

Abstract
Background
Dendritic cells (DC) pulsed with MHC class I-restricted tumour associated antigen (TAA) peptides have been widely tested in pre-clinical models and early clinical studies for their ability to prime cytotoxic T cell (CTL) responses. The effect of co-expression of allogeneic MHC antigens on DC immunogenicity has not been addressed, and has implications for the feasibility of clinical applications.
Objective
This study compared DC from autologous H-2b or semi-allogeneic F1 H-2bxk mice pulsed with the H-2b-restricted model ovalbumin (OVA) peptide SIINFEKL, and compared in vitro and in vivo their ability to (i) activate specific OT1 cells, (ii) prime naïve CTL, and (iii) protect against B16.OVA challenge. Peptide-pulsed autologous and allogeneic DC were also tested in naïve human CTL priming assays.
Results
Semi-allogeneic DC expressed higher levels of co-stimulatory molecules. On pulsing with SIINFEKL they triggered greater proliferation of OT1 cells in vitro and in vivo, but were less effective at naïve CTL priming and tumour protection. Autologous human DC were similarly more potent at naïve CTL priming against the melanoma-associated TAA MART-1 in vitro.
Conclusion
The expression of allogeneic MHC antigens on peptide-pulsed DC impairs naïve CTL priming and anti-tumour effects, despite effective TAA presentation both in vitro and in vivo.
Keywords: Dendritic cells, Cancer vaccines, Tumour-associated antigens, Cancer immunotherapy
Introduction
Dendritic cells are the most potent of antigen presenting cells (APC) with a unique ability to prime naïve T cell responses [1, 2]. Dendritic cells (DC) can acquire antigen in the periphery and, on appropriate activation, migrate to lymph nodes to initiate generation of antigen specific CD4 and CD8 T cell responses [3]. In vivo DC can be divided into multiple subtypes in both human and mouse, with significant plasticity between categories and function, often defined by the cellular and cytokine context in which DC find themselves. Dendritic cells can either support or suppress T cell responses, depending on the balance of multiple signals which can affect DC maturation and activation. Immature DC, presenting antigen without effective T cell co-stimulation, trigger immune tolerance or anergy, whilst fully activated mature DC support T cell expansion and subsequent cytotoxicity [4]. Dendritic cells also provide a critical link between early innate immunity and subsequent antigen-specific responses, via interaction and cross-talk with other effector cells such as NK cells [5].
Since DC can now be cultured in large numbers ex vivo from patients as well as normal donors, there is currently great interest in translational studies which seek to apply DC-mediated immune control to pathological disease states. The majority of clinical trials to date have been in cancer immunotherapy, using DC loaded with TAA to activate specific T cell-mediated responses against the tumour. In these protocols, TAA have been loaded as peptides, protein, tumour cell lysates, DNA, RNA or expressed by viral or non-viral vectors transfected into DC [6]. Alternatively, DC have been injected directly into tumours to acquire TAA in situ, or fused to tumour cells ex vivo to generate hybrids which encode the full repertoire of TAA in the context of a potent, activated APC [7, 8]. Although there have been some encouraging immunological and clinical responses to date, these early studies have yet to make an impact on standard clinical practice.
Among the many uncertainties surrounding the optimal preparation and application of DC to cancer immunotherapy is the best source of the cultured DC themselves. Dendritic cells can either be autologous (cultured individually from each patient to be treated), or allogeneic (derived from normal donors). Both approaches have potential advantages and drawbacks. Autologous DC are fully MHC matched to the recipient, maximising the breadth of their antigen presentation to potentially reactive T cells; however, preparation of DC from each individual patient to clinical grade is a technical challenge for widespread clinical practice. Although allogeneic DC are more convenient, even partially MHC matched donors will initiate a rejection response to some degree in the recipient. This may lead to early clearance of the vaccinating DC or overwhelm any specific priming against the TAA they carry. On the other hand, we and others have shown that expression of allogeneic MHC antigens, at least on tumour cells, may act as an immune “danger” signal, potentially supporting T cell priming against specific TAA [9, 10].
Whilst the majority of cancer trials to date have used autologous DC, allogeneic DC have been tested in some studies [11–13]. Preclinical data directly comparing autologous and allogeneic DC is currently limited, and has mainly involved vaccine strategies fusing allogeneic DC to autologous tumour cells. In that context, expression of allogeneic MHC antigens did not restrict priming of anti-tumour immunity in murine models or human in vitro systems [14–16]. When allogeneic and autologous DC fused to tumour cells were directly compared in mice in vivo, hybrids integrating allogeneic DC were at least as effective [17, 18]. In an alternative approach, autologous or allogeneic DC transfected with an adenovirus encoding IL-12 were injected directly into established tumours; here, expression of allogeneic MHC antigens appeared to improve therapy [19]. Hence in these specific applications, the current evidence suggests that allogeneic DC are a reasonable and practical source of APC for clinical use.
The most common approach to DC antigen loading in trials to date has not been fusion or transfection. Rather, many trials, particularly for the treatment of melanoma, have pulsed DC with single or multiple defined TAA peptide epitopes. Peptides can readily be manufactured to clinical grade, providing a practical source of antigens which are known to play a potential role in anti-tumour protection. A significant number of trials have now confirmed that peptide-pulsed DC can stimulate specific T cell expansion in patients [1, 20–22]. Since these epitopes are presented to T cells by specific MHC class I molecules, the vaccinating DC must express the appropriate class I allele, and therefore autologous DC have been widely used in this setting. However, the question of whether co-expression of allogeneic MHC antigens on partially matched DC (matched only for the allele required for presentation of the specific therapeutic peptide), might enhance or compromise the potency of peptide-pulsed DC, has not been addressed.
Materials and methods
Dendritic cell culture and characterisation
Six to eight week old female C57Bl/6 (H-2Kb) and C57BL/CBA F1 cross (H-2Kbxk) mice, purchased from Harlan laboratories (Oxon, UK) were housed under pathogen-free conditions. Dendritic cells were generated in vitro as previously described [10]; briefly, murine bone marrow was flushed from the femurs and tibias of euthanized mice, and disaggregated by passing through a 30 μm cell sieve. After washing, B and T cells and granulocytes were labelled using biotinylated anti-B220, anti-CD3ε, anti-CD4 and anti-Gr1 anti-mouse antibodies (BD Pharmingen, Cowly, Oxford, UK), then depleted using streptavidin coated magnetic beads and a midiMACS™ separator column (Miltenyi Biotech, Bisley, Surrey, UK). The enriched progenitor cells were cultured at no more than 1 × 106 cells/ml in RPMI 1640 medium supplemented with 10% v/v foetal calf serum, 2 mML-glutamine, 50 μm β-mercaptoethanol and GM-CSF as previously described [23], (we have not found any significant differences in DC cultured with or without additional IL-4 in this protocol), at which point approximately 70% were CD11c+ by flow cytometry (data not shown). Additional media was added to cultures as required. On day 7, immature DC were obtained as loosely adherent/suspension cells and matured overnight with 100 ng/ml LPS.
The MHC class I phenotype and maturity of DC was assessed by flow cytometry. Cells were pre-incubated with anti-CD16 to block non-specific FcγR binding of labelled antibodies, then incubated with phycoerythrin-conjugated anti-H-2Kb MHC I, H-2Kk MHC I, CD80, CD86 or MHC II Abs (BD Pharmingen) for 30 min at 4°C. Isotype-matched antibodies were used as negative controls. Flow cytometric analysis was performed on a FACScaliber system (Becton Dickinson, Cowly, Oxford, UK) and data analysed with CellQuest-pro software.
Mixed lymphocyte reaction
Primary mixed lymphocyte reactions were performed using mature DC from C57Bl/6 or C57Bl/CBA F1 mice, and third party allogeneic splenocytes. Splenocytes were isolated from naïve BALB/c mice, depleted of red blood cells by incubation in lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA), then plated in round-bottomed 96 well culture plates at 2 × 105 cells per well. Stimulator DC were serially diluted and added at different ratios to the responding splenocytes and co-incubated for 4 days. 3H-thymidine (0.5 μCi per well) was added for 18 h before harvesting (TOMTEC Harvester 96 Mach IIIM) onto filter mats and counting (Wallac Jet 1450 Microbeta scintillation counter and Microbeta windows software, Wallac, OY, Finland). Tests were carried out in sextuplet and results expressed as mean counts per minute. The levels of 3H-thymidine uptake by stimulator cells alone were consistently below 1000 cpm.
Presentation by peptide-pulsed DC to specific OT-1 lymphocytes in vitro
Day 8 mature autologous or semi-allogeneic DC were pulsed with SIINFEKL peptide for 1 h at 37°C before being washed and plated out in 96 well culture plates at 1 × 105 cells per well. Splenocytes from OT-1+Rag-/- mice were isolated and red blood cell depleted. CD8 T cells were purified by a CD8T cell isolation kit (Miltenyi Biotec) and stained with CFSE (5 μM). The CD8 T cells were added to the DC cultures at a 1:1 ratio for 3 days. OT-1 cell division was visualised by dilution of the CFSE staining analysed by flow cytometry.
In vivo peptide-pulsed DC vaccination
All animal work was approved by the appropriate regulatory authorities. C57BL/6 animals were vaccinated subcutaneously (s.c.) with 1 × 105 peptide-pulsed mature DC (100 μl) or vehicle (PBS) control on days 1 and 8, and challenged with live B16.OVA cells on day 15. In numerous previous experiments in our laboratory, unpulsed DC had never protected against B16.OVA in this protocol, and were therefore not included as a routine control. Animals were monitored and the tumour diameter measured thrice weekly. Animals were killed when tumour size was approximately 1.0 × 1.0 cm in two perpendicular directions. Animals were considered to have a tumour (to distinguish from a swelling or inflammation in reaction to injection) when measurement was in excess of 0.2 cm in the longest diameter; data is presented as percentage tumour free mice in individual groups over time. All groups of mice in any one individual experiment were challenged on the same occasion using the same preparation of cells; a naïve group of mice was also injected with these cells at the same time. Data from the animal studies were analysed by the log-rank test.
Priming of naïve cytotoxic T cells in vivo
Separate groups of mice were vaccinated as above and euthanized 4 weeks later. Splenocytes were harvested and red blood cells depleted as above. The cells were re-stimulated with irradiated EG7.OVA cells at a ratio of 10:1 splenocytes:EG7.OVA cells for 7 days before harvesting, and their cytotoxicity measured using a standard 4 h 51Cr release assay. Briefly, RMA-S target cells were pulsed with 4 μg/ml SIINFEKL or irrelevant peptide (Trp-2) and labelled with 100 μCi Cr51 for 1 h, then washed three times and co-cultured with splenocytes at different effector:target ratios. To reduce background/non-specific killing, the same number of unlabelled YAC-1 cells as target cells was added to each condition. After 4 h cells were spun down and 50 μl of supernatant was transferred to scintillation plates (Packard Biosciences, Groningen, Netherlands) and dried overnight before counting. Percentage lysis was calculated using the following formula:
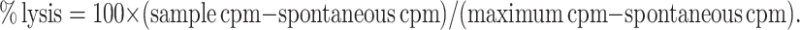 |
Proliferation of adoptively transferred OT-1 cells following vaccination in vivo
Naïve, purified, CFSE labelled OT-1 T cells as described above, were transferred via tail vein injection at 2.5 × 106 per mouse (three mice per group). On the following day, mice were given 3 × 106 peptide-pulsed autologous or semi-allogeneic DC subcutaneously. Two days later, draining lymph nodes and spleens were collected and analysed by flow cytometry for the presence of dividing OT-1 cells.
Human naïve cytotoxic T cell priming
Collection of the PBMC fraction from normal blood donors was approved by the appropriate regulatory authorities. Autologous or allogeneic HLA-A2+ mature DC were pulsed with HLA-A2-restricted MelanA/MART-127–35 (AAGIGILTV) peptide for 1 h and mixed with PBMC at a 1:10–1:30 ratio in RPMI 1640 supplemented with 2 mM l-glutamine, 1% v/v non essential amino-acids, 1 mM sodium pyruvate, 10 mM HEPES, 20 mM β-mercaptoethanol, and 7.5% FCS. 5 ng/ml IL-7 (R + D systems, Abingdon, UK) was added to cultures from day 1; 30U/ml IL-2 (R + D systems) was added on day 3 only. T cells were re-stimulated twice at weekly intervals, each time with peptide-pulsed autologous or allogeneic DC in identical proportions. At 21 days, cells were harvested and cytotoxicity of CTL measured using a standard 4 h 51Cr release assay as above with minor modifications. T2 target cells were pulsed with 4 μg/ml MART-1 or irrelevant peptide (flu matrix) and Cr51 labelled before co-culture with CTL at different effector:target ratios. To reduce background/non-specific killing, the same number of unlabelled K562 cells as target cells were added to each condition. After 4 h cells were spun down and 50 μl of supernatant was transferred to scintillation plates and analysed as above.
Elisa
Supernatant levels of IFN-γ were measured by ELISA using matched paired antibodies (BD Pharmingen) according to the manufacturer’s instructions.
Results
Characterisation of autologous and semi-allogeneic DC
To compare DC which share the H-2b class I allele capable of presenting SIINFEKL, but differ in their expression of other class I molecules allogeneic to a vaccinated H-2b host, we used DC cultured from the bone marrow of C57BL H-2b mice, or from F1 crossed C57BLxCBA, H-2bxk animals. These autologous H-2b DC, and semi-allogeneic H-2bxk DC, represent individualised or partially matched donor DC as might be used for clinical application of a peptide-loaded DC strategy. In these experiments DC were activated with LPS, as required for full maturation and effective presentation of exogenously pulsed peptide. Figure 1a shows MHC class I expression on the two sets of mature DC, confirming expression of H-2k only on DC cultured from F1 animals. Consistent with previous descriptions of murine DC phenotype [24, 25], we saw a biphasic distribution of class I expression. Although the percentage of DC expressing high levels of H-2b was greater for semi-allogeneic DC than autologous (62 vs. 54%), the overall MFI was consistently higher on autologous DC (114 vs. 74).
Fig. 1.
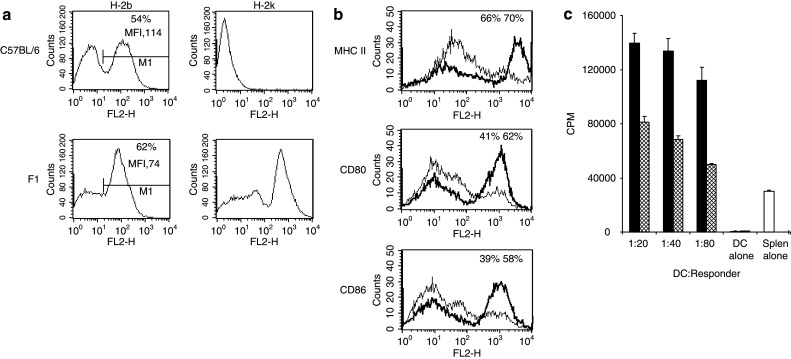
Characterisation of DC. a MHC I H-2k and H-2b expression on C57BL/6 or F1-cross derived mature DC was determined by FACS analysis. 54% of C56BL/6 DC (MFI 114) and 62% of F1 DC (MFI 74) had high levels of H-2b expression. b Markers of DC phenotypic maturation; MHC II, CD80 and CD86 were also determined by FACS analysis. Figures shown are percentages of all positively stained cells (F1 DC black; C57BL/6 DC grey). c Functionally mature DC from C57BL/6 (hatched bar) and F1 (black bar) mice were compared in their ability to stimulate a proliferative response in third party splenocytes from BALB/c mice (H-2d). Proliferation was measured by 18 h 3H-thymidine incorporation. Results shown from one representative assay from three separate experiments and are mean values from sextuplet experimental wells
We also compared further markers of phenotypic DC maturation between the two types of DC. Again, and consistent with previous data [18, 26], we saw differences between DC derived from the two strains, with semi-allogeneic mature DC expressing higher levels of the activation markers MHC class II, CD80 and CD86 (Fig. 1b). There was no difference in cytokine (IL-12) production by autologous and semi-allogeneic DC (data not shown).
On functional testing in an allogeneic MLR against a third mouse strain (BALB/c; H-2d), semi-allogeneic DC were also more potent at stimulating proliferation of responder splenocytes (Fig. 1c).
In vitro peptide presentation by autologous and semi-allogeneic DC to antigen specific T cells
Transgenic OT1 cells recognise the OVA-derived peptide SIINFEKL in the context of H-2b class I presentation. Next, we labelled OT1 cells with CSFE, and co-cultured them with SIINFEKL-pulsed mature autologous or semi-allogeneic DC in vitro. Dilution of the CFSE dye over time, as illustrated by a reduction in cell fluorescence on FACS analysis, represents T cell proliferation following effective presentation of peptide leading to T cell proliferation. Although both sets of DC were capable of triggering OT1 proliferation (Fig. 2), this effect was greater for presentation of SIINFEKL by semi-allogeneic DC.
Fig. 2.

Stimulation of antigen-specific T cells by DC in vitro. Transgenic OT1 cells were labelled with CSFE and co-cultured with either SIINFEKL-pulsed (black histogram) or unpulsed (grey histogram) mature C57BL/6 (autologous) or F1 (semi-allogeneic) derived DC in vitro. Flow cytometric analysis of the cells reflects OT1 cell proliferation by dilution of the CFSE dye 72 h later. Results shown from one representative assay from three separate experiments
SIINFEKL-pulsed autologous and semi-allogeneic DC as in vivo vaccines against OVA-expressing B16 melanoma
The data in Figs. 1 and 2 showed that SIINFEKL-pulsed semi-allogeneic DC were more effective than autologous DC at presenting peptide to specific, reactive CD8 T cells in vitro. Surprisingly however, when autologous and semi-allogeneic DC were compared as in vivo vaccines for their ability to protect mice against a subsequent challenge with live B16.OVA, autologous DC were consistently more potent (Fig. 3). Although peptide-pulsed semi-allogeneic DC protected 40% of mice against challenge with 2 × 105 live B16.OVA tumour cells (Fig. 3a), they were significantly less effective than autologous DC (P = 0.025), which protected 100% of animals in the same protocol. At a higher challenge dose of 5 × 105 cells, semi-allogeneic DC afforded no protection, whilst autologous DC still provided a statistically significant delay in tumour growth (P = 0.005 compared to semi-allogeneic DC), though with limited complete tumour rejection (Fig. 3b).
Fig. 3.

In vivo vaccination with peptide-pulsed DC. Animals were vaccinated with SIINFEKL pulsed DC or PBS control on days 1 and 8 and challenged with either 2 × 105 (a) or 5 × 105 (b) live B16.OVA tumour cells on day 15. Groups of mice (n = 6 – 8) were followed and tumour size monitored at regular intervals; data is plotted as % of tumour-free animals. Autologous DC vaccines gave significantly better protection than semi-allogeneic DC vaccines with 2 × 105 (P = 0.025) or 5 × 105 (P = 0.005) challenge doses. Autologous DC vaccine group (filled diamond); Semi-allogeneic DC vaccine group (filled square); Vehicle (PBS) vaccine group (filled triangle)
Naïve peptide-specific CTL priming in vivo following vaccination with SIINFEKL-pulsed autologous or semi-allogeneic DC
To explore potential mechanisms underlying the inferiority of semi-allogeneic DC vaccines at protecting against tumour challenge in vivo, despite their ability to effectively present TAA in vitro, we next tested priming of a peptide-specific naïve CTL response in mice following SIINFEKL-pulsed DC vaccination. As shown in Fig. 4, CTL induction correlated with the tumour protection data shown in Fig. 3, with only autologous DC priming peptide-specific cytotoxicity. No significant killing was seen by splenocytes from mice vaccinated with semi-allogeneic DC in this assay.
Fig. 4.

Priming of naïve peptide-specific CTL in vivo following vaccination with peptide-pulsed DC. Splenocytes were harvested from mice 4 weeks after vaccination with SIINFEKL-pulsed autologous (a) or semi-allogeneic (b) DC. Cell cytotoxicity was measured in a standard 4 h 51Cr release assay against SIINFEKL (filled diamond) pulsed or irrelevant peptide (open diamond) pulsed RMA-S target cells
Proliferation of adoptively transferred OT1 cells in vivo by autologous or semi-allogeneic peptide-pulsed DC
To address whether T cells which are already specific and expanded can be effectively accessed and stimulated by vaccinating DC in vivo, we next injected OT1 cells intravenously the day before subcutaneous delivery of SIINFEKL-pulsed autologous or semi-allogeneic DC. Adoptively transferred OT1 cells were again labelled with CFSE, so that lymph nodes draining the DC injection site and spleens could be harvested, and T cell proliferation analysed by dilution of CFSE fluorescence as previously. As shown in Fig. 5, peptide-pulsed semi-allogeneic DC supported division of OT1 cells more effectively than autologous DC in both lymph nodes draining the DC vaccination site, and in the spleen.
Fig. 5.

Proliferation of adoptively transferred peptide-specific OT1 cells in vivo following peptide-pulsed DC vaccination. CFSE-labelled OT1 cells were adoptively transferred into mice prior to peptide-pulsed autologous or semi-allogeneic DC vaccination. Draining lymph nodes (LN) and spleens were harvested after 48 h and OT1 cell proliferation analysed by dilution of CFSE fluorescence. The CFSE labelled OT1 cells are in the top right-hand quadrants of the analysis plots; the figure represents the percentage of labelled OT1 cells which have divided. Representative data from one of three mice is shown
Naïve CTL priming against a melanoma-associated antigenic epitope by autologous or allogeneic human peptide-pulsed DC
To address the relevance of the superiority of autologous peptide-pulsed DC in naïve CTL priming to human immunotherapy, we next compared DC cultured from pairs of HLA-A2 positive normal donors. Mature DC from each were pulsed with the A2-restricted epitope MART-1, and then cultured either with their own lymphocytes, or with lymphocytes from the other donor. This crossover allowed direct comparison of CTL induction by autologous and allogeneic DC, in a system where A2 expression and the ability to present the defined MART-1 epitope remained common to both. The CTL assays from a representative pair of donors are shown in Fig. 6a and confirm, consistent with our murine in vivo model, that autologous DC were more effective at priming a naïve cytotoxic response. Although significant levels of specific CTL killing on priming with allogeneic DC were observed, these remained consistently lower than when autologous DC were used for priming, at all effector:target ratios tested. This superior CTL priming with autologous DC occurred despite greater overall T cell proliferation (Fig. 6b) and IFN-γ production (Fig. 6c) in allogeneic cultures, consistent with a non-specific allogeneic MLR response. These data suggest that in human systems, as in mouse, autologous peptide-pulsed DC are more effective at naïve T cell priming, with non-specific reactivity against alloantigens potentially restricting optimal generation of specific responses against a defined TAA over the course of CTL priming.
Fig. 6.

Naïve human CTL priming by peptide-pulsed DC. a Naïve human CTL were primed and restimulated with MART-1 peptide-pulsed HLA-A2+ autologous or allogeneic (i.e. matched only for A2) DC. CTL cytotoxicity was measured using a standard 4 h 51Cr release assay against target cells were pulsed with MART-1 or irrelevant peptide. Representative pairs of donors are shown from three paired experiments. Autologous DC priming against MART-1 (filled diamond), against irrelevant target (open triangle); allogeneic DC priming against MART-1 (filled square), against irrelevant target (open square). Total cell culture counts (b) and IFN-γ secretion (c) during naïve priming and restimulation of CTL by autologous (hatched histogram) or allogeneic DC (black histogram) were also measured. A representative pair of donors is shown from three separate paired experiments with similar results
Discussion
Vaccination using peptide-pulsed DC is an attractive strategy for cancer immunotherapy. With an increasing number of defined TAA epitopes recognised as capable of eliciting CD8 T cell reactivity in patients, the potential for specific vaccination against tumours in patients, with minimal toxicity, is rapidly expanding. Although mature DC pulsed with TAA epitopes have been clearly shown to activate specific responses in a range of pre-clinical and clinical settings, the optimal strategy for practical delivery to patients associated with therapeutic gain, has not currently been defined.
In this study we have addressed, for the first time, the question of whether co-expression of allogeneic MHC antigens enhances or impairs priming of anti-tumour immunity by peptide-pulsed DC. Potentially, allogeneic MHC antigens on vaccinating DC may be beneficial or detrimental with regard to T cell priming. The presence of allogeneic MHC antigens could act as a “danger signal” [27] to enhance protective immunity against tumours; alternatively, the associated rejection response might swamp critical therapeutic priming against a less dominant TAA. Data comparing autologous and allogeneic DC in other vaccination strategies (tumour cell/DC hybrids and intratumoural DC injection), has suggested that allogeneic DC are more effective [11, 14, 18, 28]. Since non-individualised DC are more practical for clinical preparation and delivery, a direct comparison of autologous and allogeneic DC (though matched for the class I allele presenting the therapeutic epitope ie “semi-allogeneic”) impacts both on choice of best therapy, and the feasibility of widespread application for patients.
Using a murine model of the ovalbumin H-2b-restricted SIINFEKL peptide pulsed onto autologous H-2b or semi-allogeneic F1 H-2bxk DC, we found unexpected discrepancies between in vitro presentation to specific OT1 cells, and in vivo naïve priming and anti-tumour protection. Characterisation of the two DC populations showed that the semi-allogeneic DC expressed lower overall levels of H-2b by MFI, but higher levels of class II and the co-stimulatory molecules required for effective T cell activation (Fig. 1). Such variation in phenotype between DC derived from different mouse strains has been reported previously [18, 26], and in our system was associated with greater semi-allogeneic DC-mediated proliferation of both allogeneic splenocytes (Fig. 1c) and, on SIINFEKL pulsing, of OT1 cells (Fig. 2). This data suggests that the presence of allogeneic class I molecules on DC does not prevent effective T cell activation in vitro, both non-specifically in an MLR, and specifically on peptide presentation to OT1 cells. The higher levels of co-stimulatory molecules on semi-allogeneic DC seem consistent with the greater observed activity in the MLR; the greater proliferation of OT1 cells, despite lower DC expression of H-2b, suggests that even in an antigen specific setting, facets of an APC other than expression levels of the specific, peptide-presenting class I molecule may be critical. In view of this in vitro data indicating that semi-allogeneic DC would be superior as peptide-pulsed in vivo vaccines, it was surprisingly the autologous DC that afforded greater protection against B16.OVA at two separate doses of live tumour cell challenge (Fig. 3).
This unexpected result raised the question of whether interactions between DC and T cells at different stages of T cell priming were critical in directing in vivo anti-tumour protection. Effective in vitro cross-talk between DC and OT1 cells may not translate to the naive in vivo setting, where DC must access rare naive antigen-specific T cell precursors and trigger their activation and expansion to a degree sufficient to protect against tumour challenge. The next set of experiments addressed this hypothesis, and confirmed a differential effect in the ability of autologous and semi-allogeneic DC to (i) prime a naïve CTL response (Fig. 4), and (ii) trigger proliferation of adoptively transferred OT1 cells in vivo (Fig. 5). Autologous DC were superior for CTL priming, correlating with anti-tumour protection, whilst semi-allogeneic DC supported greater OT1 in vivo proliferation, in line with their in vitro potency. Hence the early stage of naïve T cell priming appears to be the critical point at which autologous DC are most effective at initiating a response which can eventually protect against tumour challenge. One possible explanation for this effect is that during early DC/T cell interaction following vaccination with semi-allogeneic DC, the high frequency of allo-reactive T cells dominates the response, overwhelming any expansion of rare TAA-specific precursors; we are currently addressing this question further by monitoring the emergence over time of SIINFEKL-specific CTL in naïve vaccinated animals. The rapid elimination of semi-allogeneic DC may also restrict contact with naïve SIINFEKL-restricted responders, as suggested by a recent study which showed that semi-allogeneic DC persist for shorter periods of time in draining lymph nodes than autologous DC following vaccination [18].
It is tempting to speculate on why semi-allogeneic DC should be less effective for peptide-pulsed vaccination, but more potent when intratumoural injection of DC [19], or tumour cell/DC hybrids [17, 18, 28], are used for immunotherapy. One possibility is that in the context of intratumoural or hybrid vaccination, allogeneic MHC antigens do indeed provide a necessary “danger” signal, to overcome the recognised immunosuppressive effects of the whole tumour cells used as a source of TAA in these protocols. In contrast, on vaccination of naïve animals with peptide-pulsed DC, there is no such inhibition to overcome, potentially shifting the balance between tumour-induced suppression and non-specific allogeneic activation to the detriment of TAA-specific T cell priming.
Finally in this study, we also tested priming of a naïve TAA-specific CTL response in a human in vitro system. Using HLA-A2-restricted MART-1 as the TAA, we compared CTL priming using peptide-pulsed DC which were either fully autologous to responding T cells, or derived from a different A2 positive donor. Again, specific CTL priming in this naïve context was superior in a fully autologous system, despite greater overall proliferation of T cells and IFN-γ production in the allogeneic co-cultures (Fig. 6). Hence, the detrimental effects of allogeneic MHC antigen expression by peptide-pulsed DC on murine in vivo CTL and tumour protection also apply to naïve human T cell priming in vitro. This suggests that autologous human DC are preferable not only for peptide-pulsed vaccination strategies, but also for in vitro priming and expansion of T cells for subsequent therapeutic adoptive transfer.
In summary, we have shown that peptide-pulsed DC derived from an allogeneic donor are less effective than autologous DC for naïve T cell priming and anti-tumour protection in vivo. The contrasting potency of allogeneic DC at stimulating T cells which are already expanded and antigen-specific, suggests that during early interactions between DC and naïve precursors, co-expression of allogeneic MHC antigens on DC prevents successful TAA-specific T cell activation and growth. These data suggest that, despite the potential convenience of allogeneic DC for clinical application, they are suboptimal for peptide-pulsed vaccination strategies.
Acknowledgments
Grant support: Supported by the Medical Research Council, Cancer Research UK, Mayo Foundation and NIH grants RO1 CA85931 and RO1 CA94180.
We are grateful to Tim Kottke, Jill Thompson and staff at Biological Resources, Cancer Research UK laboratories (Clare Hall), for technical assistance.
Abbreviations
- TAA
Tumour associated antigens
- OVA
Ovalbumin
- APC
Antigen presenting cell
- MLR
Mixed lymphocyte reaction
- CFSE
Carboxyfluorescein diacetate succinamidyl ester
- GM-CSF
Granulocyte macrophage colony stimulating factor
Footnotes
Alison Merrick and Rosa Maria Diaz contributed equally to this work.
References
- 1.Banchereau J, et al. Immune and clinical outcomes in patients with stage IV melanoma vaccinated with peptide-pulsed dendritic cells derived from CD34+ progenitors and activated with type I interferon. J Immunother. 2005;28(5):505–516. doi: 10.1097/01.cji.0000171292.79663.cb. [DOI] [PubMed] [Google Scholar]
- 2.Guermonprez P, et al. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 2002;20:621–667. doi: 10.1146/annurev.immunol.20.100301.064828. [DOI] [PubMed] [Google Scholar]
- 3.Rescigno M, et al. Dendritic cell maturation is required for initiation of the immune response. J Leukoc Biol. 1997;61(4):415–421. [PubMed] [Google Scholar]
- 4.Smits HH, et al. Different faces of regulatory DCs in homeostasis and immunity. Trends Immunol. 2005;26(3):123–129. doi: 10.1016/j.it.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Ferlazzo G. Natural killer and dendritic cell liaison: recent insights and open questions. Immunol Lett. 2005;101(1):12–17. doi: 10.1016/j.imlet.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 6.Melcher A, et al. Dendritic cells for the immunotherapy of cancer. Clin Oncol (R Coll Radiol) 2002;14(3):185–192. doi: 10.1053/clon.2001.0038. [DOI] [PubMed] [Google Scholar]
- 7.Gong J, et al. Induction of antitumor activity by immunization with fusions of dendritic and carcinoma cells. Nat Med. 1997;3(5):558–561. doi: 10.1038/nm0597-558. [DOI] [PubMed] [Google Scholar]
- 8.Phan V, et al. A new genetic method to generate and isolate small, short-lived but highly potent dendritic cell-tumor cell hybrid vaccines. Nat Med. 2003;9(9):1215–1219. doi: 10.1038/nm923. [DOI] [PubMed] [Google Scholar]
- 9.Plautz GE, et al. Immunotherapy of malignancy by in vivo gene transfer into tumors. Proc Natl Acad Sci USA. 1993;90(10):4645–4649. doi: 10.1073/pnas.90.10.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Errington F, et al. Allogeneic tumor cells expressing fusogenic membrane glycoproteins as a platform for clinical cancer immunotherapy. Clin Cancer Res. 2006;12(4):1333–1341. doi: 10.1158/1078-0432.CCR-05-1113. [DOI] [PubMed] [Google Scholar]
- 11.Neves AR, et al. Dendritic cells derived from metastatic cancer patients vaccinated with allogeneic dendritic cell-autologous tumor cell hybrids express more CD86 and induce higher levels of interferon-gamma in mixed lymphocyte reactions. Cancer Immunol Immunother. 2005;54(1):61–66. doi: 10.1007/s00262-004-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hus I, et al. Allogeneic dendritic cells pulsed with tumor lysates or apoptotic bodies as immunotherapy for patients with early-stage B-cell chronic lymphocytic leukemia. Leukemia. 2005;19(9):1621–1627. doi: 10.1038/sj.leu.2403860. [DOI] [PubMed] [Google Scholar]
- 13.Holtl L, et al. Allogeneic dendritic cell vaccination against metastatic renal cell carcinoma with or without cyclophosphamide. Cancer Immunol Immunother. 2005;54(7):663–670. doi: 10.1007/s00262-004-0629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gong J, et al. Fusions of human ovarian carcinoma cells with autologous or allogeneic dendritic cells induce antitumor immunity. J Immunol. 2000;165(3):1705–1711. doi: 10.4049/jimmunol.165.3.1705. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka Y, et al. Vaccination with allogeneic dendritic cells fused to carcinoma cells induces antitumor immunity in MUC1 transgenic mice. Clin Immunol. 2001;101(2):192–200. doi: 10.1006/clim.2001.5112. [DOI] [PubMed] [Google Scholar]
- 16.Siders WM, et al. Induction of specific antitumor immunity in the mouse with the electrofusion product of tumor cells and dendritic cells. Mol Ther. 2003;7(4):498–505. doi: 10.1016/S1525-0016(03)00044-3. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki T, et al. Vaccination of dendritic cells loaded with interleukin-12-secreting cancer cells augments in vivo antitumor immunity: characteristics of syngeneic and allogeneic antigen-presenting cell cancer hybrid cells. Clin Cancer Res. 2005;11(1):58–66. [PubMed] [Google Scholar]
- 18.Wells JW, et al. Semi-allogeneic dendritic cells can induce antigen-specific T-cell activation, which is not enhanced by concurrent alloreactivity. Cancer Immunol Immunother. 2007;56(12):1861–1873. doi: 10.1007/s00262-007-0328-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirapu I, et al. Improving efficacy of interleukin-12-transfected dendritic cells injected into murine colon cancer with anti-CD137 monoclonal antibodies and alloantigens. Int J Cancer. 2004;110(1):51–60. doi: 10.1002/ijc.20093. [DOI] [PubMed] [Google Scholar]
- 20.Chakraborty NG, et al. Regulatory T-cell response and tumor vaccine-induced cytotoxic T lymphocytes in human melanoma. Hum Immunol. 2004;65(8):794–802. doi: 10.1016/j.humimm.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 21.Babatz J, et al. Induction of cellular immune responses against carcinoembryonic antigen in patients with metastatic tumors after vaccination with altered peptide ligand-loaded dendritic cells. Cancer Immunol Immunother. 2006;55(3):268–276. doi: 10.1007/s00262-005-0021-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wierecky J, et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer Res. 2006;66(11):5910–5918. doi: 10.1158/0008-5472.CAN-05-3905. [DOI] [PubMed] [Google Scholar]
- 23.Melcher A, et al. Adoptive transfer of immature dendritic cells with autologous or allogeneic tumor cells generates systemic antitumor immunity. Cancer Res. 1999;59(12):2802–2805. [PubMed] [Google Scholar]
- 24.Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5(11):1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- 25.Inaba K, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petersen MS, et al. Strain-specific variations in the development of dendritic cells in murine bone-marrow cultures. Scand J Immunol. 2000;51(6):586–594. doi: 10.1046/j.1365-3083.2000.00732.x. [DOI] [PubMed] [Google Scholar]
- 27.Gallucci S, Matzinger P. Danger signals: SOS to the immune system. Curr Opin Immunol. 2001;13(1):114–119. doi: 10.1016/S0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 28.Yasuda T, et al. Superior anti-tumor protection and therapeutic efficacy of vaccination with allogeneic and semiallogeneic dendritic cell/tumor cell fusion hybrids for murine colon adenocarcinoma. Cancer Immunol Immunother. 2007;56(7):1025–1036. doi: 10.1007/s00262-006-0252-5. [DOI] [PMC free article] [PubMed] [Google Scholar]