

The authors report relevant clinical information regarding the U.S. Food and Drug Administration approval of a new drug, omacetaxine mepesuccinate (Synribo), for the treatment of adult patients with chronic or accelerated phase chronic myeloid leukemia with resistance and/or intolerance to two or more tyrosine kinase inhibitors. Omacetaxine mepesuccinate has shown activity and a favorable benefit to risk profile for the studied population. Further evidence of response durability to verify clinical benefit is pending.
Keywords: FDA, Omacetaxine mepesuccinate, CML, Chronic myeloid leukemia
Abstract
On October 26, 2012, the U.S. Food and Drug Administration (FDA) granted accelerated approval to omacetaxine mepesuccinate (Synribo; Teva Pharmaceuticals USA, Inc., North Wales, PA, http://www.tevausa.com) for the treatment of adult patients with chronic phase (CP) or accelerated phase (AP) chronic myeloid leukemia (CML) with resistance and/or intolerance to two or more tyrosine kinase inhibitors (TKIs). The approval was based on the FDA review of data from 111 patients with CML in CP or in AP who had received two or more prior TKIs, including imatinib. Major cytogenetic response was achieved in 18% of patients with CP, with a median response duration of 12.5 months. Major hematologic response was achieved in 14% of patients with AP, with a median response duration of 4.7 months. The FDA safety evaluation was based on submitted data from 163 patients with CP or AP CML who had received at least one dose of omacetaxine mepesuccinate. The safety evaluation was limited by the single-arm design of the clinical trials as conducted in a small number of previously treated patients. The most common (≥20%) adverse reactions of any grade in enrolled patients included thrombocytopenia, anemia, neutropenia, diarrhea, nausea, fatigue, asthenia, injection site reaction, pyrexia, and infection. The FDA concluded that omacetaxine mepesuccinate has shown activity and a favorable benefit-to-risk profile for the studied population of adult patients with CML (CP or AP) with resistance and/or intolerance to two or more TKIs. Further evidence of response durability to verify clinical benefit is pending.
Implications for Practice:
We report relevant clinical information regarding the approval of a new drug, omacetaxine mepesuccinate (Synribo), for the treatment of adult patients with chronic or accelerated phase chronic myeloid leukemia with resistance and/or intolerance to two or more tyrosine kinase inhibitors. We discuss the disease, available treatment options, proposed mechanisms of action of omacetaxine mepesuccinate, clinical trial data supporting the accelerated approval of omacetaxine mepesuccinate, the approved indication as derived from trials conducted, drug storage and handling limitations, recommended dosing, method of administration, and expected toxicities.
Introduction
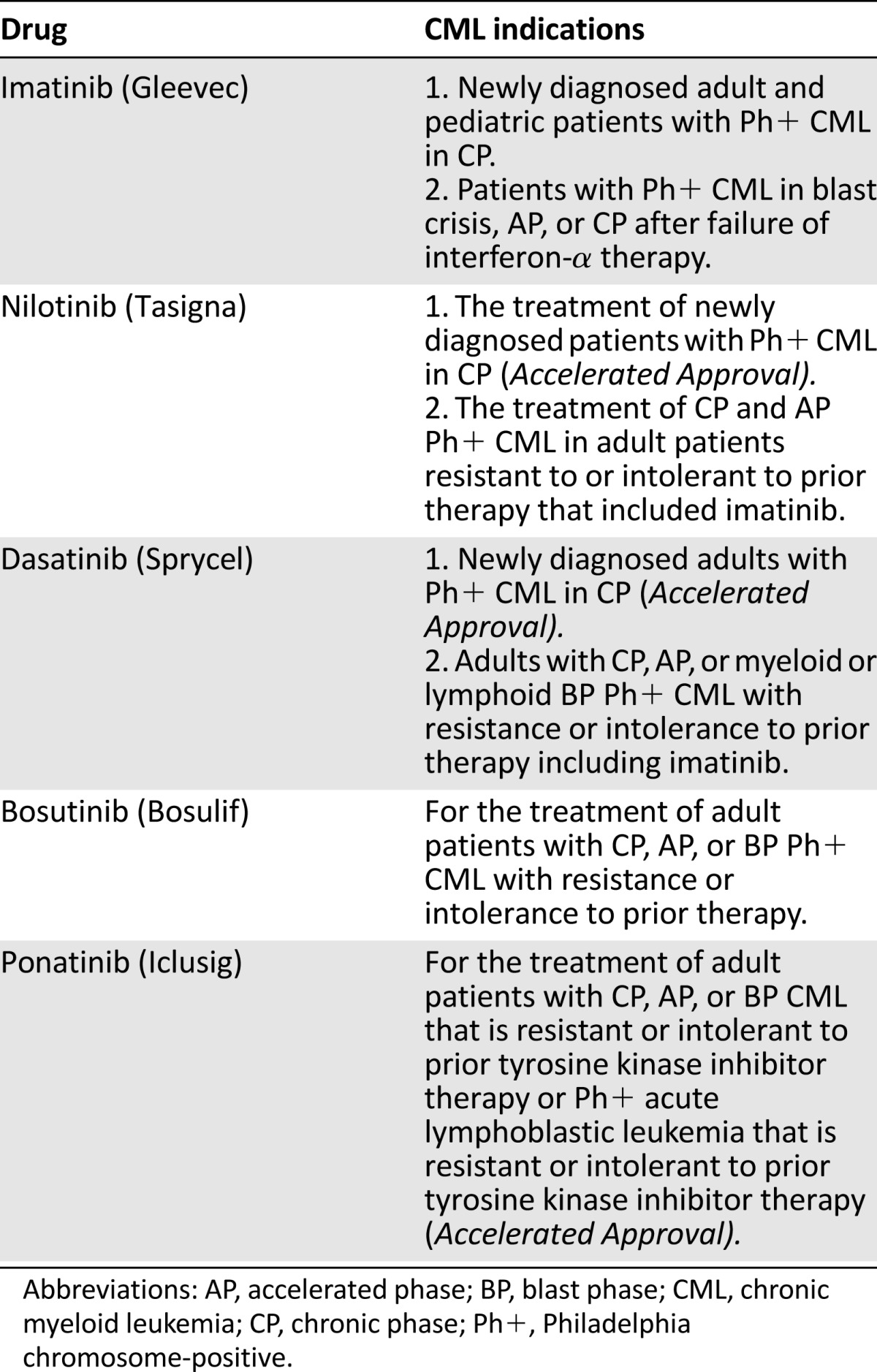
Chronic myeloid leukemia (CML) results from the neoplastic transformation of a hematopoietic stem cell, affecting all hematopoietic cell lineages. CML is characterized by the presence of the Philadelphia chromosome (a reciprocal translocation between the long arms of chromosomes 9 and 22, leading to formation of a BCR-ABL gene). The product of this translocation, BCR-ABL protein, is a constitutively active tyrosine kinase that results in the abnormal myelopoiesis in CML [1]. It is expected that in the U.S., there will be approximately 5,920 new cases of CML and 610 deaths in 2013. In the years between 2000 and 2009, the largest annual decline in death rate from cancer was for CML (8.4%) [2]. CML has an incidence of approximately 1–2 cases per 100,000 adults, accounting for approximately 15% of newly diagnosed leukemia in adults [2]. CML has three phases, representing a disease continuum from chronic phase (CP) to accelerated phase (AP) and to a final blast phase (BP). Transition from CP to AP and BP usually occurs gradually over a period of 1 year or longer, but a blast crisis may occur more rapidly. The disease is usually diagnosed in CP and later progresses to AP and then BP as the number of blasts in the blood and bone marrow increases [1]. Before the approval of the tyrosine kinase inhibitor (TKI) imatinib, the median survival of patients following a diagnosis of CP CML was 4-6 years. Survival after development of AP was typically <1 year and only a few months after BP. Eight-year follow-up data from the imatinib (IRIS) controlled trial demonstrated a survival rate of 85% for patients treated with imatinib [3]. Despite these improvements in CML treatment, approximately 30%–40% of patients receiving imatinib discontinue treatment after 5–8 years because of drug resistance or toxicity. Five TKIs are now approved for CML: imatinib, nilotinib, dasatinib, bosutinib, and ponatinib. Table 1 shows the approved TKI drugs that have received an indication for CML.
Table 1.
Approved treatments for CML
Patients with CML who are relapsed, refractory, or intolerant of at least two of the approved TKIs have limited treatment options and poor prognosis. Omacetaxine has been studied in this population.
On October 26, 2012, the U.S. Food and Drug Administration (FDA) granted accelerated approval for omacetaxine mepesuccinate (Synribo for injection, for subcutaneous use; Teva Pharmaceuticals USA, Inc., North Wales, PA, http://www.tevausa.com/) for the treatment of adult patients with chronic or AP CML with resistance and/or intolerance to two or more TKIs.
Based on the data available from the sponsor at the time of approval, the prescribing information for omacetaxine mepesuccinate states that the drug should be prepared in a health care facility and should be administered by a health care professional. Stability and sterility data require that the drug be administered within 12 hours of reconstitution when stored at room temperature and within 24 hours of reconstitution if stored at 2°C to 8°C.
We describe the FDA review process and the trial results supporting the accelerated approval.
Materials and Methods
Omacetaxine mepesuccinate is a semisynthetic formulation of homoharringtonine, a cytotoxic plant alkaloid extracted from various Cephalotaxus species (Fig. 1). The mechanism of action of omacetaxine mepesuccinate is distinct from TKIs [4] and includes inhibition of protein synthesis (by interfering with the initial protein elongation step), leading to cell death. Results from in vitro studies demonstrated that omacetaxine mepesuccinate treatment decreased levels of proteins important for leukemia cell survival, including both native and mutated forms of BCR-ABL and MCL1 (an antiapoptotic member of the BCL2 family of proteins). The action of omacetaxine mepesuccinate is not targeted, however, and can affect both normal and malignant hematopoietic cell types.
Figure 1.

Chemical structure of omacetaxine.
Synribo for injection is supplied as a sterile, preservative-free lyophilized powder in a single-use vial, containing 3.5 mg of omacetaxine mepesuccinate and mannitol. It is reconstituted with 1.0 mL of 0.9% sodium chloride and administered by subcutaneous injection.
Clinical Trial Population
The new drug application (NDA) consisted of data from a subset of patients with CP CML (CML-CP) and AP CML (CML-AP) who had already received two or more approved TKIs and had documented evidence of resistance or intolerance to dasatinib and/or nilotinib therapy from previous phase II nonrandomized, open-label, multicenter trials in adults with CML. The subset of patients from the two trials included 81 patients with CML-CP and 41 patients with CML-AP, combined to form the applicant’s efficacy evaluable population. This subset analysis was discussed with and agreed to by the FDA prior to the NDA submission. After excluding 3 patients from two sites because of data unreliability and an additional 8 patients for being characterized as responders at Synribo study entry, the final NDA study population consisted of a total of 76 patients with CML-CP and 35 patients with CML-AP. The safety-evaluable population consisted of all patients with CML-CP and CML-AP who received at least one dose of omacetaxine mepesuccinate in single-arm trials.
Therapy
Treatment in the clinical trials consisted of omacetaxine mepesuccinate 1.25 mg/m2 by subcutaneous injection given twice daily for 14 consecutive days every 28 days for up to six induction cycles. In the trials, some patients received omacetaxine mepesuccinate injections in their homes by a nurse or self-administered (after training on the procedure). Injections were to be rotated so that no site was used twice, with at least 3 cm of separation between sites. Those patients who achieved hematologic or cytogenetic response then received maintenance omacetaxine at 1.25 mg/m2 by subcutaneous injection twice daily for 7 consecutive days every 28 days. Modifications for toxicities included dosing schedule modification (delay and reduction of number of dosing days per cycle) without modification of the dose of omacetaxine mepesuccinate.
The primary efficacy endpoint for patients with CML-CP was the proportion of patients who achieved major cytogenetic response (MCyR), consisting of complete cytogenetic response (defined as evidence of 0% Philadelphia chromosome-positive cells) and partial cytogenetic response (defined as evidence of >0%–35% Philadelphia chromosome-positive cells). The primary efficacy endpoint for patients with CML-AP was the proportion of patients who achieved major hematologic response (MaHR), consisting of complete hematologic response (defined as absolute neutrophil count ≥1.5 × 109/L, platelet count ≥100 × 109/L, no blood blasts, bone marrow blasts <5%, and no extramedullary disease), or no evidence of leukemia (defined as morphologically leukemia-free state with <5% bone marrow blasts but lacking adequate recovery of neutrophils or platelets). In patients achieving complete hematologic response, complete cytogenetic response, or MCyR during the induction phase, the response was to be confirmed by a repeat complete blood count, bone marrow aspiration (for patients with a hematologic response), cytogenetics of the bone marrow aspirate (for patients with a cytogenetic response), and BCR-ABL transcript levels by quantitative reverse transcription polymerase chain reaction of peripheral blood at prespecified intervals. The efficacy results were adjudicated by a data monitoring committee.
Results
Efficacy
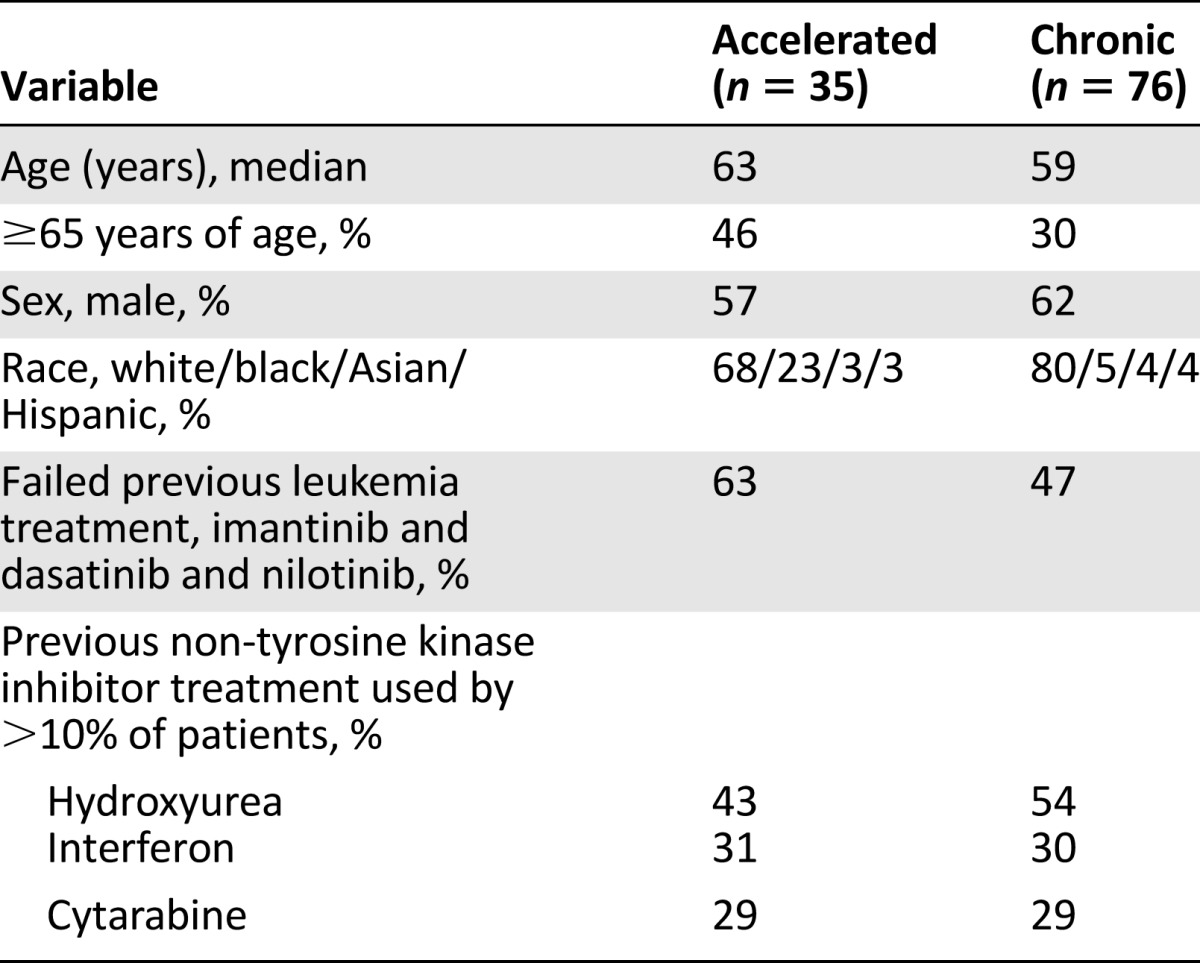
A total of 76 patients with CP CML and a total of 35 patients with AP CML were included in the efficacy analysis. Patient demographics are shown in Table 2.
Table 2.
Patient demographics
CML-CP
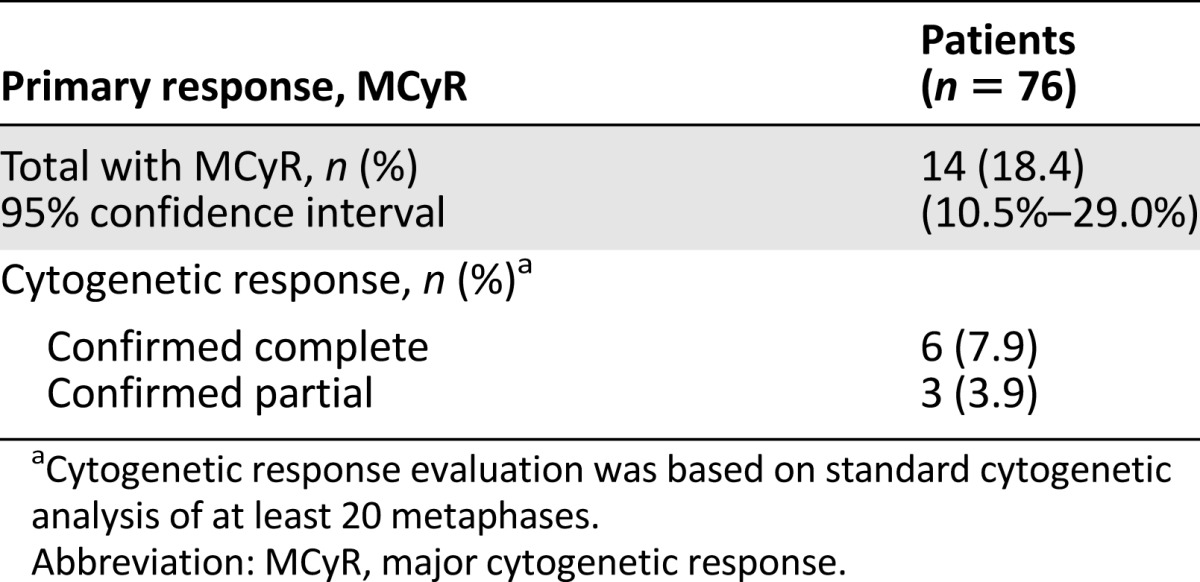
In the patients with CML-CP, 14 patients (18%) achieved MCyR. The mean time to MCyR response was 3.5 months, and the median duration of MCyR was 12.5 months by Kaplan-Meier estimate (Table 3). The median age of patients with CML-CP was 59 years; 30% were aged ≥65 years, 62% were male, 80% were white, 5% were black, 4% were Asian, and 4% were Hispanic. Thirty-six patients (47%) had failure of treatment with imatinib, dasatinib, and nilotinib. Most patients had also received prior non-TKI treatments, most commonly hydroxyurea (54%), interferon (30%), and/or cytarabine (29%).
Table 3.
Efficacy in chronic phase chronic myeloid leukemia
The median duration of exposure for the 108 patients with CML-CP was 7.4 months (range: 0–43 months). The median total cycles of exposure was 6 (range: 1–41 cycles), and the median total dose delivered during the trials was 131 mg/m2 (range: 1.2–678 mg/m2). During cycle 1, 87% of the patients with CML-CP received 14 days of treatment. By cycles 2 and 3, the percentage of patients receiving 14 days of treatment decreased to 42% and 16%, respectively. Ninety-one patients received at least two cycles of treatment, and of these, 79 patients (87%) had at least one cycle delay during the clinical trials. The median duration of cycle delay was greatest when more patients were receiving induction cycles, specifically prior to cycle 2 (17 days) and cycle 3 (25 days).
CML-AP
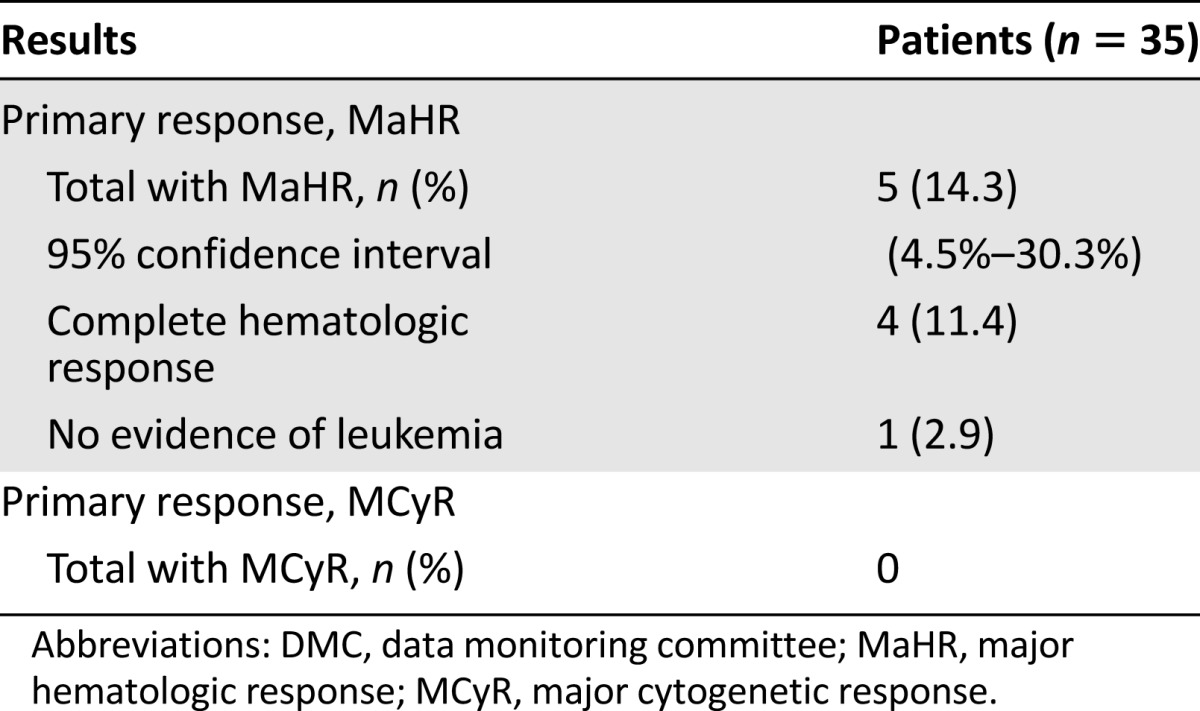
In patients with CML-AP, 5 patients (14%) experienced MaHR, with a mean time to response of 2.3 months and a median duration of response of 4.7 months by Kaplan-Meier estimate (Table 4). The efficacy population demographics for patients with CML-AP were median age of 63 years, 46% were aged ≥65 years, 57% were male, 68% were white, 23% were black, 3% were Asian, and 3% were Hispanic. Twenty-two (63%) of 35 patients with CML-AP had failure of treatment with imatinib, dasatinib, and nilotinib. Most patients had also received prior non-TKI treatments, most commonly hydroxyurea (43%), interferon (31%), and/or cytarabine (29%).
Table 4.
Efficacy results evaluated by DMC for patients with advanced phase chronic myeloid leukemia
The median duration of exposure for the 55 patients with CML-AP was 1.9 months (range: 0–30 months). Median total cycles of exposure was 2 (range: 1–29 cycles), and the median total dose delivered during the trials was 70 mg/m2. During cycle 1, 86% of the patients with CML-AP received 14 days of treatment. By cycles 2 and 3, the percentage of patients receiving 14 days of treatment decreased to 55% and 44%, respectively. Forty patients received at least two cycles of treatment, and of these, 27 patients (68%) had at least one cycle delay during the clinical trials. The median duration of cycle delay was greatest for cycle 3 (31 days) and cycle 8 (36 days).
Safety
The safety evaluation was based on 163 patients with CML-CP and CML-AP who had received at least one dose of omacetaxine mepesuccinate. Analyses of safety were conducted separately for the CP and AP patients. The safety profile of omacetaxine mepesuccinate in this population could not be fully evaluated because of the lack of a control arm in these single-arm trials. The lack of a control arm precluded a conclusive determination of the relationship of adverse events to omacetaxine mepesuccinate versus underlying disease.
CML-CP
Adverse reactions, regardless of investigator attribution, were reported for 99% of the patients with CML-CP. The most frequently occurring adverse reactions of any grade (>30%) were thrombocytopenia (74%), anemia (61%), neutropenia (50%), infections and infestations (46%), diarrhea (42%), infusion and injection site-related reactions (34%), and nausea (32%). A total of 87% of patients reported at least one grade 3 or grade 4 treatment-emergent adverse reaction. The most frequently reported grade 3 or 4 adverse reactions (≥10%) were thrombocytopenia (67%), neutropenia (45%), anemia (36%), lymphopenia (16%), infections and infestations (11%), bone marrow failure (10%), and febrile neutropenia (10%). A complete list of adverse reactions reported in at least 10% of patients is presented in Table 5. Serious adverse reactions were reported for 51% of patients. Serious adverse reactions reported for at least 5% of patients were bone marrow failure and thrombocytopenia (each 10%), infections (8%), and febrile neutropenia (6%).
Table 5.
Adverse reactions occurring in 15% of patients with CP CML
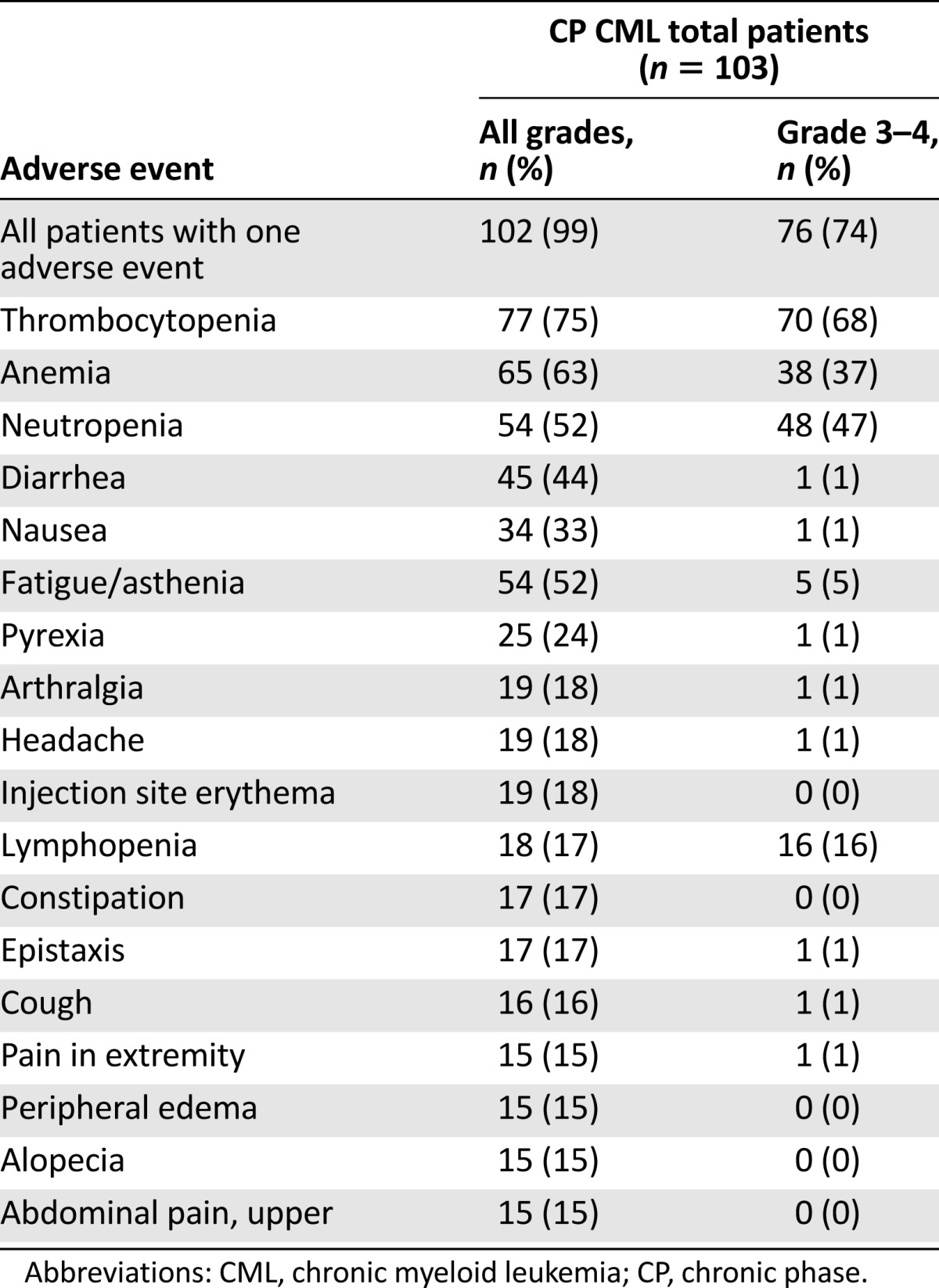
A total of 87% of patients reported at least one grade 3 or grade 4 treatment-emergent adverse reaction. The most frequently reported grade 3 or 4 adverse reactions (≥10%) were thrombocytopenia (67%), neutropenia (45%), anemia (36%), lymphopenia (16%), infections and infestations (11%), bone marrow failure (10%), and febrile neutropenia (10%).
The most frequently occurring adverse reactions leading to discontinuation were pancytopenia, thrombocytopenia, and increased alanine aminotransferase (each 2%). A total of 18% of patients had adverse reactions leading to withdrawal from omacetaxine mepesuccinate treatment. Five patients (5%) with CML-CP died within 30 days of last omacetaxine mepesuccinate dose. Of these deaths, two were from cerebral hemorrhage, one was from multiorgan failure, one was from disease progression, and one was from unknown causes.
CML-AP
Adverse reactions regardless of investigator attribution were reported for 100% of patients with CML-AP. The most frequently reported adverse reactions of any grade (>30%) were thrombocytopenia (56%), infections and infestations (56%), anemia (51%), diarrhea (35%), and fatigue (31%). A total of 84% of patients reported at least one grade 3 or grade 4 treatment-emergent adverse reaction. The most frequently reported grade 3 or grade 4 adverse reactions reported in at least 10% of patients were thrombocytopenia (49%), anemia (36%), infections and infestations (20%), neutropenia (18%), and febrile neutropenia (16%). A complete list of adverse reactions reported in at least 10% of patients is presented in Table 6.
Table 6.
Adverse reactions occurring in 15% of patients with AP CML
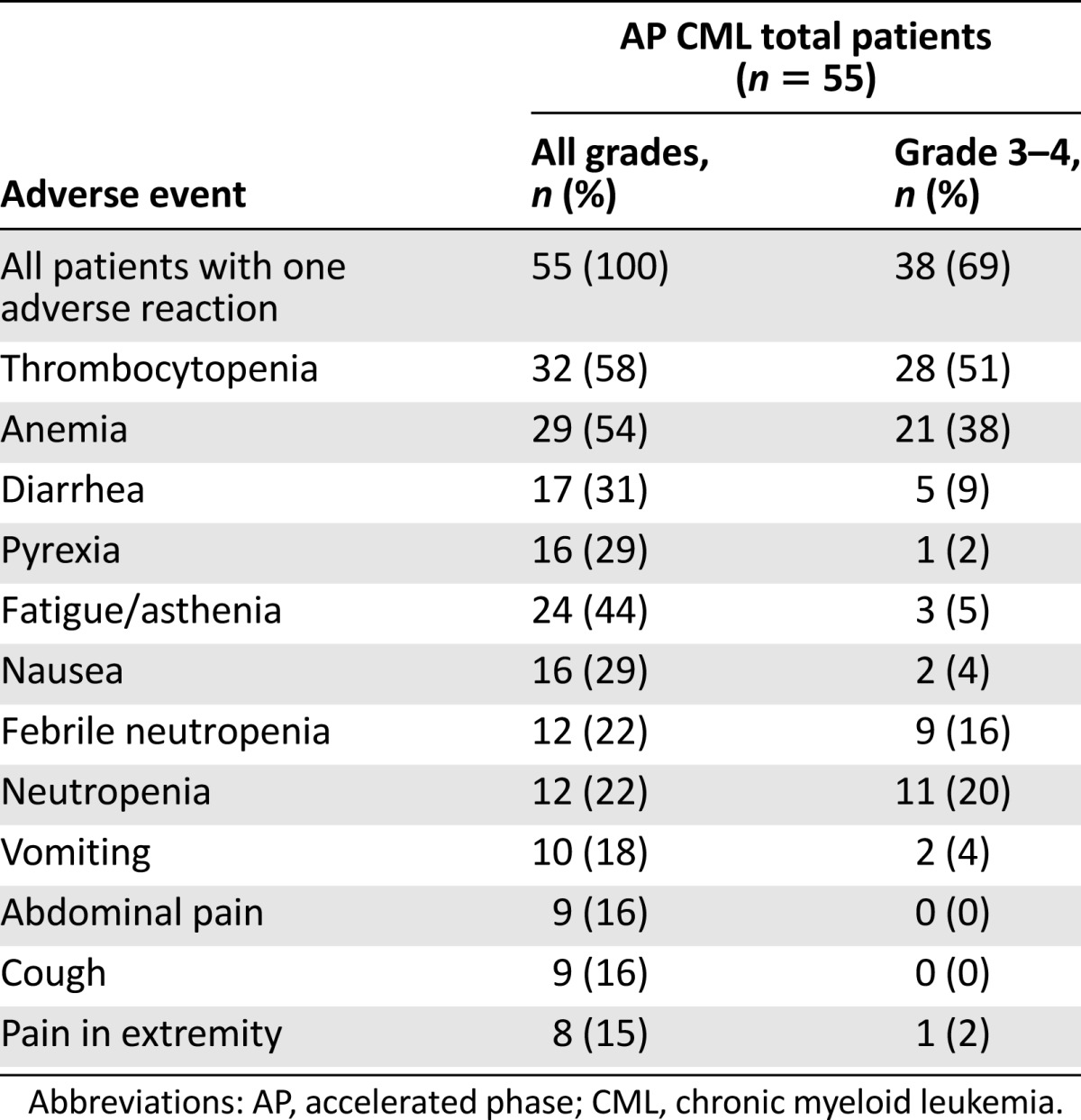
Serious adverse reactions were reported for 60% of patients with CML-AP. Serious adverse reactions reported for at least 5% of patients were febrile neutropenia (18%), infections (11%), thrombocytopenia (9%), anemia (7%), and diarrhea and convulsions (6% each). A total of 33% of patients had adverse reactions leading to discontinuation of omacetaxine mepesuccinate treatment. The most frequently occurring adverse reactions leading to withdrawal were leukocytosis (6%) and thrombocytopenia (4%). Five patients (9%) with CML-AP died within 30 days of the last omacetaxine mepesuccinate dose. Of these deaths, two were attributed to cerebral hemorrhage and three were attributed to disease progression.
Clinical Pharmacology
An exposure-response or -safety relationship was not established for omacetaxine mepesuccinate, and dose proportionality is unknown. Peak plasma concentrations of omacetaxine mepesuccinate were reached approximately 30 minutes after subcutaneous injection. In vitro, omacetaxine mepesuccinate is primarily hydrolyzed to an inactive metabolite via plasma esterases with little hepatic microsomal oxidative and/or esterase-mediated metabolism. On average, <15% of omacetaxine mepesuccinate is excreted unchanged in the urine. The mean half-life of omacetaxine mepesuccinate is approximately 6 hours following multiple dosing. Omacetaxine mepesuccinate is a substrate of P-glycoprotein. Omacetaxine mepesuccinate and its metabolite do not inhibit major cytochrome P450 enzymes or P-glycoprotein. The potential of omacetaxine mepesuccinate or its metabolite to induce cytochrome P450 enzymes has not been determined conclusively. Additional clinical drug-interaction trials were not warranted. No QT-prolonging effects of omacetaxine mepesuccinate (1.25 mg/m2 twice daily) were detected in an open-label single-arm trial in patients with advanced cancer.
Special Safety Studies
The applicant proposed in-home patient reconstitution and self-injection for omacetaxine mepesuccinate. Successful and safe administration of omacetaxine mepesuccinate requires correct performance of the following steps: preparation, reconstitution, injection, and disposal. The applicant developed Instructions for Use (IFU) to communicate the details of the four steps to patients and sponsored a labeling comprehension study to evaluate the readability and understanding of the IFU, to measure the ability of patients to correctly prepare a dose of omacetaxine mepesuccinate, and to identify potential areas of improvement if necessary for the IFU. The labeling comprehension study was conducted in 25 subjects of similar age and gender distribution to the proposed population of patients with CML. In this study, subjects were given a demonstration of proper omacetaxine mepesuccinate dose preparation by a chemotherapy-certified registered oncology nurse, after which subjects were to prepare a dose with the aid of the IFU. The research moderator intervened if needed to correct the subjects. In this study, 72% of subjects successfully performed all key steps of the dose preparation without intervention or assistance. An additional 8% performed the dose preparation with one corrective intervention by the research moderator. Twelve percent (three subjects) could not perform the dose preparation (including two of the three low-literacy subjects). An additional study was proposed but could not be completed and reviewed during the review cycle.
The study also evaluated whether proper dose preparation actually occurred as prepared by the subjects. The results identified that 15 of 21 prepared doses (71%) would have an effective dose equivalent to 11% or more than prescribed—a potential overdose of unknown consequences. The percent deviation of the prepared dose ranged from −9% to +25%. The review team did not agree that the results from this study supported the safety of in-home reconstitution and administration of omacetaxine mepesuccinate.
There is no precedent for approval of home reconstitution and self-administration of a parenteral cytotoxic chemotherapeutic agent. The results of the labeling comprehension study raised concerns that home reconstitution and self-administration could introduce further risk to patients as well as to other residents in the home (including pregnant women and children). These risks involved improper dosing to patients or any exposure at all to family members through the necessary refrigeration or spillage onto home surfaces during reconstitution or dosing steps.
There is no precedent for approval of home reconstitution and self-administration of a parenteral cytotoxic chemotherapeutic agent. The results of the labeling comprehension study raised concerns that home reconstitution and self-administration could introduce further risk to patients as well as to other residents in the home (including pregnant women and children).
Discussion
The accelerated approval of omacetaxine mepesuccinate for treatment of adult patients with CML-CP or CML-AP with resistance and/or intolerance to two or more TKIs was based on review of a median of 19.5 months (95% confidence interval: 14.4–23.1) and 11.5 months (95% confidence interval: 6.8–16) of follow-up data for the primary endpoints for the CML-CP and CML-AP populations, respectively. This approval was based on response rate, and there are no trials verifying an improvement in disease-related symptoms or increased survival with omacetaxine mepesuccinate. On availability of 24 months of follow-up data, the FDA will consider conversion of this accelerated approval to a regular approval. This approach is consistent with prior approvals of other drugs for the treatment of CML.
Parenteral chemotherapy agents are typically reconstituted in a ventilated cabinet that prevents harmful fumes from sickening the preparer (typically a pharmacist or pharmacy technician) and containing spills that could contaminate the environment [4]. Patient self-reconstitution and administration of omacetaxine mepesuccinate require further data in support of the safety of home reconstitution, the ability of patients to prepare the correct dosing, and the safety of drug self-administration by the patient in the home setting. The review team did not agree that parenteral chemotherapeutic agents should be prepared in the patient’s home by the patient (or other lay caregiver) without the benefit of specialized training and a ventilated cabinet to prevent exposure by anyone present in the household to potentially cytotoxic agents [5]. These concerns resulted in product labeling that specifies that omacetaxine mepesuccinate should be prepared in a health care facility and administered by a health care professional. Labeling the product in this way meant that patients would be required to come to a medical facility daily for up to 14 days for the reconstitution and first dose as well as having the second dose of the day administered by a home care nurse. These limitations may be revised in the future on review of data that support an alternative administration method.
Conclusion
The FDA concluded that omacetaxine mepesuccinate has shown activity and a favorable benefit to risk profile for the population of adult patients with CML-CP or CML-AP who are resistant and/or intolerant to two or more TKIs. Omacetaxine mepesuccinate represents the first drug approved for CML-CP and CML-AP patients who are resistant and/or intolerant to two or more TKIs.
Author Contributions
Conception/Design: Virginia E. Kwitkowski, Firoozeh Alvandi, Chia-Wen Ko, Mark D. Rothmann, Edvardas Kaminskas, Ann T. Farrell, Richard Pazdur
Collection and/or assembly of data: Firoozeh Alvandi, Chia-Wen Ko, Mark D. Rothmann, Edvardas Kaminskas, Ann T. Farrell, Richard Pazdur, Stacey Ricci, Debasis Ghosh, Janice Brown, Erika Pfeiler, Elsbeth Chikhale, Joseph Grillo, Julie Bullock
Data analysis and interpretation: Virginia E. Kwitkowski, Firoozeh Alvandi, Chia-Wen Ko, Mark D. Rothmann, Edvardas Kaminskas, Ann T. Farrell, Richard Pazdur, Robert Kane, Debasis Ghosh, Janice Brown, Erika Pfeiler, Elsbeth Chikhale, Joseph Grillo, Julie Bullock
Manuscript writing: Virginia E. Kwitkowski, Firoozeh Alvandi, Chia-Wen Ko, Mark D. Rothmann, Edvardas Kaminskas, Ann T. Farrell, Richard Pazdur, Stacey Ricci, Haleh Saber, Robert Kane
Final approval of manuscript: Virginia E. Kwitkowski, Firoozeh Alvandi, Chia-Wen Ko, Mark D. Rothmann, Edvardas Kaminskas, Ann T. Farrell, Richard Pazdur, Haleh Saber, Robert Kane
Disclosures
The authors indicated no financial relationships.
References
- 1.Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2012 update on diagnosis, monitoring, and management. Am J Hematol. 2012;87:1037–1045. doi: 10.1002/ajh.23282. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 3.Deininger M, O’Brien S, Guilhot F, et al. International randomized study of interferon vs ST1571 (IRIS) 8-year follow up: Sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood. 2009;114:1126a. [Google Scholar]
- 4.Quintás-Cardama A, Kantarjian H, Cortes J. Homoharringtonine, omacetaxine mepesuccinate, and chronic myeloid leukemia circa 2009. Cancer. 2009;115:5382–5393. doi: 10.1002/cncr.24601. [DOI] [PubMed] [Google Scholar]
- 5. Occupational Safety and Health Administration. OSHA technical manual (OTM), section IV: Chapter 2. Controlling occupational exposure to hazardous drugs. Available at http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html. Accessed March 1, 2013.