding kinetics of NC (71,85) as opposed to the slow dissociation of the other two SSB proteins from ssDNA (86), which would block access of A3A to the DNA substrate.
DISCUSSION
In the present work, we report biochemical and mutagenesis studies designed to elucidate the molecular properties of A3A and obtain mechanistic insights into A3A function in light of the recently determined A3A NMR structure (16). Although it is well known that A3A deaminates ssDNA substrates, it was of interest to evaluate, from a structural perspective, whether A3A could also use an ssRNA substrate. Within the human APOBEC family, A1 can deaminate RNA, as required for tissue-specific editing of apolipoprotein B mRNA (87) and activation-induced deaminase (AID), a known ssDNA deaminase that generates antibody diversity (9), has been implicated in C to U editing of HBV nucleocapsid RNA (88). In contrast, A3G is not able to deaminate ssRNA (58).
Here, we directly demonstrate by NMR that A3A is incapable of deaminating RNA (Figure 2A), in agreement with an earlier observation (24). In addition, it appears from mapping the A3A-RNA binding interface (Figure 2C) that the cytosine ring in a 9-nt RNA does not permit optimal positioning for catalysis. Some of the interactions that were observed with a 9-nt ssDNA (having the same sequence as the 9-nt RNA) (16) are absent, suggesting a less intimate interaction between A3A and ssRNA (Figure 2C) compared with its interaction with ssDNA (Figure 2D). While this article was under review, Nabel et al. published results showing that differential puckering of the sugar moiety in ssDNA and ssRNA substrates influences selective deamination of ssDNA by AID (89). Thus, it seems possible that the inability of A3A to accommodate the cytosine in ssRNA in the active site and the less intimate contact with ssRNA are related to the sugar pucker of the cytosine ribose ring.
We have extended the previous analysis on the interaction with short ssDNA oligonucleotides to include ssDNA substrates of varying lengths (up to 80 nt) and show that longer oligonucleotides interact more avidly with A3A (Figure 4A). This could be due to the greater contribution of non-specific interactions as the length of the oligonucleotide is increased, resulting in increased binding with the longer ssDNAs. NMR analysis demonstrated that although A3A binding to a 9-mer ssDNA is specific, binding to a 15-mer ssDNA involves both specific and non-specific interactions (16).
Comparison of the nucleic acid binding activities of A3A with A3G in EMSA studies illustrates an interesting difference between the properties of the two proteins. In contrast to one major band seen with A3A (Figure 4A), multiple product bands are observed in A3G gel-shift assays, likely due to formation of higher order complexes, even at an A3G concentration of 20 nM (58). These observations may be related to the fact that A3G multimerizes, especially in the presence of nucleic acids (54,80–83), whereas A3A is monomeric in solution, at least at concentrations <0.2 mM (14,16).
The relationship between ssDNA length and deaminase activity is also different for A3A and A3G. At an A3A concentration of 100 nM, deaminase activity is reduced as the length of the ssDNA substrate is increased (Figure 4B). Non-specific interactions of A3A with these longer ssDNAs at non-specific binding sites may contribute to this inhibitory effect. In addition, the weak sliding and jumping activities of A3A on nucleic acid substrates (14) may interfere with its ability to locate the active site motif (TTCA) in the longer ssDNAs. At a higher A3A concentration (e.g. 200 nM), however, deaminase activity is independent of substrate length, as A3A activity should be saturated under conditions where the A3A:substrate concentration is ∼1. In contrast to A3A, A3G exhibits increasing deaminase activity as the length of CCC-containing ssDNAs is increased from 15 nt to 60 nt (90). This result presumably reflects the fact that A3G has two Zn-binding domains, one of which (the N-terminal domain) exhibits strong nucleic acid binding activity (58,91), whereas A3A is a single-domain protein.
Structure-guided mutagenesis, based not only on the A3A NMR structure but also on a model of the complex with the DNA target T−2T−1CA+1 (16), was used to create alanine mutants of several A3A residues that interact with either T−1 (P134), T−2 (W104 and E138) or both (F102 and L135). These mutants exhibit reduced deaminase activity, but the activity is not totally abolished (Figures 3A and 6A). Thus, individual amino acid changes may not be sufficient for disrupting interactions that are important for substrate positioning during catalysis. However, changing G105, which contacts T−2, to alanine, results in complete abrogation of catalytic activity (Figure 3A), either due to an unfavorable conformational change in the binding site or loss of the overall structural integrity of the protein. Interestingly, deletion of W104 and G105 results in only partial loss of A3A catalytic activity (17,33).
Studies of A3G-CD2 have shown that loop 7 (313-RIYDDQGR-320) (Figure 1A) is an important determinant of deaminase specificity (preference for CCCA motif) (6,51,69,70). In our A3A/ssDNA model (16), several residues (D131, Y132, D133, P134 and L135) that contact bases adjacent to the catalytic C, reside in loop 7 (127-ARIYDYDPL-135) (Figure 1A). Although changes of the last two amino acids in this loop (P134 and L135) have only small effects on deaminase activity (Figures 6D and 3A, respectively), substitutions of residues D131 and Y132 (Figure 6A) and D133 (13) lead to complete loss of catalytic function. This indicates that the contributions of individual loop 7 residues to A3A deaminase activity are not equivalent. Interestingly, replacing all A3G loop 7 residues with those found in A3A yields an active protein (92), whereas an A3A with an A3G loop 7 (Loop 7 mutant) exhibits complete loss of enzyme activity (Figure 6A). This is most likely explained by the fact that changing Y132 of A3A to the corresponding residue in A3G (D317) is detrimental to the deaminase activity of A3A (Y132D mutant, Figure 6A), although A3G with Y317 is active (92).
Mutation of the Zn-binding residues (H70R, C101S and C106S) and residues that directly contact the C in the TTCA motif (N57A, E72A, E72D and E72Q) abolishes A3A deaminase activity (Figure 5A) (10,13,33), either by destroying the structural integrity of the protein or by interfering with the correct positioning of the cytosine in the active site. Interestingly, changing the catalytic E72 to Q does not affect structural integrity (Supplementary Figure S1) (16) or ability to bind ssDNA [Figure 4C; (16)]; only deaminase activity is affected. Changes in the flexible loop 3 (F66A and G65A) only partially abrogate the activity of A3A, suggesting that the plasticity of this loop is sufficient to allow binding and/or catalysis.
A key question that arises in studies of A3 proteins is how deaminase activity is related to biological function. One area of interest is the role of deamination in A3A’s inhibition of LINE-1 retrotransposition. The data presented here (Figures 5 and 6) indicate that A3A mutations result in three different phenotypes. In some instances, LINE-1 restriction activity is present in the absence of deaminase activity. For example, E72D (Figure 5A and B) and D131E (Figure 6A and B) have no enzymatic activity, but exhibit close to WT levels of anti-LINE-1 activity. Other deaminase-negative mutants (e.g. Y130F and Y132D [Figure 6A and B]) still restrict LINE-1, but to a lesser extent; however, increased expression of A3A increases restriction activity. These findings are in accord with earlier studies showing that no A3A-induced deamination signatures could be detected in LINE-1 DNA (10,26,27).
We have also characterized A3A mutants with the following properties: (i) presence of both deaminase and anti-LINE-1 activities (e.g. F102A and P134A [Figure 6D and E]); and (ii) absence of both activities (e.g. C101S and E72Q [Figure 5A and B]). Some examples of mutants with phenotypes (i) and (ii) have also been reported by others (10,13,27). Importantly, these results strongly suggest that although the determinants of deaminase and anti-LINE-1 activities can overlap, they are not identical. Deaminase activity is sensitive to even small changes in the active site geometry and its immediate environment. However, such changes may not significantly affect the anti-LINE-1 activity, as in the case of the E72D and D131E mutants. Thus, taken together, our data support the conclusion that deaminase and LINE-1 restriction activities are not linked. We also considered the possibility that anti-LINE-1 activity might be related to the ability to bind ssDNA. However, the ssDNA binding experiments with A3A mutant proteins show that despite their different anti-LINE-1 activities, binding efficiency in each case was equivalent to that of WT (Figure 4C). Thus, under our conditions, the data suggest that inhibition of retrotransposition is not directly correlated with nucleic acid binding activity.
In contrast to LINE-1 retrotransposition, there is strong evidence that the deamination activity of A3A is required for blocking replication of human retroviruses such as HIV-1 and HTLV-1. A3A deamination signatures were observed in HIV-1 (14,40) and HTLV-1 DNA (36), and it appears that DNA editing is the primary mode of inhibition of these retroviruses. As A3A binds only weakly to nucleic acids (Figure 2 and 4) (16) and does not multimerize (14,16), it is not surprising that A3A fails to block movement of RT during DNA synthesis (Figure 7).
In addition to deamination of viral ssDNA, A3A has also been shown to edit cytosines present in genomic DNA (15,44,45) and in foreign DNA plasmids transfected into mammalian cells (12,15). How could an ssDNA deaminase like A3A mutate ds genomic or foreign DNA? Here we show that intact dsDNA clearly is not a good substrate for A3A (Figure 8B) (10) and that A3A acts only on ss regions present in otherwise ds nucleic acid (Figure 8B). This suggests that genomic and foreign DNA in cells would only be accessible to A3A during transient strand opening during processes such as transcription and DNA replication (93).
APOBEC protein-mediated genomic DNA mutations have been implicated in carcinogenesis (94–101) and a recent report has suggested a role for A3A in DNA break-associated mutations, which are associated with breast cancer (101). Therefore, one may pose the question: ‘how is A3A regulated inside a cell so as not to cause undesirable damage to genomic DNA?’ One scenario that is shown here is the possible involvement of SSB proteins, with strong ss nucleic acid binding affinity and slow on-off rates, that could block access to genomic DNA (Figure 8C). Interestingly, there is a precedent for the mechanism we suggest: in a recent report, it was shown that replication protein A, a nuclear SSB protein in yeast, strongly inhibits A3G deaminase activity as well as enzyme processivity (102).
Other mechanisms have also been proposed to explain how genomic DNA mutations by A3A might be prevented. One possibility involves reduction of A3A levels by TRIB 3 (47). However, this has been questioned in a recent study by Land et al. (48), where TRIB 3 was reported to be ineffective in preventing DNA damage by A3A. Interestingly, in the same study, localization of A3A within the cytoplasm of monocytic cells was shown to prevent genotoxicity, suggesting that these cells have developed robust mechanisms to protect genomic DNA integrity. However, these mechanisms might be compromised in pathological conditions such as cancer (103), and therefore need to be evaluated individually for different conditions and cells.
In summary, the present study provides new insights into the biological activities of human A3A by examination of its deaminase and nucleic acid binding properties in the context of its 3D structure. These properties define the nature of A3A’s interaction with its nucleic acid substrates and are critical for its role as a host restriction factor and a genomic DNA mutator. How cells regulate undesirable DNA editing by A3A and other human A3 proteins is a major question for future investigation.
NOTE ADDED IN PROOF
During the proof stage of this manuscript, we became aware of a paper by Pham et al., which also reported A3A deaminase activity on partially single-stranded DNA substrates (Pham,P., Landolph, A., Mendez, C., Li, N. and Goodman, M.F. A biochemical analysis linking APOBEC3A to disparate HIV-1 restriction and skin cancer. J. Biol. Chem. doi: 10.1074/jbc.M113.504175).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Intramural Research Program at the National Institutes of Health [Eunice Kennedy Shriver National Institute of Child Health and Human Development (to M.M., K.H., K.H-R., D.S., G.N. and J.G.L.) and the National Cancer Institute, HIV Drug Resistance Program and Center for Cancer Research (to S.H. and G.H.)]; and the National Institutes of Health [Grant P50GM082251 to J.A., I-L.B., C-H.B., L.M.C. and A.M.G.]. Funding for open access charge: Intramural Program of NIH/NICHD.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Tiyun Wu for valuable advice on the minus-strand transfer assay and for providing the RNA and DNA constructs, Klara Post for helpful advice on EMSA gels, Robert J. Gorelick for purified NC protein, Klaus Strebel for antiserum used to detect A3A, Jason Concel and Maria DeLucia for assistance in purification of recombinant A3A, Phil Greer and Doug Bevan for computer technical support and Michael J. Delk for NMR instrumental support.
REFERENCES
- 1.Harris RS, Liddament MT. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 2004;4:868–877. doi: 10.1038/nri1489. [DOI] [PubMed] [Google Scholar]
- 2.Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008;26:317–353. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- 3.Malim MH. APOBEC proteins and intrinsic resistance to HIV-1 infection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009;364:675–687. doi: 10.1098/rstb.2008.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duggal NK, Emerman M. Evolutionary conflicts between viruses and restriction factors shape immunity. Nat. Rev. Immunol. 2012;12:687–695. doi: 10.1038/nri3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 2007;32:118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Bransteitter R, Prochnow C, Chen XS. The current structural and functional understanding of APOBEC deaminases. Cell. Mol. Life Sci. 2009;66:3137–3147. doi: 10.1007/s00018-009-0070-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- 8.Betts L, Xiang S, Short SA, Wolfenden R, Carter CW., Jr Cytidine deaminase. The 2.3 Å crystal structure of an enzyme: transition-state analog complex. J. Mol. Biol. 1994;235:635–656. doi: 10.1006/jmbi.1994.1018. [DOI] [PubMed] [Google Scholar]
- 9.Jaszczur M, Bertram JG, Pham P, Scharff MD, Goodman MF. AID and Apobec3G haphazard deamination and mutational diversity. Cell. Mol. Life Sci. 2013;70:3089–3108. doi: 10.1007/s00018-012-1212-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 2006;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 11.Aguiar RS, Lovsin N, Tanuri A, Peterlin BM. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 2008;283:2518–2525. doi: 10.1074/jbc.M706436200. [DOI] [PubMed] [Google Scholar]
- 12.Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010;17:222–229. doi: 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulliard Y, Narvaiza I, Bertero A, Peddi S, Röhrig UF, Ortiz M, Zoete V, Castro-Diaz N, Turelli P, Telenti A, et al. Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J. Virol. 2011;85:1765–1776. doi: 10.1128/JVI.01651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Love RP, Xu H, Chelico L. Biochemical analysis of hypermutation by the deoxycytidine deaminase APOBEC3A. J. Biol. Chem. 2012;287:30812–30822. doi: 10.1074/jbc.M112.393181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinohara M, Io K, Shindo K, Matsui M, Sakamoto T, Tada K, Kobayashi M, Kadowaki N, Takaori-Kondo A. APOBEC3B can impair genomic stability by inducing base substitutions in genomic DNA in human cells. Sci. Rep. 2012;2:806. doi: 10.1038/srep00806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byeon I-J, Ahn J, Mitra M, Byeon C-H, Hercík K, Hritz J, Charlton LM, Levin JG, Gronenborn AM. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 2013;4:1890. doi: 10.1038/ncomms2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carpenter MA, Li M, Rathore A, Lackey L, Law EK, Land AM, Leonard B, Shandilya SMD, Bohn M-F, Schiffer CA, et al. Methylcytosine and normal cytosine deamination by the foreign DNA restriction enzyme APOBEC3A. J. Biol. Chem. 2012;287:34801–34808. doi: 10.1074/jbc.M112.385161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wijesinghe P, Bhagwat AS. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res. 2012;40:9206–9217. doi: 10.1093/nar/gks685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suspène R, Aynaud M-M, Vartanian J-P, Wain-Hobson S. Efficient deamination of 5-methylcytidine and 5-substituted cytidine residues in DNA by human APOBEC3A cytidine deaminase. PloS One. 2013;8:e63461. doi: 10.1371/journal.pone.0063461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peng G, Greenwell-Wild T, Nares S, Jin W, Lei KJ, Rangel ZG, Munson PJ, Wahl SM. Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood. 2007;110:393–400. doi: 10.1182/blood-2006-10-051763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koning FA, Newman ENC, Kim E-Y, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J. Virol. 2009;83:9474–9485. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38:4274–4284. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rasmussen HH, Celis JE. Evidence for an altered protein kinase C (PKC) signaling pathway in psoriasis. J. Invest. Dermatol. 1993;101:560–566. doi: 10.1111/1523-1747.ep12365986. [DOI] [PubMed] [Google Scholar]
- 24.Madsen P, Anant S, Rasmussen HH, Gromov P, Vorum H, Dumanski JP, Tommerup N, Collins JE, Wright CL, Dunham I, et al. Psoriasis upregulated phorbolin-1 shares structural but not functional similarity to the mRNA-editing protein apobec-1. J. Invest. Dermatol. 1999;113:162–169. doi: 10.1046/j.1523-1747.1999.00682.x. [DOI] [PubMed] [Google Scholar]
- 25.Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006;34:89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O'Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl Acad. Sci. USA. 2006;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muckenfuss H, Hamdorf M, Held U, Perković M, Löwer J, Cichutek K, Flory E, Schumann GG, Münk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 2006;281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- 28.Kinomoto M, Kanno T, Shimura M, Ishizaka Y, Kojima A, Kurata T, Sata T, Tokunaga K. All APOBEC3 family proteins differentially inhibit LINE-1 retrotransposition. Nucleic Acids Res. 2007;35:2955–2964. doi: 10.1093/nar/gkm181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niewiadomska AM, Tian C, Tan L, Wang T, Sarkis PTN, Yu X-F. Differential inhibition of long interspersed element 1 by APOBEC3 does not correlate with high-molecular-mass-complex formation or P-body association. J. Virol. 2007;81:9577–9583. doi: 10.1128/JVI.02800-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lovšin N, Peterlin BM. APOBEC3 proteins inhibit LINE-1 retrotransposition in the absence of ORF1p binding. Ann. N. Y. Acad. Sci. 2009;1178:268–275. doi: 10.1111/j.1749-6632.2009.05006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koito A, Ikeda T. Intrinsic restriction activity by AID/APOBEC family of enzymes against the mobility of retroelements. Mob. Genet. Elements. 2011;1:197–202. doi: 10.4161/mge.1.3.17430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koito A, Ikeda T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol. 2013;4:28. doi: 10.3389/fmicb.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Narvaiza I, Linfesty DC, Greener BN, Hakata Y, Pintel DJ, Logue E, Landau NR, Weitzman MD. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 2009;5:e1000439. doi: 10.1371/journal.ppat.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vartanian J-P, Guétard D, Henry M, Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science. 2008;320:230–233. doi: 10.1126/science.1153201. [DOI] [PubMed] [Google Scholar]
- 35.Wiegand HL, Cullen BR. Inhibition of alpharetrovirus replication by a range of human APOBEC3 proteins. J. Virol. 2007;81:13694–13699. doi: 10.1128/JVI.01646-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ooms M, Krikoni A, Kress AK, Simon V, Münk C. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 2012;86:6097–6108. doi: 10.1128/JVI.06570-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho S-J, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004;14:1392–1396. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 38.Goila-Gaur R, Khan MA, Miyagi E, Kao S, Strebel K. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology. 2007;4:61. doi: 10.1186/1742-4690-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hultquist JF, Lengyel JA, Refsland EW, LaRue RS, Lackey L, Brown WL, Harris RS. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011;85:11220–11234. doi: 10.1128/JVI.05238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berger G, Durand S, Fargier G, Nguyen X-N, Cordeil S, Bouaziz S, Muriaux D, Darlix J-L, Cimarelli A. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog. 2011;7:e1002221. doi: 10.1371/journal.ppat.1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koning FA, Goujon C, Bauby H, Malim MH. Target cell-mediated editing of HIV-1 cDNA by APOBEC3 proteins in human macrophages. J. Virol. 2011;85:13448–13452. doi: 10.1128/JVI.00775-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thielen BK, McNevin JP, McElrath MJ, Hunt BVS, Klein KC, Lingappa JR. Innate immune signaling induces high levels of TC-specific deaminase activity in primary monocyte-derived cells through expression of APOBEC3A isoforms. J. Biol. Chem. 2010;285:27753–27766. doi: 10.1074/jbc.M110.102822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lackey L, Law EK, Brown WL, Harris RS. Subcellular localization of the APOBEC3 proteins during mitosis and implications for genomic DNA deamination. Cell Cycle. 2013;12:762–772. doi: 10.4161/cc.23713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011;12:444–450. doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suspène R, Aynaud M-M, Guétard D, Henry M, Eckhoff G, Marchio A, Pineau P, Dejean A, Vartanian J-P, Wain-Hobson S. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc. Natl Acad. Sci. USA. 2011;108:4858–4863. doi: 10.1073/pnas.1009687108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mussil B, Suspène R, Aynaud M-M, Gauvrit A, Vartanian J-P, Wain-Hobson S. Human APOBEC3A Isoforms Translocate to the Nucleus and Induce DNA Double Strand Breaks Leading to Cell Stress and Death. PloS One. 2013;8:e73641. doi: 10.1371/journal.pone.0073641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aynaud M-M, Suspène R, Vidalain PO, Mussil B, Guétard D, Tangy F, Wain-Hobson S, Vartanian J-P. Human Tribbles 3 protects nuclear DNA from cytidine deamination by APOBEC3A. J. Biol. Chem. 2012;287:39182–39192. doi: 10.1074/jbc.M112.372722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Land AM, Law EK, Carpenter MA, Lackey L, Brown WL, Harris RS. Endogenous APOBEC3A DNA cytosine deaminase is cytoplasmic and nongenotoxic. J. Biol. Chem. 2013;288:17253–17260. doi: 10.1074/jbc.M113.458661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kitamura S, Ode H, Nakashima M, Imahashi M, Naganawa Y, Kurosawa T, Yokomaku Y, Yamane T, Watanabe N, Suzuki A, et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012;19:1005–1010. doi: 10.1038/nsmb.2378. [DOI] [PubMed] [Google Scholar]
- 50.Chen KM, Harjes E, Gross PJ, Fahmy A, Lu Y, Shindo K, Harris RS, Matsuo H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature. 2008;452:116–119. doi: 10.1038/nature06638. [DOI] [PubMed] [Google Scholar]
- 51.Holden LG, Prochnow C, Chang YP, Bransteitter R, Chelico L, Sen U, Stevens RC, Goodman MF, Chen XS. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature. 2008;456:121–124. doi: 10.1038/nature07357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furukawa A, Nagata T, Matsugami A, Habu Y, Sugiyama R, Hayashi F, Kobayashi N, Yokoyama S, Takaku H, Katahira M. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009;28:440–451. doi: 10.1038/emboj.2008.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harjes E, Gross PJ, Chen K-M, Lu Y, Shindo K, Nowarski R, Gross JD, Kotler M, Harris RS, Matsuo H. An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J. Mol. Biol. 2009;389:819–832. doi: 10.1016/j.jmb.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shandilya SMD, Nalam MNL, Nalivaika EA, Gross PJ, Valesano JC, Shindo K, Li M, Munson M, Royer WE, Harjes E, et al. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure. 2010;18:28–38. doi: 10.1016/j.str.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vasudevan AAJ, Smits SHJ, Höppner A, Häussinger D, Koenig BW, Münk C. Structural features of antiviral DNA cytidine deaminases. Biol. Chem. 2013;394:1357–1370. doi: 10.1515/hsz-2013-0165. [DOI] [PubMed] [Google Scholar]
- 56.Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 2003;77:11398–11407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 58.Iwatani Y, Takeuchi H, Strebel K, Levin JG. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 2006;80:5992–6002. doi: 10.1128/JVI.02680-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Curcio MJ, Garfinkel DJ. Single-step selection for Ty1 element retrotransposition. Proc. Natl Acad. Sci. USA. 1991;88:936–940. doi: 10.1073/pnas.88.3.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heidmann O, Heidmann T. Retrotransposition of a mouse IAP sequence tagged with an indicator gene. Cell. 1991;64:159–170. doi: 10.1016/0092-8674(91)90217-m. [DOI] [PubMed] [Google Scholar]
- 61.Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH., Jr High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87:917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 62.Levin JG, Guo J, Rouzina I, Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 2005;80:217–286. doi: 10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- 63.Levin JG, Mitra M, Mascarenhas A, Musier-Forsyth K. Role of HIV-1 nucleocapsid protein in HIV-1 reverse transcription. RNA Biol. 2010;7:754–774. doi: 10.4161/rna.7.6.14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Piekna-Przybylska D, Bambara RA. Requirements for efficient minus strand strong-stop DNA transfer in human immunodeficiency virus 1. RNA Biol. 2011;8:230–236. doi: 10.4161/rna.8.2.14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heilman-Miller SL, Wu T, Levin JG. Alteration of nucleic acid structure and stability modulates the efficiency of minus-strand transfer mediated by the HIV-1 nucleocapsid protein. J. Biol. Chem. 2004;279:44154–44165. doi: 10.1074/jbc.M401646200. [DOI] [PubMed] [Google Scholar]
- 66.Wu T, Datta SAK, Mitra M, Gorelick RJ, Rein A, Levin JG. Fundamental differences between the nucleic acid chaperone activities of HIV-1 nucleocapsid protein and Gag or Gag-derived proteins: biological implications. Virology. 2010;405:556–567. doi: 10.1016/j.virol.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu T, Heilman-Miller SL, Levin JG. Effects of nucleic acid local structure and magnesium ions on minus-strand transfer mediated by the nucleic acid chaperone activity of HIV-1 nucleocapsid protein. Nucleic Acids Res. 2007;35:3974–3987. doi: 10.1093/nar/gkm375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- 69.Carpenter MA, Rajagurubandara E, Wijesinghe P, Bhagwat AS. Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair. 2010;9:579–587. doi: 10.1016/j.dnarep.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kohli RM, Maul RW, Guminski AF, McClure RL, Gajula KS, Saribasak H, McMahon MA, Siliciano RF, Gearhart PJ, Stivers JT. Local sequence targeting in the AID/APOBEC family differentially impacts retroviral restriction and antibody diversification. J. Biol. Chem. 2010;285:40956–40964. doi: 10.1074/jbc.M110.177402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iwatani Y, Chan DSB, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007;35:7096–7108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bishop KN, Verma M, Kim E-Y, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008;4:e1000231. doi: 10.1371/journal.ppat.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adolph MB, Webb J, Chelico L. Retroviral restriction factor APOBEC3G delays the initiation of DNA synthesis by HIV-1 reverse transcriptase. PloS One. 2013;8:e64196. doi: 10.1371/journal.pone.0064196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chaurasiya KR, McCauley MJ, Wang W, Qualley DF, Wu T, Kitamura S, Geertsema H, Chan DSB, Hertz A, Iwatani Y, et al. Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid binding protein. Nat. Chem. 2013;6:28–33. doi: 10.1038/nchem.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bishop KN, Holmes RK, Malim MH. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 2006;80:8450–8458. doi: 10.1128/JVI.00839-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- 77.Luo K, Wang T, Liu B, Tian C, Xiao Z, Kappes J, Yu XF. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 2007;81:7238–7248. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bélanger K, Savoie M, Rosales Gerpe MC, Couture J-F, Langlois MA. Binding of RNA by APOBEC3G controls deamination-independent restriction of retroviruses. Nucleic Acids Res. 2013;41:7438–7452. doi: 10.1093/nar/gkt527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gillick K, Pollpeter D, Phalora P, Kim E-Y, Wolinsky SM, Malim MH. Suppression of HIV-1 infection by APOBEC3 proteins in primary human CD4+ T cells is associated with inhibition of processive reverse transcription as well as excessive cytidine deamination. J. Virol. 2013;87:1508–1517. doi: 10.1128/JVI.02587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chelico L, Sacho EJ, Erie DA, Goodman MF. A model for oligomeric regulation of APOBEC3G cytosine deaminase-dependent restriction of HIV. J. Biol. Chem. 2008;283:13780–13791. doi: 10.1074/jbc.M801004200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huthoff H, Autore F, Gallois-Montbrun S, Fraternali F, Malim MH. RNA-dependent oligomerization of APOBEC3G is required for restriction of HIV-1. PLoS Pathog. 2009;5:e1000330. doi: 10.1371/journal.ppat.1000330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.McDougall WM, Okany C, Smith HC. Deaminase activity on single-stranded DNA (ssDNA) occurs in vitro when APOBEC3G cytidine deaminase forms homotetramers and higher-order complexes. J. Biol. Chem. 2011;286:30655–30661. doi: 10.1074/jbc.M111.269506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shlyakhtenko LS, Lushnikov AY, Li M, Lackey L, Harris RS, Lyubchenko YL. Atomic force microscopy studies provide direct evidence for dimerization of the HIV restriction factor APOBEC3G. J. Biol. Chem. 2011;286:3387–3395. doi: 10.1074/jbc.M110.195685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Richard DJ, Bolderson E, Khanna KK. Multiple human single-stranded DNA binding proteins function in genome maintenance: structural, biochemical and functional analysis. Crit. Rev. Biochem. Mol. Biol. 2009;44:98–116. doi: 10.1080/10409230902849180. [DOI] [PubMed] [Google Scholar]
- 85.Cruceanu M, Gorelick RJ, Musier-Forsyth K, Rouzina I, Williams MC. Rapid kinetics of protein-nucleic acid interaction is a major component of HIV-1 nucleocapsid protein's nucleic acid chaperone function. J. Mol. Biol. 2006;363:867–877. doi: 10.1016/j.jmb.2006.08.070. [DOI] [PubMed] [Google Scholar]
- 86.Pant K, Karpel RL, Rouzina I, Williams MC. Mechanical measurement of single-molecule binding rates: kinetics of DNA helix-destabilization by T4 gene 32 protein. J. Mol. Biol. 2004;336:851–870. doi: 10.1016/j.jmb.2003.12.025. [DOI] [PubMed] [Google Scholar]
- 87.Teng B, Burant CF, Davidson NO. Molecular cloning of an apolipoprotein B messenger RNA editing protein. Science. 1993;260:1816–1819. doi: 10.1126/science.8511591. [DOI] [PubMed] [Google Scholar]
- 88.Liang G, Kitamura K, Wang Z, Liu G, Chowdhury S, Fu W, Koura M, Wakae K, Honjo T, Muramatsu M. RNA editing of hepatitis B virus transcripts by activation-induced cytidine deaminase. Proc. Natl Acad. Sci. USA. 2013;110:2246–2251. doi: 10.1073/pnas.1221921110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nabel CS, Lee JW, Wang LC, Kohli RM. Nucleic acid determinants for selective deamination of DNA over RNA by activation-induced deaminase. Proc. Natl Acad. Sci. USA. 2013;110:14225–14230. doi: 10.1073/pnas.1306345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chelico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA deaminase acts processively 3′ –> 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 2006;13:392–399. doi: 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
- 91.Navarro F, Bollman B, Chen H, König R, Yu Q, Chiles K, Landau NR. Complementary function of the two catalytic domains of APOBEC3G. Virology. 2005;333:374–386. doi: 10.1016/j.virol.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 92.Rathore A, Carpenter MA, Demir Ö, Ikeda T, Li M, Shaban NM, Law EK, Anokhin D, Brown WL, Amaro RE, et al. The local dinucleotide preference of APOBEC3G can be altered from 5′-CC to 5′-TC by a single amino acid substitution. J. Mol. Biol. 2013;425:4442–4454. doi: 10.1016/j.jmb.2013.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chahwan R, Wontakal SN, Roa S. Crosstalk between genetic and epigenetic information through cytosine deamination. Trends Genet. 2010;26:443–448. doi: 10.1016/j.tig.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 94.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell. 2012;46:424–435. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 2013;494:366–370. doi: 10.1038/nature11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burns MB, Temiz NA, Harris RS. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013;45:977–983. doi: 10.1038/ng.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nowarski R, Kotler M. APOBEC3 cytidine deaminases in double-strand DNA break repair and cancer promotion. Cancer Res. 2013;73:3494–3498. doi: 10.1158/0008-5472.CAN-13-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013;45:970–976. doi: 10.1038/ng.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Taylor BJ, Nik-Zainal S, Wu YL, Stebbings LA, Raine K, Campbell PJ, Rada C, Stratton MR, Neuberger MS. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife. 2013;2:e00534. doi: 10.7554/eLife.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lada AG, Waisertreiger IS-R, Grabow CE, Prakash A, Borgstahl GEO, Rogozin IB, Pavlov YI. Replication protein A (RPA) hampers the processive action of APOBEC3G cytosine deaminase on single-stranded DNA. PloS One. 2011;6:e24848. doi: 10.1371/journal.pone.0024848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hung M-C, Link W. Protein localization in disease and therapy. J. Cell Sci. 2011;124:3381–3392. doi: 10.1242/jcs.089110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.