


Abstract
Determining the composition of messenger ribonucleoprotein (mRNP) particles is essential for a comprehensive understanding of the complex mechanisms underlying mRNA regulation, but is technically challenging. Here we present an RNA-based method to identify RNP components using a modified streptavidin (SA)-binding RNA aptamer termed S1m. By optimizing the RNA aptamer S1 in structure and repeat conformation, we improved its affinity for SA and found a 4-fold repeat of S1m (4×S1m) to be more efficient than the established MS2 and PP7 systems from bacteriophages. We then attached the AU-rich element (ARE) of tumor necrosis factor alpha (TNFα), a well-known RNA motif that induces mRNA degradation, via 4×S1m to a SA matrix, and used the resulting RNA affinity column to purify ARE-binding proteins (BPs) from cellular extracts. By quantitative mass spectrometry using differential dimethyl labeling, we identified the majority of established ARE-BPs and detected several RNA-BPs that had previously not been associated with AREs. For two of these proteins, Rbms1 and Roxan, we confirmed specific binding to the TNFα ARE. The optimized 4×S1m aptamer, therefore, provides a powerful tool for the discovery of mRNP components in a single affinity purification step.
INTRODUCTION
Messenger (m)RNAs are controlled by trans-acting factors at virtually every step of their complex lives, from nuclear capping, cleavage, polyadenylation, splicing and export to cytoplasmic localization, translation and degradation (1,2). The fate of any given mRNA is essentially determined by the messenger ribonucleoprotein (mRNP) complex, i.e. the ensemble of proteins and regulatory RNAs an mRNA interacts with. Although decades of research have uncovered specific functions of numerous RNA-binding proteins (RNA-BPs), surprisingly little is known about the composition, heterogeneity and dynamics of whole mRNPs.
There are essentially two strategies to explore the content of mRNPs. On one hand, protein-based approaches rely on the purification of a particular RNA-BP by immunoprecipitation together with its associated mRNAs, whose identity can then be determined by cDNA cloning, microarray analysis (RIP-Chip) or deep sequencing (RIP-Seq) (3). RNA-sequencing technologies in combination with cross-linking protocols, e.g. (i) CLIP, PAR-CLIP and HITS-CLIP, recently emerged as efficient tools to identify protein recognition sites at a transcriptome-wide level (4–6). These protein-based approaches provide the spectrum of target mRNAs a particular RNA-BP interacts with, yet they do not reveal the complexity and dynamics of mRNPs, i.e. whether different proteins associate with the same mRNA in a cooperative, independent or competitive manner.
These limitations call for alternative RNA-centric strategies for mRNP isolation. Here, a single mRNA species is purified together with its associated proteins, which can be identified by mass spectrometry (MS). The advantage of this alternative approach is that one obtains a less biased picture of the mRNP, including proteins that were not known to interact with the RNA of interest. Ideally, RNA-based purification should reveal changes that occur during the lifetime of an mRNP or in response to signaling events. However, this approach faces major difficulties: finding a suitable way to purify a specific mRNA in the context of its chaperoning proteins; the generally low abundance of mRNAs compared to proteins in the cell; and the scale of purification needed for successful identification of proteins by MS. Moreover, some RNA-BPs bind to mRNAs transiently with high off-rates, which calls for cross-linking before RNP-purification, a step that comes along with its own set of disadvantages.
One way to purify endogenous RNAs, together with associated proteins, is by using antisense oligonucleotides, as exemplified by the isolation of the telomerase RNP (7) or bulk poly(A)-mRNPs (8,9). A different approach exploits naturally occurring, bacteriophage-derived RNA-protein interactions of high specificity and affinity such as the boxB RNA motif, which binds to the λN peptide of the Escherichia coli bacteriophage λ antiterminator protein N (10,11). Another example is the coat protein (cp) from the E. coli bacteriophage MS2, which binds tightly to an RNA stem-loop in the MS2 RNA (12). The Pseudomonas aeroginosa bacteriophage PP7cp is related to the MS2cp and binds with high specificity to a distinct stem-loop in the PP7 RNA (13). These systems have proven powerful for visualizing RNAs in living cells (14) and tethering proteins of interest to reporter mRNAs inside cells (15–18). The MS2 system was used for purification of highly stable RNPs such as the U1 snRNP (19), less stable mRNPs (20) and RNPs associated with non-coding regulatory RNAs (21–24). Similarly, the PP7 system has been exploited for the isolation of both stable 7SK RNPs (25) and mRNPs (26). Recently, the Csy4 endoribonuclease of the P. aeroginosa CRISPR system has been engineered for RNP affinity purification, making use of its catalytic activity for the elution step (27).
A third possibility is to use aptamers, structured RNA motifs selected in vitro for the ability to bind proteins or small molecule ligands. In contrast to the described bacteriophage systems, they do not require synthesis of recombinant proteins. Examples include a streptomycin-binding aptamer that was used for U1 snRNP isolation (28) and a tobramycin-binding aptamer that was used for purification of pre-spliceosomal RNP complexes (29). Moreover, Srisawat and Engelke developed a streptavidin (SA)-binding aptamer termed S1 by which yeast and human RNase P-associated RNPs could be purified (30,31). After cross-linking of bound proteins to an S1-tagged mRNA inside cells, the S1 aptamer was further used for the isolation of an mRNP that mediates translation activation (32). Finally, mRNA-interacting proteins were affinity purified from human cells and Drosophila embryo extracts using in vitro transcribed RNAs attached to SA via the S1 tag (33–35).
Here we present an optimized SA-binding aptamer, S1m (modified), which has higher affinity for SA than the original S1, binds with highest avidity to SA in a four-repeat conformation (4×S1m) and whose efficiency in our hands is superior to that of the MS2 and PP7 systems. As a model mRNP, we used a reporter mRNA containing the AU-rich element (ARE) of mouse tumor necrosis factor α (TNFα). AREs are located in the 3′ untranslated region (UTR) of many short-lived mRNAs and serve as regulatory elements for mRNA degradation and translation (36,37). Numerous ARE-binding proteins (ARE-BPs) have been identified since the initial description of the ARE (38). Using our optimized S1m tag fused to the TNFα ARE, we generated RNA affinity columns and purified most of the known ARE-BPs as well as novel ones, establishing S1m as an efficient tool for the isolation of mRNP-associated proteins.
MATERIALS AND METHODS
Plasmid construction
The following plasmids have been described previously: pTet-7B-ARE (pTet-7B-TNFα-ARE53, p2260) containing a Tet-inducible promoter and a T7-tagged rabbit β-globin reporter gene with the 53 nt ARE of mouse TNFα in its 3′UTR (39); puroMXβglobin (p2220, also referred to as puroMXβ) containing the β-globin gene driven by the Moloney murine leukemia virus promoter (40); puroMXβ-TNFα-ARE (p2225) (40) and puroMXβ-TNFα-CDE37-V3 (p2823) (41). pcDNA3-hygro-T7tag (p3203) and pcDNA3-hygro-T7tag-Roxan (p3204) (42) were provided by Didier Poncet (CNRS, Gif sur Yvette, France), and pGEX-2T-MS2cp (p2809) was a gift from Stephen Wax and Paul Anderson (Brigham and Women’s Hospital, Boston, USA). pTet-Off (p2249) was purchased from Clontech.
For the S1 and S1m-containing pTet-7B-TNFα-ARE reporter constructs, oligonucleotides containing the S1 (G1779/G1780) or S1m (G1850/G1851) aptamer sequence (see Supplementary Figure S1A) were annealed, concatemerized via their 5′BamHI- and 3′BglII-compatible ends and ligated. Head-to-head and tail-to-tail conformations were suppressed by cleavage with BglII and BamHI, whereas BglII/BamHI fusion sites in head-to-tail conformations gave rise to multimers. Fragments containing 1–7 consecutive repeats of the S1 or S1m aptamer were then cloned into the BglII site of pTet-7B-ARE (p2260). Thereby, the following series of pTet vectors was generated: pTet-7B-TNFα-ARE-1×S1 (p2775), -2×S1 (p2776), -3×S1 (p2777), -4×S1 (p2778), -5×S1 (p2779), -6×S1 (p2780), -7×S1 (p2788); pTet-7B-TNFα-ARE-1×S1m (p2781), -2×S1m (p2782), -3×S1m (p2783), -4×S1m (p2784), -5×S1m (p2785), -6×S1m (p2786), -7×S1m (p2787).
Plasmid puroMXβ-4×S1m (p2824) was generated by PCR-amplifying 4×S1m from pTet-7B-TNFα-ARE-4×S1m (p2784) using the forward BglII-mismatch repair primer G1919, which reconstitutes a BglII site from the BglII/BamHI fusion site and reverse primer G1918. The amplicon was then digested with BglII and inserted into the BglII site of puroMXβ (p2220). The same 4×S1m fragment was cloned into the BglII site of puroMXβ-TNFα-ARE (p2225) to generate puroMXβ-TNFα-ARE-4×S1m (p2891) and into the BglII site of pSP73 (Promega) to generate pSP73-4×S1m (p2880) (see Supplementary Figures S1 and S2). For pSP73-4×S1 (p3278), a 4×S1 fragment with flanking BglII sites was PCR-amplified from pTet-7B-TNFα-ARE-4×S1 (p2778) with primers G1919/G1918, BglII-digested and cloned into the BglII site of pSP73. For pSP73-TNFα-ARE-4×S1m (p2881), the ARE-4×S1m fragment was PCR- amplified from puroMXβ-TNFα-ARE-4×S1m (p2891) using the forward BamHI-mismatch repair primer G2247, which reconstitutes a BamHI site from the BglII/BamHI fusion site and reverse primer G2235. Thereupon, the 5′BamHI- and 3′BglII-flanked insert was cloned into the BglII site of pSP73. In analogy, for pSP73-TNFα-ARE-4×S1 (p3279), an ARE-4×S1 fragment with flanking BamHI/BglII sites was PCR amplified from pTet-7B-TNFα-ARE-4×S1 (p2778) with primers G2247/G2235, BamHI/BglII-digested and inserted into the BglII site of pSP73.
The PP7bs was synthesized by annealing oligonucleotides G95 and G96 and concatemerized using the same BamHI/BglII strategy as described earlier in text for S1/S1m. A 6-fold head-to-tail repeat of PP7bs was then cloned into pSP73 and transferred as a BamHI-BglII fragment into the BglII site of pTet-7B-ARE (p2260) (39) to generate pTet-7B-TNFα-ARE-6×PP7bs (p2259). For pTet-7B-TNFα-ARE-6×MS2bs (p2258), a 6-fold repeat of MS2bs was amplified by PCR using primers G37 and G38 from plasmid b-6bs (15) and cloned as a BamHI-BglII fragment into the BglII site of pTet-7B-ARE (p2260) (39).
For the expression of GST-tagged recombinant proteins, a 5′BamHI-, 3′EcoRI-linkered TEV sequence encoding ENLYFQG was first synthesized by annealing oligos G1875/G1876 and ligated into the BamHI/EcoRI sites of pGEX-4T-3 to generate pGEX-4T-3-TEV (p2772). The PP7cp sequence was then PCR-amplified with oligos G1859/G1860 from pc-HA-PP7cp (p2211) (43) and inserted into the EcoRI/XhoI sites of pGEX-4T-3-TEV (p2772) to generate pGEX-4T-3-TEV-PP7cp (p2773). Using the same strategy, oPP7cp (25) was PCR-amplified from pET28-ZZ-TEV-oPP7cp-His6 (p2770), a gift from Kathy Collins (University of California Berkeley, USA), using oligos G1860/G1861, and inserted into the EcoRI/XhoI sites of pGEX-4T-3-TEV (p2772) to generate pGEX-4T-3-TEV-oPP7cp (p2774). A TEV-linked version of GST-MS2cp was generated by inserting two consecutive BamHI/BglII-linked TEV sites (G1852/G1853) into the BamHI site of pGEX-2T-MS2cp (p2809), resulting in pGEX-2T-TEV-TEV-MS2cp (p2810). All oligonucleotide sequences are provided in Supplementary Table S1. Mutations, cloning boundaries and coding sequences were systematically verified by DNA sequencing (GATC Biotech).
Recombinant protein purification
For expression of the recombinant fusion proteins GST-T-PP7cp, GST-T-oPP7cp, GST-MS2cp and GST-TT-MS2cp, pGEX-based plasmids described earlier in text were transformed into BL21 (DE3) codon+ E. coli. Cells were cultured in 1 l Luria–Bertani medium supplemented with 100 µg/ml ampicillin at 37°C to an OD600 of 0.5, and protein expression was induced by addition of 1 mM IPTG. The culture was incubated for additional 16 h at 21°C before cells were collected at 4°C, washed once with ice-cold phosphate-buffered saline (PBS), resuspended in 10 ml PBS-MEC [PBS, 0.1% (v/v) β-mercaptoethanol (ME), 1 tablet/10 ml Mini Complete Protease Inhibitors, EDTA-free (Roche)] and lysed in PBS-ME using a microfluidizer (Emulsiflex, Avestin). The lysate was supplemented with 1% Triton-X-100, insoluble debris was removed by centrifugation and the supernatant was incubated under rotation with Glutathione (GSH) Sepharose High Performance 4B (GE Healthcare) beads for 1 h at 4°C. Beads were washed five times for 10 min with PBS-ME and transferred to a Poly-Prep column (Biorad) with a frit. PBS-ME was drained by gravity, and recombinant protein was eluted in 2 ml of elution buffer [50 mM glutathione, 50 mM Tris-HCl (pH 8.0), 10% glycerol, 1 tablet/10 ml Mini Complete Protease Inhibitors, EDTA-free (Roche), 0.1% (v/v) β-ME] for 2 h at 4°C under rotation. Eluted protein was dialyzed against dialysis buffer [50 mM Tris-HCl (pH 8.0), 0.1% (v/v) β-ME] for 6 h at 4°C, changing the buffer every 2 h. The eluate was recovered and supplemented with 20% glycerol, and protein concentration was determined by Nanodrop measurement and SDS-PAGE using a BSA standard.
Cell culture and transfection
COS7, NIH3T3 and HEK293T cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco) supplemented with 10% fetal calf serum (PAA Laboratories), 2 mM l-glutamine, 100 U/ml penicillin and 0.1 mg/ml streptomycin (all PAN Biotech) at 37°C in 5% CO2. NIH3T3 B2A2 cells (44) were a gift from Ann-Bin Shyu (University of Texas-Houston Medical School, USA). Cells were seeded in 10-cm dishes and transfected the following day with 9–12 μg of plasmid using polyethyleneimine [PEI, Polysciences Europe, 1 mg/ml (pH 7.0)] at a ratio of 1:2 (DNA:PEI) in serum-free and antibiotic-free DMEM. In case Tet-inducible vectors were transfected, 6 µg pTet reporter plasmid was co-transfected with 3 µg pTet-Off (p2249), and medium containing 10% (v/v) Tet-negative fetal calf serum (PAA) was used. The medium was changed to regular DMEM, 4–6 h after transfection. Stable NIH3T3 cell lines were generated by PEI transfection and subsequent selection with puromycin (4–6 μg/ml, Applichem). Mass cultures were used for mRNA decay assays.
Cellular RNP purification via SA-binding aptamers S1/S1m
One day after transient transfection of 1.2 × 106 COS7 cells per 10-cm dish, cells were washed once with PBS and the cell pellet was snap frozen in liquid nitrogen. By addition of a 5-mm steel bead, the cell pellet was homogenized by cryomilling using a tissue lyser (QIAgen TissueLyser II) at 25 Hz for 15 s.
For SA-pulldown of S1 and S1m-tagged RNAs, the homogenate was solubilized in 500 μl ice-cold SA-RNP lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 2 mM DTT, 2 mM vanadylribonucleosid complex RNase inhibitor (NEB), 1 tablet/10 ml Mini Complete Protease Inhibitors, EDTA-free (Roche)]. Lysates were first incubated with avidin agarose beads (Thermo Pierce) for 10 min at 4°C, as a blocking step, before cell debris was removed by centrifugation for 5 min at 2000 rpm (∼400 × g) at 4°C. The pre-cleared lysate was transferred to a fresh tube and 1:10 was saved as RNA input, mixed with 500 µl TriFast (PeqLab) and stored at -20°C for later RNA extraction. Lysates were incubated with Streptavidin Sepharose High Performance (GE Healthcare) beads for 4 h at 4°C under rotation before washing five times for 5 min at 4°C with SA-RNP wash buffer [20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 5 mM MgCl2, 2 mM DTT]. For the last wash, beads were transferred to a fresh tube, and RNA was eluted from the beads using 500 µl TriFast reagent (PeqLab) according to the manufacturer’s instructions. 15 µg GlycoBlue (Ambion) was added to the RNA samples before precipitation. Subsequently, RNA was resolved on denaturing agarose gels for northern blot analysis to determine RNA-binding efficiency to SA beads.
Cellular RNP purification via GST
COS7 cells were transfected, harvested and homogenized as described for SA-pulldown earlier in text. Homogenates were solubilized in 500 µl ice-cold GSH-RNP lysis buffer [50 mM Tris-HCl (pH 8.0 at 25°C), 150 mM NaCl, 1 mM MgCl2, 10% v/v glycerol, 2 mM DTT, 1 tablet/10 ml Mini Complete Protease Inhibitors, EDTA-free (Roche), 2 mM vanadylribonucleosid complex RNase inhibitor (NEB)], tumbled for 10 min at 4°C and cell debris was removed by centrifugation. The lysate was transferred to a fresh tube and 1:10 was saved for extraction of input RNA using TriFast (PeqLab). Lysates were pre-cleared with Glutathione Sepharose High Performance 4B (GE Healthcare) beads for 2 h at 4°C. The supernatant was then transferred to a new tube and 1:10 was saved for extraction of pre-clear RNA. In all, 2 µg of recombinant GST-tagged proteins was added to the pre-cleared lysates for 3 h at 4°C before incubation with Glutathione Sepharose High Performance 4B (GE Healthcare) for another 3 h. The beads were then washed six times for 5 min at 4°C with GSH-RNP lysis buffer. For the last wash, beads were transferred to a fresh tube, and protein was eluted from 10% of the beads using SDS sample buffer. RNA was eluted from 90% of the beads using 500 μl TriFast (PeqLab).
In vitro RNP purification via SA-binding S1m aptamer
Four RNAs were synthesized by in vitro transcription: ARE-4×S1 and ARE-4×S1m RNA containing a single copy of the 53-nt long TNFα ARE attached to four copies of the S1 or S1m aptamer, respectively, as well as 4×S1 and 4×S1m alone as negative control RNAs. As amplification of the highly structured 4×S1 and 4×S1m tag by PCR was problematic, linearized pSP73 plasmids or excised fragments from plasmids containing the ARE and 4×S1/4×S1m sequences served as templates. To this end, plasmids pSP73-4×S1 (p3278), pSP73-4×S1m (p2880), pSP73-TNFα-ARE-4×S1 (p3279) and pSP73-TNFα-ARE-4×S1m (p2881) were linearized at the EcoRV site downstream of the 4×S1m sequence, purified with the QIAquick PCR Purification Kit (QIAgen) and used as DNA templates for run-off in vitro transcription using SP6 RNA polymerase. Alternatively, plasmids were digested for 6 h with NdeI and EcoRV to release SP6-tagged DNA templates that were gel-extracted with the QIAquick Gel Extraction Kit (QIAgen). A 50 µl transcription reaction contained 8 µg linear DNA template, 4 mM of each NTP (Fermentas), 50 U RNasin (Promega), 400 U SP6 RNA polymerase (Fermentas), 1× SP6 Transcription Buffer (Fermentas) and 0.5 U yeast pyrophosphatase (Sigma). After incubation for 4 h at 37°C, DNA was digested by addition of 5 U RQ1 DNase (Promega) for 15 min at 37°C. In cases where higher RNA yields were needed, the MEGAscript SP6 Kit (Invitrogen, Life Technologies) was used for transcription and DNase digestion according to the manufacturer’s instructions. Synthesized RNA was purified by gel filtration using pre-packed G-50 Mini Quick Spin Sephadex RNA columns (Roche) according to the manufacturer’s instructions, and RNA concentration was determined by Nanodrop. One reaction typically yielded 25–50 µg of RNA.
For the following procedures, DNA/RNA LoBind tubes (Eppendorf) were used to reduce the degree of unspecific binding. Per sample, 100 µl 50% slurry of Streptavidin Sepharose High Performance (GE Healthcare) beads were washed three times with 1 ml of SA-RNP lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 2 mM DTT, 2 mM vanadylribonucleosid complex RNase inhibitor (NEB), 1 tablet/10 ml Mini Complete Protease Inhibitors, EDTA-free (Roche)]. At each step, beads were gently pelleted at 500 rpm (∼20 × g) for 1 min at 4°C. 30 µg of the in vitro transcribed 4×S1m or ARE-4×S1m RNAs was renatured in 50 µl SA-RNP lysis buffer by heating at 56°C for 5 min, 10 min at 37°C and incubation at room temperature for several minutes to refold RNA structures. The RNA was added to the 100 µl SA Sepharose slurry together with 3 µl RNasin (120 U, Promega), and 10 µl of the supernatant (10%) was saved for extraction of input RNA using TriFast (PeqLab). The mixture was incubated at 4°C for 2–3 h under rotation to permit binding of the RNA to the column. After that, beads were sedimented and a 10 µl sample of the supernatant (10%) was saved for extraction of unbound RNA using TriFast (PeqLab), and the remaining supernatant was discarded. Input and unbound RNA samples were compared side by side through 6% polyacrylamide/TBE/urea gel electrophoresis and methylene blue staining to assess the efficiency of RNA coupling.
Cellular extracts were prepared from 40 confluent 15-cm dishes of untransfected NIH3T3 B2A2 cells. A total of 3.5 g cells was collected, washed once in PBS, divided into 300 mg portions and aliquoted in 2 ml safe-lock tubes (Eppendorf). The cell pellets were then snap frozen in liquid nitrogen, homogenized by cryomilling after addition of a 5-mm steel bead using a tissue lyser (QIAgen TissueLyser II) at 25 Hz for 15 s 4–6 times, and the powder was either processed directly or stored in liquid nitrogen. The frozen homogenate of one aliquot (300 mg) was solubilized by the addition of 300 µl ice-cold RNP lysis buffer and allowed to thaw for 5 min at room temperature. Cell debris was removed by centrifugation for 10 min at 17.000 × g at 4°C, resulting in a supernatant of ∼ 500 µl. The protein concentration in the extract was determined by Nanodrop to be ∼70 mg/ml.
Next, the extract (∼500 µl) was pre-cleared by addition of 25 µl of a 50% slurry of Avidin Agarose (Thermo Pierce) beads and tumbling for 30 min at 4°C. The supernatant was transferred to a fresh tube and tumbled with 50 µl of a 50% slurry of SA Sepharose beads for an additional 2–3 h. Beads were discarded, and the pre-cleared lysate was supplemented with 1.5 µl (60 U) of RNasin (Promega), added onto the freshly prepared RNA-coupled SA Sepharose matrix and incubated 4°C for 3–4 h under rotation. Beads were washed 6 times for 2–5 min with 1 ml SA-RNP wash buffer [20 mM Tris-HCl (pH 7.5), 300 mM NaCl, 5 mM MgCl2, 2 mM DTT] supplemented with 2 mM vanadylribonucleosid complex RNase inhibitor (NEB) and 1 tablet/50 ml Complete Protease Inhibitors, EDTA-free (Roche). After the last wash, beads were transferred to a fresh tube and RNA-bound proteins were eluted by addition of 5 µg RNase A (Genomed) in 100 µl low salt buffer [20 mM Tris-HCl (pH 7.5), 30 mM NaCl, 5 mM MgCl2, 2 mM DTT, 1 tablet/10 ml Mini Complete Protease Inhibitors, EDTA-free (Roche)] for 10 min at 4°C. The RNase A eluate was recovered and concentrated by vacuum centrifugation from 100 to 30 µl. In all, 10 µl of the eluate was analyzed by SDS-PAGE and colloidal Coomassie staining, whereas 20 µl was used for subsequent LC-MS/MS analysis. After RNase A elution, the beads were extracted with 30 µl 2× SDS sample buffer, 10 µl of which were analyzed by SDS-PAGE.
Where indicated, elution of RNA-protein complexes with biotin was carried out using 10 mM biotin (50 mM stock, Invitrogen, Life Technologies) in SA-RNP lysis buffer for 1 h at 4°C. For initial small-scale experiments aimed at quantifying binding and elution efficiencies, 20% of the RNA at each step was analyzed by 6% polyacrylamide/TBE/urea gel electrophoresis and methylene blue staining.
Mass spectrometry
MS was carried out at the Mass Spectrometry Core Facility of the ZMBH (Zentrum für Molekulare Biologie der Universität Heidelberg). Proteins eluted from the RNA affinity matrix were shortly run into a 10% polyacrylamide Tris-glycine gel (NuPAGE, Novex/Invitrogen) and stained by colloidal Coomassie blue. For each sample, the entire lane was cut into four gel slices including the wells. After in-gel digestion with trypsin, the supernatant was dried in a speed vac and solubilized in 55 µl of dimethyl labeling reaction mixture (50 µl 100 mM TEAB, 2 µl 150 mM NaBH3CN) and 2 µl formaldehyde for light labeling and 2 µl deuterated formaldehyde for medium labeling (45). After 1 h at 25°C, the reaction was stopped with 1.6 µl 5% NH4OH and acidified with 4 µl 50% formic acid 10 min later. NanoUPLC-MS/MS analysis (LTQ Orbitrap XL, Thermo Scientific) was carried out as described in more detail previously (46). Database search was done using Mascot in the framework of ProteomeDiscoverer (PD1.3; ThermoScientific) with carbamidomethyl cysteine as fixed modification, and deamidation and oxidation, as variable modification against the NCBI database (taxonomy: mus). Light and medium dimethylation at K and the N-terminus were searched as static modifications in two different nodes of PD1.3.
RNA-IP
RNA-IP from HEK293T cells was performed as described recently (41). Briefly, HEK293T cells grown in 10-cm dishes were disrupted by cryomilling as described earlier in text, solubilized in 400 μl RNA-IP lysis buffer (41) and pre-cleared with Protein A/G UltraLink Resin (Thermo Pierce) for 1 h at 4°C. For RNA-IP of endogenous Rbms1, 3 μg of Rbms1 antibody (ab150353, Abcam) or myc control antibody (Santa Cruz sc-789) was added for 2 h at 4°C before incubation with protein A/G beads for another 2 h. For RNA-IP of T7-tagged Roxan, 1 µg of T7 antibody (Novagen, 69522-3) was used instead. After washing twice with RNA-IP lysis buffer and four times at 300 mM NaCl concentration with RNA-IP wash buffer (41), protein was eluted from 1:5 of the beads using SDS sample buffer and RNA was eluted from 4:5 of the beads using 500 μl TriFast reagent (PeqLab) as described earlier in text.
Electromobility shift assay (EMSA)
Recombinant SA protein was purchased from Life Technologies (43-4301) and reconstituted to 1 mg/ml in PBS. S1 (44 nt, ACCGACCAGAAUCAUGCAAGUGCGUAAGAUAGUCGCGGGCCGGG) and S1m (60 nt, AUGCGGCCGCCGACCAGAAUCAUGCAAGUGCGUAAGAUAGUCGCGGGUCGGCGGCCGCAU) RNAs were commercially synthesized (IDT Inc) and 5′ end-labeled using [γ-32P]-ATP and T4 polynucleotide kinase (NEB). The 23-nt-long ARE control RNA and the EMSA protocol have been described before (41), except that ZnSO4 was omitted in the binding buffer, the polyacrylamide gel and the TBE running buffer.
mRNA decay and northern blot analysis
For mRNA decay experiments, NIH3T3 B2A2 cells were stably or transiently transfected with puroMXβglobin reporter plasmids. mRNA decay assays were performed by the addition of actinomycin D (5 μg/ml, Applichem) and subsequent extraction of total RNA using the GeneMatrix Universal RNA Purification Kit (EurX, Roboklon) at regular intervals. Northern blot analysis was performed as described earlier in (18) using digoxigenin-labeled RNA probes against globin and nucleolin.
Western blot analysis and antibodies
Western blot analysis was performed as described previously (41) using the following antibodies: monoclonal mouse anti-G3BP1 (TT-Y, sc-81940, Santa Cruz), anti-HuR (3A2, sc-5261, Santa Cruz) and anti-T7-Tag (Novagen, 69522-3); monoclonal rabbit anti-RBMS1 (ab150353, Abcam); polyclonal rabbit anti-myc (Santa Cruz sc-789, A-14); and polyclonal rabbit anti-GST antibody was provided by Ludger Hengst (Innsbruck Medical University, Austria) and Frauke Melchior (ZMBH, University of Heidelberg, Germany).
RESULTS
Development of S1m, an optimized SA-binding aptamer
The S1 aptamer was originally developed by Srisawat and Engelke (2001) through in vitro selection of an SA-binding aptamer from a random RNA library by SELEX (systematic evolution of ligands by exponential enrichment) (30). The minimal S1 motif consists of a 44-nt-long structured RNA containing two double-stranded (ds) RNA helices, a 13-nt internal loop between the two helices and a 9-nt terminal loop (Figure 1A) (47). To test the efficiency of the S1 aptamer for mRNP purification, we introduced a single copy of S1 into the 3′UTR of a β-globin reporter gene that contains the 53-nt long ARE of mouse TNFα (40) (Figure 1B). The globin-ARE-1×S1 reporter gene was then transiently transfected into COS7 cells, which were harvested the following day by rapid freezing in liquid nitrogen. Cellular lysates were prepared by detergent-free cryomilling as previously suggested for RNP purification (48). Compared to detergent-based lysis, cryolysis was equally efficient with regard to RNA recovery (data not shown). After pre-clearing with avidin beads that do not bind the aptamer (47) but remove biotin and biotinylated proteins due to the high affinity of egg white avidin for biotin, lysates were incubated with SA Sepharose to capture the mRNP. Beads were then washed and RNA was eluted by phenol/chloroform extraction to assess the efficiency of mRNP recovery. Northern blot analysis of input and purified (SA pulldown) RNA showed only minimal binding of globin-ARE-1×S1 mRNA (Figure 1C, lane 7). Quantification indicated that only 0.3% of globin-ARE-1×S1 mRNA was retained on SA Sepharose compared to 0.2% background binding of globin-ARE mRNA lacking the S1 aptamer (Figure 1E).
Figure 1.

Improved SA-binding efficiency of the S1m aptamer. (A) Left: secondary structure model of the original S1 aptamer published by Srisawat and Engelke (30). Right: secondary structure model of the modified S1m aptamer with a longer stem of perfect complementarity. Nucleotides that were mutated are shown in red and those added to prolong the stem are shown in yellow. (B) Schematic representation of globin reporter mRNAs encoding rabbit β-globin fused to the T7 epitope. The ARE of TNFα was inserted into the β-globin 3′UTR, upstream of S1 or S1m. (C) COS7 cells were transiently transfected with globin-TNFα-ARE, globin-TNFα-ARE-1×S1, -1×S1m, -4×S1 or -4×S1m. One day later, cells were lysed by cryomilling, and cleared supernatants were incubated with SA Sepharose beads. RNA was isolated from the cleared supernatants (input) and from the beads after five washing steps (SA pulldown). The RNA was resolved on 1.1% agarose gels and subjected to northern blot analysis using a β-globin probe against the globin reporter mRNA. Nucleolin (ncl) mRNA serves as a loading control. (D) COS7 cells were transiently transfected with globin reporter genes containing the ARE alone or the TNFα ARE together with 1–7 copies of the S1m aptamer. Retention of the mRNAs by SA pulldown was analyzed as in C. (E) Quantification of SA pulldown efficiencies. Different reporter mRNAs were expressed in COS7 cells as in C and D, and the amount of mRNA retained by SA Sepharose was expressed as % of input. Shown are average values ± SD, n is indicated above the graph. (F) Globin reporter genes containing no insert in the 3′UTR (−), 4×S1m or ARE-4×S1m were stably transfected into NIH3T3 cells. Reporter mRNA decay was measured by treatment with actinomycin D (actD), extraction of total RNA at 2 h intervals followed by northern blot analysis. Globin mRNA signals normalized to nucleolin (ncl) were used for calculation of average mRNA half-lives (t1/2) ± SD, n = 3. (G) After transient transfection of NIH3T3 cells, the decay of globin-ARE and globin-ARE-4×S1m reporter mRNAs was measured as in F; shown are average mRNA half-lives (t1/2) ± SD, n = 3.
With the aim to improve binding efficiency, we generated a modified aptamer termed S1m (Figure 1A and Supplementary Figure S1) in which the terminal S1 RNA structure was stabilized by introducing perfect strand complementarity in the basal stem (shown in red), as was done previously for a modified S1 aptamer in the design of an RNA-based antibody mimic (49). Similar to Xu and Shi (49), we also extended the basal stem of S1, yet only by 8 bp (Figure 1A, in yellow), to a total length of 15 bp. When the modified S1m aptamer was inserted into the reporter mRNA (globin-TNFα-ARE-1×S1m), we observed 3–4-fold higher binding efficiency (Figure 1C, lane 8) with 1.1% of the mRNA retained on SA Sepharose (Figure 1E).
We then introduced multiple copies of the S1m aptamer into the reporter gene, speculating that a synergistic effect might further increase binding efficiency. A linear 21-nt spacer sequence lacking secondary structure (Supplementary Figure S2) was inserted between the individual aptamers to increase flexibility between consecutive motifs. We observed steadily increasing binding efficiencies with 2, 3 and 4 copies of S1m (Figure 1D, lanes 11–13). Additional copies of S1m (up to 7) did not further improve mRNA recovery; in fact, these longer repeats were less efficient than 4×S1m (lanes 14–16). Using four copies of the original S1 aptamer also improved RNA binding compared to the single copy 1×S1 construct (2.3 versus 0.3%, Figures 1C and 1E). Quantification of several repeat experiments showed that of all constructs tested, 4×S1m has the highest binding efficiency: with 4.5% of mRNA retained on SA Sepharose, it was 15 times more efficient than the original S1 aptamer.
To assess functionality of the aptamer-tagged mRNPs, we tested whether insertion of 4×S1m would interfere with ARE-mediated mRNA decay. NIH3T3 cells were stably transfected with globin-TNFα-ARE-4×S1m, globin-4×S1m or globin alone, and reporter mRNA stability was measured by actinomycin D chase experiments (Figure 1F). Northern blot analysis showed that the globin and globin-4×S1m control mRNAs were stable over time with half-lives >9 h, whereas the globin-TNFα-ARE-4×S1m mRNA was rapidly degraded with a half-life of 1.9 ± 0.1 h. As demonstrated in transiently transfected NIH3T3 cells (Figure 1G), there was no difference between the decay rate of globin-TNFα-ARE (1.8 ± 0.3 h) and globin-TNFα-ARE-4×S1m (1.8 ± 0.5 h). Thus, ARE-mRNA decay is not affected by placing 4×S1m in the vicinity of the ARE, a prerequisite for purifying functional mRNPs.
Comparison to the PP7 and MS2 systems
Our next goal was to compare the S1m aptamer with the PP7 and MS2 systems. To this end, we generated globin reporter mRNAs containing the TNFα ARE and six copies of either the PP7 binding site (bs) or the MS2bs (Figure 2A). From tethering assays, it is know that inserting 6–8 copies of the MS2bs strongly increases binding efficiency to the MS2cp (15). To capture the mRNA, PP7cp and MS2cp were expressed in E. coli as glutathione S transferase (GST)-fusion proteins. In addition, one or two cleavage sites for the tobacco etch virus protease (TEV, T in Figure 2) were engineered between GST and the coat proteins (Figure 2A). The globin-TNFα-ARE-6×PP7bs and globin-TNFα-ARE-6×MS2bs reporter genes were transiently transfected into COS7 cells, and lysates were prepared by cryomilling as described for the S1 aptamer earlier in text. After pre-clearing with GSH Sepharose beads, lysates were incubated with the recombinant GST-tagged coat proteins before capture on GSH Sepharose beads. Northern blot analysis showed that only 0.7% of globin-ARE-6×PP7bs mRNA was retained by GST-T-PP7cp on GSH Sepharose (Figure 2B, lane 6, quantification in Figure 2C). We also tested an optimized (o)PP7cp, a previously published PP7cp mutant with higher affinity to its cognate RNA and reduced tendency to multimerize (25). With oPP7cp, 1.0% of the globin-ARE-6×PP7bs mRNA was retained on GSH Sepharose, slightly more than with the wild-type PP7cp protein.
Figure 2.

Binding efficiencies of the MS2 and PP7 systems. (A) Schematic representation of globin reporter mRNAs containing in their 3′UTR the TNFα ARE upstream of six copies of either the PP7-binding site (bs) or the MS2bs. GST-tagged coat proteins used for mRNP purification are depicted on the right: GST-T-PP7cp containing the wild-type PP7cp cDNA, GST-T-oPP7cp containing an optimized (o)PP7cp with higher RNA-binding affinity (25), GST-MS2cp and GST-TT-MS2cp. T stands for TEV cleavage site. (B) COS7 cells were transiently transfected with either globin-TNFα-ARE-6×PP7bs or globin-TNFα-ARE-6×MS2bs. Cell lysates were pre-cleared and incubated with recombinant GST-T-PP7cp, GST-T-oPP7cp, GST-MS2cp or GST-TT-MS2cp before addition of GSH Sepharose beads. RNA was extracted from the lysates before (input) and after pre-clearing (pre-clear), and from the beads after five washing steps (GSH pulldown). The RNA was subjected to northern blot analysis as in Figure 1C. Protein was extracted from the beads and detected by western blot analysis using an anti-GST antibody. (C) Quantification of GSH pulldown efficiencies from B and repeat experiments. The amount of mRNA retained by the different GST fusion proteins was expressed as percentage of input mRNA. Shown are average values ± SD, n = 3.
MS2cp was either fused to GST directly or via a linker containing two consecutive TEV protease cleavage sites (TT, Figure 2A). With both GST-MS2cp and GST-TT-MS2cp, more globin-ARE-MS2bs mRNA was recovered than with PP7cp or oPP7cp (Figure 2B, lanes 9 and 10, quantification in Figure 2C). GST-MS2cp gave the best result with 3.2% of the input mRNA retained on the beads. When compared to the 4×S1m aptamer, however, the PP7 and MS2 systems performed less well, so we decided to pursue mRNP purification using 4×S1m.
Higher affinity of S1m for SA
To further explore to which extent the S1m aptamer performs better than S1, we compared their binding affinities to SA in vitro. EMSA showed that one copy of S1 RNA binds to recombinant SA protein with an apparent dissociation constant (Kd) of 59 nM (Figure 3A), similar to the Kd of 70 nM reported previously (30,31). S1m showed a 2-fold higher affinity with an apparent Kd of 29 nM (Figure 3A). We noticed that a considerable amount of the RNA remained unbound even at high SA concentrations. This did not affect calculation of the Kd, as we achieved saturated binding, but suggested that a proportion of the RNA was incorrectly folded. An unrelated unstructured 23-nt-long ARE RNA did not form a complex with SA by EMSA (Supplementary Figure S3), confirming that the complex we observed between SA and S1/S1m is due to specific binding. Taken together, these data revealed that S1m has higher affinity toward SA than S1. This, together with the synergistic effect of connecting four copies of the aptamer, explains the improved efficiency of the 4×S1m tag in capturing a cellular mRNA (Figure 1C–E).
Figure 3.

Binding affinities and elution of the S1 and S1m aptamers (A) In vitro binding of recombinant SA protein to S1 and S1m RNA was measured by EMSA. Radiolabeled S1 and S1m RNAs were incubated with increasing concentrations of SA. Free RNA (black arrow) was separated from RNA-protein complexes (white arrow) by native 6% PAGE. Bottom graph: the apparent dissociation constant Kd ± SD was calculated from quantification of three independent experiments. (B) Schematic representation of DNA templates used for in vitro transcription of 4×S1, 4×S1m, ARE-4×S1 and ARE-4×S1m RNAs. (C) Elution by biotin was examined by in vitro SA pulldown experiments. In vitro transcribed RNAs were coupled to SA-Sepharose beads, incubated with NIH3T3 cell extracts, washed and eluted using 10 mM biotin (EB). Residual RNA bound to beads was subsequently eluted with Trizol (ET). RNA was resolved by 6% denaturing urea-PAGE and visualized by methylene blue staining. Each lane represents 20% of the starting amount, low range ssRNA ladder (NEB) was loaded for reference. On the right, the percentage of RNA recovered at each step was quantified based on three independent repeat experiments, average values ± SD are shown. (D) The same experiment was carried out as in C, except that the first elution was carried out with 0.05 µg/µl RNase A. (E) Upon RNA affinity purification using an NIH3T3 cell lysate as in C, proteins were eluted with RNase A followed by subsequent elution with SDS to determine residual protein bound to the beads. Top: 10% of each RNA fraction was resolved by denaturing urea-PAGE and stained with methylene blue. Bottom: 23% of the RNase A and SDS eluates was resolved by PAGE and proteins were monitored by western blot analysis. The ARE-BP HuR serves as positive control, whereas G3BP is a negative control.
Elution with biotin and RNase
We next wanted to compare the ability of biotin to elute 4×S1- and 4×S1m-tagged RNAs from SA Sepharose in the presence of a cellular lysate. ARE-4×S1 and ARE-4×S1m RNAs were transcribed in vitro, subjected to a folding step and coupled to SA beads (Figure 3C). As expected from the higher affinity of S1m, binding of ARE-4×S1m (59%, input minus unbound) was more efficient than binding of ARE-4×S1 (27%). The RNA affinity resin was then incubated with a highly concentrated pre-cleared NIH3T3 cell extract (30–50 mg/ml), washed three times and eluted with 10 mM biotin. Residual RNA on the beads was recovered by a second elution using Trizol. This analysis showed that 70% of the bound ARE-4×S1 RNA and 47% of the bound ARE-4×S1m RNA was eluted with biotin (Figure 3C). In all, 28% (ARE-4×S1) and 17% (ARE-4×S1m) of the RNA remained on the beads and was recovered only after Trizol elution.
As an alternative for eluting proteins bound to the captured RNA, we observed that RNase A degraded the SA-bound ARE-4×S1 and ARE-4×S1m RNAs completely (Figure 3D). We also saw that RNase A was efficient in eluting protein from mRNPs recovered from intact cells (data not shown). Thus, cellular proteins do not interfere with access of the RNase to RNA-protein complexes. We then examined the release of proteins bound to the RNA by RNase A elution (Figure 3E). HuR, a well-established ARE-BP, was captured specifically by ARE-4×S1 and ARE-4×S1m but not by the corresponding control RNAs lacking the ARE. RNase A was able to release most of the bound HuR, whereas only small amounts remained bound to the SA beads, visualized by SDS elution. Notably, ARE-4×S1m RNA was able to recover more HuR protein than ARE-4×S1. G3BP, an RNA-BP not related to AREs, did not interact with any of the RNA-affinity resins, demonstrating specificity of our purification method. In all, this was a first indication that an ARE-BP can be specifically purified via 4×S1m-tagged ARE-RNA.
Taken together, elution with biotin appears somewhat less efficient, releasing only about half of the bound ARE-4×S1m RNA, yet might be chosen as an alternative if the goal is to recover native RNP complexes from the column i.e. for a second purification step. However, our results suggested that elution by RNase A is the method of choice if the goal is to recover proteins from the RNA-affinity column.
Purification of ARE-associated proteins via 4×S1m
In our initial experiments, we attempted to identify proteins associated with the globin-ARE-4×S1m mRNA stably expressed in NIH3T3 cells. However, we mostly detected cytoskeletal proteins in both the ARE and control purifications, and were not able to capture specific RNA-BPs (data not shown). Therefore, we turned to the in vitro RNA affinity chromatography approach established in Figure 3C–E (schematically depicted in Figure 4) and optimized the protocol (details are given in the ‘Methods’ section).
Figure 4.
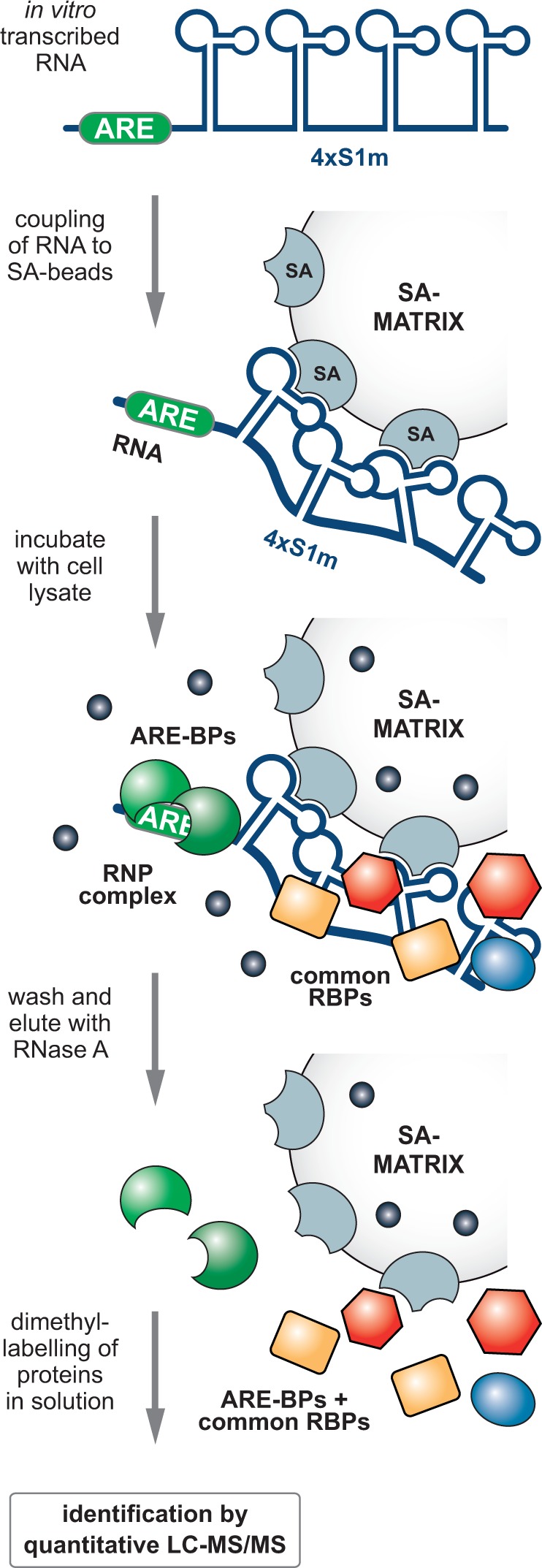
Schematic representation of S1m-mediated in vitro RNP purification. In vitro transcribed ARE-4×S1m and control 4×S1m RNAs are coupled to SA Sepharose beads and incubated with a highly concentrated cellular extract. RNP complexes formed contain proteins specific to the ARE and RNA-BPs that interact with aptamers and linkers. Following high-salt washes, elution by RNase A releases RNA-associated proteins. Proteins enriched in the ARE-4×S1m eluate over the 4×S1m eluate are identified by quantitative MS using dimethyl-labeling after tryptic digestion.
The ARE-4×S1m and negative control 4×S1m RNAs were transcribed in vitro, refolded and coupled to SA beads. In this scaled-up approach, RNA binding to the column was found to be highly efficient (77–95%, Figure 5A). Then, highly concentrated cell extracts (70 mg protein/ml) were obtained by cryomilling of NIH3T3 cells, cleared from cellular debris by centrifugation at 17.000 × g, pre-blocked with avidin and SA beads and incubated with the RNA affinity columns. After washing with a buffer containing 300 mM NaCl, proteins bound to the RNA were eluted with RNase A. One-third of the eluate was resolved by PAGE and visualized by colloidal Coomassie blue staining (Figure 5B, left). Subsequent elution of the columns with SDS showed that vast amounts of protein adhered unspecifically to the SA Sepharose resin (right), whereas RNA-associated proteins represented a minority. Thus, elution by RNase provides an important advantage in the purification scheme because many of the unspecifically bound proteins remain on the column.
Figure 5.

Identification of ARE-BPs by S1m-mediated RNA affinity chromatography. (A) In vitro transcribed RNAs were coupled to SA beads. To monitor coupling efficiency, 10% of the input and unbound RNA fraction were resolved on a 6% polyacrylamide/TBE/urea gel and stained with methylene blue. Low range ssRNA ladder (NEB) was loaded for reference. (B) Extracts of NIH3T3 cells were incubated with the RNA-coupled SA Sepharose matrix, washed and eluted using RNase A. SDS was used for a second elution of the beads. One-third of the eluates was resolved on a 5–20% polyacrylamide gradient gel and proteins were stained with colloidal Coomassie blue. The black arrow indicates the RNase A band. (C) Two-thirds of the RNase A elution samples were resolved over a short distance on a 10% polyacrylamide gel and stained with colloidal Coomassie blue. The entire lanes including the wells were cut into four gel slices and in-gel trypsin-digested. Peptides from the 4×S1m (light) and ARE-4×S1m (medium) eluate were differentially dimethyl-labeled and processed for MS; MW, molecular weight marker. (D) List of proteins identified by MS in ARE-RNPs purified via 4×S1m. The ratio of medium/light reflects the specific enrichment of proteins in the ARE-4×S1m eluate over the 4×S1m eluate. All proteins enriched >8-fold are depicted and categorized as established ARE-BPs (green), putative ARE-BPs identified previously (blue) or other RNA-BPs (yellow).
Two-thirds of the RNase A eluates were vacuum-concentrated and resolved by PAGE over a short distance (Figure 5C). The pattern of protein bands was similar in the 4×S1m and ARE-4×S1m samples, suggesting that most of the purified proteins were associated with the 4×S1m aptamer or the spacer sequence. To detect ARE-specific proteins enriched in the ARE-4×S1m sample we used quantitative MS. The two lanes were cut into four slices each and proteins were in-gel digested with trypsin, followed by peptide dimethyl labeling (45) using either formaldehyde (4×S1m sample, light) or deuterated formaldehyde (ARE-4×S1m sample, medium). The light and medium samples were then combined for liquid chromatography-MS/MS analysis. This approach allowed us to calculate the enrichment of low abundance proteins in one sample despite the presence of highly abundant proteins in both samples.
Figure 5D lists proteins that were enriched by >8-fold in the ARE-4×S1m sample. Notably, our purification yielded most of the established ARE-BPs (in green) including HuR (Elav1), BRF2 (Zfp36L2), TIAR (Tial1), BRF1 (Zfp36L1) and AUF1 (Hnrnpd). We also found two proteins (in blue), Fubp3 and Dazap1, which had previously been purified with the TNFα ARE (50,51), although their function in regulating ARE-mRNAs has not been established. An additional five RNA-BPs (in yellow) were specifically enriched in the ARE-4×S1m purification including two proteins of the U2 snRNP (A’ and B’’), a general snRNP protein (Sm-D2), an RRM-containing protein (Rbms1) and a zinc finger protein termed Roxan (Zc3h7b).
Confirmation of Rbms1 and Roxan as novel ARE-BPs
To confirm binding of novel candidate ARE-BPs, we chose Rbms1 and Roxan from the list of enriched proteins (Figure 5D), both of which had previously not been associated with the ARE. Rbms1 (RNA-binding motif, single-stranded-interacting protein 1), also known as MSSP (c-myc single-strand-binding protein), contains two RNA recognition motif (RRM) domains. It was initially identified as a DNA-binding protein that interacts with a region upstream of the c-Myc gene (52) and proposed to function in DNA replication (53). Rbms1 was further shown to bind to the c-Myc protein and stimulate its transforming activity (54), yet the RNA-binding properties of Rbms1 have not been investigated so far. The second candidate, Roxan (rotavirus X protein associated with NPS3), also termed Zc3h7b (zinc finger CCCH domain-containing protein 7B), is a 110 kDa cellular protein of unknown function that was shown to bind the rotavirus non-structural protein NSP3. NSP3 interacts with both the 3′ consensus sequence of non-polyadenylated viral mRNAs and eukaryotic translation initiation factor eIF4GI, suggesting that Roxan may function in the regulation of viral mRNA translation (42). Moreover, Roxan was shown to interact with polyA-binding protein (PABP-C1) and to promote its nuclear localization during rotavirus infection (55).
First, we confirmed the specific interaction of Rbms1 with the ARE in an in vitro SA pulldown experiment (as in Figure 3E) using an antibody against endogenous Rbms1 (Figure 6A). Rbms1 was found to be specifically enriched by RNase A elution in the ARE-4×S1 and ARE-4×S1m samples. Notably, the second elution with SDS revealed additional unspecific binding of Rbms1 to the beads, underlining the importance of elution by RNase. By an inverse approach, i.e. RNA-immunoprecipitation (IP) of endogenous Rbms1 from HEK293 cells, we confirmed that Rbms1 strongly associates with globin-TNFα-ARE mRNA but not with globin mRNA alone (Figure 6B). Likewise, Rbms1 did not bind globin-TNFα-CDE mRNA, which contains the constitutive decay element (CDE), a stem-loop motif that confers ARE-independent mRNA decay (41). As a negative control antibody against myc did not pulldown any of the reporter mRNAs, this experiment demonstrates that Rbms1 is a specific ARE-BP.
Figure 6.
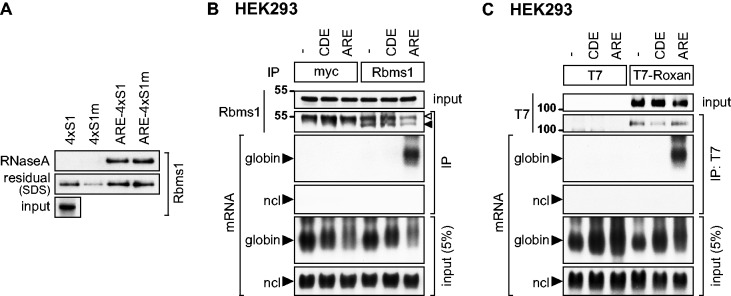
Rbms1 and Roxan specifically bind to the ARE. (A) Binding of Rbms1 to the ARE was analyzed by in vitro SA pulldown, as in Figure 3D. Rbms1 was detected in elution and input samples using an antibody against endogenous Rbms1. (B) Binding of endogenous Rbms1 to the TNFα ARE was analyzed by RNA-IP. HEK293 cells were transiently transfected with the globin reporter lacking an insert (−), globin-TNFα-CDE or globin-TNFα-ARE. After IP of endogenous Rbms1 with an Rbms1-specific antibody, the protein was monitored by western blot analysis; the white arrow indicates the immunoglobulin heavy chain, the black arrow Rbms1. Anti-myc antibody was used for the negative control IP. Globin reporter mRNAs were visualized by northern blot analysis, ncl mRNA serves as negative control. (C) Binding of Roxan to the TNFα ARE was analyzed by RNA-IP. HEK293 cells were transiently transfected with T7 or T7-Roxan together with the globin reporter lacking an insert (−), globin-TNFα-CDE or globin-TNFα-ARE. After IP with anti-T7 antibody, T7-Roxan was monitored by western blot analysis. Globin reporter mRNAs were visualized by northern blot analysis, ncl mRNA serves as negative control.
To assess binding of Roxan to ARE-RNA, RNA-IP was then carried out with T7-tagged Roxan transiently expressed in HEK293 cells. Similar to Rbms1, we observed that T7-Roxan specifically interacts with globin-TNFα-ARE, but not with the globin or globin-TNFα-CDE mRNAs (Figure 6C). The T7-tag alone did not bind to any of the reporter mRNAs. These results illustrate that 4×S1m-mediated RNA affinity chromatography, and quantitative MS successfully identified at least two novel ARE-BPs in addition to a large number of known ARE-BPs.
DISCUSSION
In this study, we improved a SA-binding aptamer by elongating the basal stem of the original S1 aptamer (30), stabilizing its structure similar to the design in (49) and concatemerizing four aptamers in a single tag. With the optimized 4×S1m aptamer, the recovery of a reporter mRNA expressed in cells could be increased by 15-fold (Figure 1). In our hands, 4×S1m was also more efficient than the MS2 and PP7 systems (Figure 2). An SA-binding aptamers offer several advantages such as a straightforward one-step purification scheme without the need for recombinant protein production, readily available matrices and the option to either tag in vitro transcribed RNAs or RNAs expressed in cells. A previous report has compared S1 to the use of biotin-labeled ribonucleotides incorporated during in vitro transcription and found the S1 aptamer to be superior (56). Other investigators also found that elongating the basal stem of S1 increases binding to SA (49,57). Moreover, stabilizing S1 by addition of a tRNA scaffold increased binding efficiency 10-fold (34). These authors also tested six tandemly repeated S1 motifs, yet found the 6-fold repeat to be much less efficient than the single S1 aptamer. In contrast, our most efficient construct, 4×S1m, clearly benefits from an avidity effect. In addition, the S1m RNA as a single motif binds with 2-fold higher affinity to SA than the original S1 aptamer (Figure 3). Thus, elongation of the basal stem in S1m might induce a sterically favorable conformation that stabilizes the apical binding portion of the aptamer, and further helps the binding portion to protrude from the mRNP: both would facilitate retention on SA Sepharose beads.
Our next goal was to purify mRNPs from cells using the 4×S1m aptamer. However, MS of these purifications showed high levels of contaminating proteins and did not yield the expected pattern of RNA- and ARE-BPs (data not shown). One important limitation is that even with the optimized 4×S1m aptamer, we could only retrieve 4.5% of the tagged reporter mRNA from cells (Figure 1E). We are aware of one example where the S1 aptamer was used successfully for purification of a cellular mRNP (32). In that case, additional cross-linking and gel filtration steps increased specificity of the purification scheme. We envision that the improved 4×S1m aptamer may offer an advantage in such a combined protocol.
We decided to use the 4×S1m aptamer for purification of ARE-BPs from cellular extracts, as the binding efficiency of in vitro transcribed 4×S1m RNA to SA is high (Figure 3C and 5A). After incubating cell lysates with the ARE-4×S1m-coupled matrix and subsequent washing, RNA-associated proteins were eluted with RNase A. This was more efficient than elution with biotin (Figure 3C) and much more specific than elution with SDS (Figure 5B). Key to successful identification of ARE-BPs was the direct comparison of the ARE-4×S1m and 4×S1m samples by quantitative MS using differential dimethyl peptide-labeling. About 200 proteins were co-purified with similar efficiencies in both samples (enrichment 0.5–2.0-fold, peptide coverage >5%), which is also reflected in the similar patterns of protein bands in the eluates of the two purifications (Figure 5C). In contrast, only 16 proteins were associated preferentially with the TNFα ARE (enrichment >8-fold, Figure 5D) including most of the established ARE-BPs: HuR, TIAR, AUF1, BRF1 and BRF2. Only two of the major ARE-BPs, TTP and KSRP, were not enriched in our purification. For TTP, the reason might be that its expression is low in resting fibroblasts (58). Taken together, our results demonstrate that purification via the 4×S1m tag was efficient at identifying specific bona fide RNA-interacting proteins.
In addition to known ARE-BPs, we also identified five RNA-BPs that had previously not been associated with AREs. They included two proteins of the to U2 snRNP (A’ and B’’) and a general snRNP protein (Sm-D2), suggesting that the ARE-4×S1m RNA might base-pair with U2 snRNA and retain U2 snRNPs. Alternatively, ARE-4×S1m may fold into a motif with similarity to U2 snRNA, and thus bind U2-associated proteins directly. Although the interaction of these proteins was not studied further, we did confirm binding of Rbms1 and Roxan to the ARE by RNA-IP (Figure 6). Rbms1 has so far not been studied with respect to its RNA-binding function, yet the reported connections to c-Myc (52,54) pose the question whether Rbms1 might bind to the ARE located in the 3′UTR of c-Myc mRNA. Roxan has been implicated in rotavirus mRNA translation (42), and it will be interesting to determine whether Roxan might also contribute to the ability of AREs to repress translation of cellular mRNAs.
Recently, we applied 4×S1m-mediated RNA affinity chromatography to identify Roquin as the major protein that interacts with the CDE, a stem-loop RNA degradation motif located downstream of the ARE in the TNFα 3′UTR (41). We anticipate that SA-binding aptamers will continue to be useful for identifying RNA-protein interactions and help uncover the intricate nature of mRNP complexes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft [STO 859/3-1 and SFB 1036/TP07 to G.S.]; PhD stipend from the Helmholtz International Graduate School for Cancer Research (to K.L.). Funding for open access charge: Deutsche Forschungsgemeinschaft [STO 859/3-1].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Kathleen Collins (University of California Berkeley, USA), David Peabody (University of New Mexico School of Medicine, USA), Jens Lykke-Andersen (University of California San Diego, USA), Didier Poncet (CNRS, Gif sur Yvette, France), as well as Stephen Wax and Paul Anderson (Brigham and Women’s Hospital, Boston, USA) for generously providing plasmids and cDNAs. The authors are grateful to Ludger Hengst (Innsbruck Medical University, Austria) and Frauke Melchior (ZMBH, University of Heidelberg, Germany) for providing the anti-GST antibody, and Ann-Bin Shyu (University of Texas-Houston Medical School) for the NIH3T3 B2A2 cell line. The authors also thank Thomas Ruppert from the ZMBH Mass Spectrometry Core Facility (University of Heidelberg, Germany) for extensive support, Matthias Mayer and Christine Clayton (both ZMBH, University of Heidelberg, Germany) for critical comments on the article and all members of the Stoecklin laboratory for support.
REFERENCES
- 1.Hieronymus H, Silver PA. A systems view of mRNP biology. Genes Dev. 2004;18:2845–2860. doi: 10.1101/gad.1256904. [DOI] [PubMed] [Google Scholar]
- 2.Moore MJ. From birth to death: the complex lives of eukaryotic mRNAs. Science. 2005;309:1514–1518. doi: 10.1126/science.1111443. [DOI] [PubMed] [Google Scholar]
- 3.Keene JD. Minireview: global regulation and dynamics of ribonucleic acid. Endocrinology. 2010;151:1391–1397. doi: 10.1210/en.2009-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lingner J, Cech TR. Purification of telomerase from Euplotes aediculatus: requirement of a primer 3′ overhang. Proc. Natl Acad. Sci. USA. 1996;93:10712–10717. doi: 10.1073/pnas.93.20.10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mitchell SF, Jain S, She M, Parker R. Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 2013;20:127–133. doi: 10.1038/nsmb.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 10.Mogridge J, Mah TF, Greenblatt J. A protein-RNA interaction network facilitates the template-independent cooperative assembly on RNA polymerase of a stable antitermination complex containing the lambda N protein. Genes Dev. 1995;9:2831–2845. doi: 10.1101/gad.9.22.2831. [DOI] [PubMed] [Google Scholar]
- 11.Czaplinski K, Kocher T, Schelder M, Segref A, Wilm M, Mattaj IW. Identification of 40LoVe, a Xenopus hnRNP D family protein involved in localizing a TGF-beta-related mRNA during oogenesis. Dev. Cell. 2005;8:505–515. doi: 10.1016/j.devcel.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Carey J, Cameron V, de Haseth PL, Uhlenbeck OC. Sequence-specific interaction of R17 coat protein with its ribonucleic acid binding site. Biochemistry. 1983;22:2601–2610. doi: 10.1021/bi00280a002. [DOI] [PubMed] [Google Scholar]
- 13.Lim F, Downey TP, Peabody DS. Translational repression and specific RNA binding by the coat protein of the Pseudomonas phage PP7. J. Biol. Chem. 2001;276:22507–22513. doi: 10.1074/jbc.M102411200. [DOI] [PubMed] [Google Scholar]
- 14.Bertrand E, Chartrand P, Schaefer M, Shenoy SM, Singer RH, Long RM. Localization of ASH1 mRNA particles in living yeast. Mol. Cell. 1998;2:437–445. doi: 10.1016/s1097-2765(00)80143-4. [DOI] [PubMed] [Google Scholar]
- 15.Lykke-Andersen J, Shu MD, Steitz JA. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell. 2000;103:1121–1131. doi: 10.1016/s0092-8674(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 16.Baron-Benhamou J, Gehring NH, Kulozik AE, Hentze MW. Using the lambdaN peptide to tether proteins to RNAs. Methods Mol. Biol. 2004;257:135–154. doi: 10.1385/1-59259-750-5:135. [DOI] [PubMed] [Google Scholar]
- 17.Coller J, Wickens M. Tethered function assays using 3′ untranslated regions. Methods. 2002;26:142–150. doi: 10.1016/S1046-2023(02)00016-6. [DOI] [PubMed] [Google Scholar]
- 18.Ozgur S, Chekulaeva M, Stoecklin G. Human Pat1b Connects Deadenylation with mRNA Decapping and Controls the Assembly of Processing-Bodies. Mol. Cell. Biol. 2010;30:4308–4323. doi: 10.1128/MCB.00429-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bardwell VJ, Wickens M. Purification of RNA and RNA-protein complexes by an R17 coat protein affinity method. Nucleic Acids Res. 1990;18:6587–6594. doi: 10.1093/nar/18.22.6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slobodin B, Gerst JE. A novel mRNA affinity purification technique for the identification of interacting proteins and transcripts in ribonucleoprotein complexes. RNA. 2010;16:2277–2290. doi: 10.1261/rna.2091710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee N, Pimienta G, Steitz JA. AUF1/hnRNP D is a novel protein partner of the EBER1 noncoding RNA of Epstein-Barr virus. RNA. 2012;18:2073–2082. doi: 10.1261/rna.034900.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoon JH, Srikantan S, Gorospe M. MS2-TRAP (MS2-tagged RNA affinity purification): tagging RNA to identify associated miRNAs. Methods. 2012;58:81–87. doi: 10.1016/j.ymeth.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong C, Popp MW, Maquat LE. Biochemical analysis of long non-coding RNA-containing ribonucleoprotein complexes. Methods. 2012;58:88–93. doi: 10.1016/j.ymeth.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Said N, Rieder R, Hurwitz R, Deckert J, Urlaub H, Vogel J. In vivo expression and purification of aptamer-tagged small RNA regulators. Nucleic Acids Res. 2009;37:e133. doi: 10.1093/nar/gkp719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hogg JR, Collins K. RNA-based affinity purification reveals 7SK RNPs with distinct composition and regulation. RNA. 2007;13:868–880. doi: 10.1261/rna.565207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hogg JR, Goff SP. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell. 2010;143:379–389. doi: 10.1016/j.cell.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee HY, Haurwitz RE, Apffel A, Zhou K, Smart B, Wenger CD, Laderman S, Bruhn L, Doudna JA. RNA-protein analysis using a conditional CRISPR nuclease. Proc. Natl Acad. Sci. USA. 2013;110:5416–5421. doi: 10.1073/pnas.1302807110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bachler M, Schroeder R, von Ahsen U. StreptoTag: a novel method for the isolation of RNA-binding proteins. RNA. 1999;5:1509–1516. doi: 10.1017/s1355838299991574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartmuth K, Urlaub H, Vornlocher HP, Will CL, Gentzel M, Wilm M, Luhrmann R. Protein composition of human prespliceosomes isolated by a tobramycin affinity-selection method. Proc. Natl Acad. Sci. USA. 2002;99:16719–16724. doi: 10.1073/pnas.262483899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srisawat C, Engelke DR. Streptavidin aptamers: affinity tags for the study of RNAs and ribonucleoproteins. RNA. 2001;7:632–641. doi: 10.1017/s135583820100245x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Altman S. Partial reconstitution of human RNase P in HeLa cells between its RNA subunit with an affinity tag and the intact protein components. Nucleic Acids Res. 2002;30:3706–3711. doi: 10.1093/nar/gkf499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasudevan S, Steitz JA. AU-rich-element-mediated upregulation of translation by FXR1 and argonaute 2. Cell. 2007;128:1105–1118. doi: 10.1016/j.cell.2007.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dienstbier M, Boehl F, Li X, Bullock SL. Egalitarian is a selective RNA-binding protein linking mRNA localization signals to the dynein motor. Genes Dev. 2009;23:1546–1558. doi: 10.1101/gad.531009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iioka H, Loiselle D, Haystead TA, Macara IG. Efficient detection of RNA-protein interactions using tethered RNAs. Nucleic Acids Res. 2011;39:e53. doi: 10.1093/nar/gkq1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dix CI, Soundararajan HC, Dzhindzhev NS, Begum F, Suter B, Ohkura H, Stephens E, Bullock SL. Lissencephaly-1 promotes the recruitment of dynein and dynactin to transported mRNAs. J. Cell. Biol. 2013;202:479–494. doi: 10.1083/jcb.201211052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schott J, Stoecklin G. Networks controlling mRNA decay in the immune system. Wiley Interdiscip. Rev. RNA. 2010;1:432–456. doi: 10.1002/wrna.13. [DOI] [PubMed] [Google Scholar]
- 37.Ronkina N, Menon MB, Schwermann J, Tiedje C, Hitti E, Kotlyarov A, Gaestel M. MAPKAP kinases MK2 and MK3 in inflammation: complex regulation of TNF biosynthesis via expression and phosphorylation of tristetraprolin. Biochem. Pharmacol. 2010;80:1915–1920. doi: 10.1016/j.bcp.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 38.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stoecklin G, Stoeckle P, Lu M, Muehlemann O, Moroni C. Cellular mutants define a common mRNA degradation pathway targeting cytokine AU-rich elements. RNA. 2001;7:1578–1588. [PMC free article] [PubMed] [Google Scholar]
- 41.Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell. 2013;153:869–881. doi: 10.1016/j.cell.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 42.Vitour D, Lindenbaum P, Vende P, Becker MM, Poncet D. RoXaN, a novel cellular protein containing TPR, LD, and zinc finger motifs, forms a ternary complex with eukaryotic initiation factor 4G and rotavirus NSP3. J. Virol. 2004;78:3851–3862. doi: 10.1128/JVI.78.8.3851-3862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ozgur S, Stoecklin G. Role of Rck-Pat1b binding in assembly of processing-bodies. RNA. Biol. 2013;10:528–539. doi: 10.4161/rna.24086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu N, Loflin P, Chen CY, Shyu AB. A broader role for AU-rich element-mediated mRNA turnover revealed by a new transcriptional pulse strategy. Nucleic Acids Res. 1998;26:558–565. doi: 10.1093/nar/26.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protoc. 2009;4:484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 46.Sandler H, Kreth J, Timmers HT, Stoecklin G. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic Acids Res. 2011;39:4373–4386. doi: 10.1093/nar/gkr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Srisawat C, Engelke DR. RNA affinity tags for purification of RNAs and ribonucleoprotein complexes. Methods. 2002;26:156–161. doi: 10.1016/S1046-2023(02)00018-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oeffinger M, Wei KE, Rogers R, DeGrasse JA, Chait BT, Aitchison JD, Rout MP. Comprehensive analysis of diverse ribonucleoprotein complexes. Nat. Methods. 2007;4:951–956. doi: 10.1038/nmeth1101. [DOI] [PubMed] [Google Scholar]
- 49.Xu D, Shi H. Composite RNA aptamers as functional mimics of proteins. Nucleic Acids Res. 2009;37:e71. doi: 10.1093/nar/gkp243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J. 2002;21:6505–6514. doi: 10.1093/emboj/cdf639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morton S, Yang HT, Moleleki N, Campbell DG, Cohen P, Rousseau S. Phosphorylation of the ARE-binding protein DAZAP1 by ERK2 induces its dissociation from DAZ. Biochem. J. 2006;399:265–273. doi: 10.1042/BJ20060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Negishi Y, Nishita Y, Saegusa Y, Kakizaki I, Galli I, Kihara F, Tamai K, Miyajima N, Iguchi-Ariga SM, Ariga H. Identification and cDNA cloning of single-stranded DNA binding proteins that interact with the region upstream of the human c-myc gene. Oncogene. 1994;9:1133–1143. [PubMed] [Google Scholar]
- 53.Niki T, Galli I, Ariga H, Iguchi-Ariga SM. MSSP, a protein binding to an origin of replication in the c-myc gene, interacts with a catalytic subunit of DNA polymerase alpha and stimulates its polymerase activity. FEBS Lett. 2000;475:209–212. doi: 10.1016/s0014-5793(00)01679-3. [DOI] [PubMed] [Google Scholar]
- 54.Niki T, Izumi S, Saegusa Y, Taira T, Takai T, Iguchi-Ariga SM, Ariga H. MSSP promotes ras/myc cooperative cell transforming activity by binding to c-Myc. Genes Cells. 2000;5:127–141. doi: 10.1046/j.1365-2443.2000.00311.x. [DOI] [PubMed] [Google Scholar]
- 55.Harb M, Becker MM, Vitour D, Baron CH, Vende P, Brown SC, Bolte S, Arold ST, Poncet D. Nuclear localization of cytoplasmic poly(A)-binding protein upon rotavirus infection involves the interaction of NSP3 with eIF4G and RoXaN. J. Virol. 2008;82:11283–11293. doi: 10.1128/JVI.00872-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Butter F, Scheibe M, Morl M, Mann M. Unbiased RNA-protein interaction screen by quantitative proteomics. Proc. Natl Acad. Sci. USA. 2009;106:10626–10631. doi: 10.1073/pnas.0812099106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leonov AA, Sergiev PV, Bogdanov AA, Brimacombe R, Dontsova OA. Affinity purification of ribosomes with a lethal G2655C mutation in 23 S rRNA that affects the translocation. J. Biol. Chem. 2003;278:25664–25670. doi: 10.1074/jbc.M302873200. [DOI] [PubMed] [Google Scholar]
- 58.Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ. Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts. Mol. Cell. Biol. 2006;26:9196–9208. doi: 10.1128/MCB.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.